

Abstract
Objective
Neointimal hyperplasia characterized by abnormal accumulation of vascular smooth muscle cells (SMCs) is a hallmark of occlusive disorders such as atherosclerosis, post-angioplasty restenosis, vein graft stenosis, and allograft vasculopathy. Cyclic nucleotides are vital in SMC proliferation and migration, which are regulated by cyclic nucleotide phosphodiesterases (PDEs). Our goal is to understand the regulation and function of PDEs in SMC pathogenesis of vascular diseases.
Methods & Results
We performed screening for genes differentially expressed in normal contractile versus proliferating synthetic SMCs. We observed that PDE1C expression was low in contractile SMCs but drastically elevated in synthetic SMCs in vitro and in various mouse vascular injury models in vivo. Additionally, PDE1C was highly induced in neointimal SMCs of human coronary arteries. More importantly, injury-induced neointimal formation was significantly attenuated by PDE1C deficiency or PDE1 inhibition in vivo. PDE1 inhibition suppressed vascular remodeling of human saphenous vein explants ex vivo. In cultured SMCs, PDE1C deficiency or PDE1 inhibition attenuated SMC proliferation and migration. Mechanistic studies revealed that PDE1C plays a critical role in regulating the stability of growth factor receptors, such as PDGF-receptor-beta (PDGFRβ) known to be important in pathological vascular remodeling. PDE1C interacts with LDL-receptor-related-protein-1 (LRP1) and PDGFRβ, thus regulating PDGFRβ endocytosis and lysosome-dependent degradation in an LRP1-dependent manner. A transmembrane-adenylyl-cyclase (tmAC)-cAMP-PKA cascade modulated by PDE1C is critical in regulating PDGFRβ degradation.
Conclusion
These findings demonstrated that PDE1C is an important regulator of SMC proliferation, migration, and neointimal hyperplasia, in part through modulating endosome/lysosome dependent PDGFRβ protein degradation via LRP1.
Keywords: cyclic nucleotide, phosphodiesterase, smooth muscle cells, and neointimal hyperplasia
Introduction
Intimal hyperplasia and lumen stenosis are the key characteristics of a number of different vascular disorders, such as atherosclerosis, post-angioplasty restenosis, vein graft stenosis, and allograft vasculopathy 1-3. Under normal conditions, SMCs residing in the media of vessels are quiescent with a very low turnover rate and insignificant secretory activity. These SMCs are highly differentiated cells that possess a contractile phenotype by expressing large amounts of contractile proteins, and principally function to maintain vascular tone. However, SMCs also retain a degree of plasticity to allow phenotypic modulation. For example, during vascular injury, SMCs undergo profound metamorphosis changing from a quiescent/contractile phenotype to an active/synthetic phenotype 4. Synthetic SMCs down-regulate contractile proteins and up-regulate growth factors, growth factor receptors, extracellular matrix (ECM) components, ECM proteinases and inflammatory mediators 5, 6. Inhibiting intimal SMC-like cell proliferation has been used as a therapeutic strategy to antagonize pathologic vascular remodeling. To prevent restenosis after percutaneous coronary intervention, the most effective therapy is local delivery of anti-proliferative reagents via drug-eluting stents (DES), containing drugs such as sirolimus 7 and paclitaxel 8. These drugs inhibit cell proliferation by targeting mTOR or microtubule formation. However, these drugs also attenuate re-endothelization, and can lead to increased in-stent thrombosis 9. In addition, although DES is effective for focal lesions, DES cannot treat vascular disorders with diffuse neointimal lesions. Thus, developing novel and systemically safe drugs is currently in high demand.
Endothelial dysfunction or damage triggers underlying SMC phenotype transition and pathological vascular wall remodeling. Prostacyclin (PGI2) and nitric oxide (NO), two major factors released from the healthy endothelium, stimulate the production of cAMP and cGMP, respectively, in adjacent SMCs. Cyclic AMP and cGMP have a variety of biological effects in vascular SMCs, such as promoting SMC relaxation and inhibiting SMC proliferation, migration, and ECM synthesis 10, 11. Cyclic nucleotide phosphodiesterases (PDEs), by catalyzing the hydrolysis of cAMP and cGMP to 5′AMP and 5′GMP, regulate the amplitude, duration, and compartmentalization of intracellular cyclic nucleotide signaling. Alterations of PDE expression/activation have been implicated in a number of diseases 12, 13. To date, more than 60 different PDE isoenzymes derived from 22 genes have been identified and grouped into 11 broad families (PDE1-PDE11) based on distinct kinetic, regulatory, and inhibitory properties 14. PDEs are expressed in a cell/tissue-specific manner and only a few enzymes are expressed in any single cell type. Importantly, different PDE isoforms serve to control distinct cyclic nucleotide signaling and fulfill distinct functions. Over the past decades, PDEs have been proven to be ideal and feasible drug targets, as exampled by drugs such as sildenafil, milrinone, cilostazol, and roflumilast. Thus, selectively targeting individual PDE isoforms may represent a feasible and appealing strategy for modulating specific cyclic nucleotide pools without affecting global intracellular cyclic nucleotides.
To understand the specific cyclic nucleotide signaling pathway responsible for the synthetic SMC phenotype, we performed initial discovery screening for PDE isozymes that are differentially expressed in synthetic SMCs compared to contractile SMCs. We found that the PDE1C isozyme is markedly upregulated in synthetic SMCs. Consistent with in vitro findings, PDE1C is nearly undetectable in medial SMCs of normal arteries and veins in vivo, but is markedly induced in synthetic SMC-like cells of vascular lesions from various animal injury models and human disease vessels. PDE1C belongs to the calcium/calmodulin (Ca2+/CaM)-stimulated PDE family comprising 3 gene products: PDE1A, 1B, and 1C 15. In the vasculature, PDE1 activity is primarily associated with SMCs but not endothelial cells 16, 17, suggesting a specific role of PDE1C in synthetic SMCs. We also used in vitro and in vivo approaches to prove that PDE1C plays a causative role in synthetic SMC proliferation/migration and neointimal hyperplasia. Furthermore, we identified the molecular mechanism by which PDE1C promotes the protein stability of growth factor receptors via negatively regulating endosome/lysosome-mediated degradation. Our data suggest that PDE1C may represent a novel therapeutic target for treating cardiovascular diseases associated with SMC hyperplasia.
Methods
Animal care and use was in accordance with institutional guidelines. The global PDE1C knockout mice were kindly provided by Haiqing Zhao (Johns Hopkins University) and backcrossed to C57BL/6 mice for at least 9 generations. FVB/NJ mice were obtained from Jackson Laboratories. Carotid artery intima/media thickening was induced in vivo by blood flow cessation through complete ligation of the left common carotid artery for two weeks 18. For the in vivo animal study with PDE1 inhibitor (IC86340), the compound was applied perivascularly through pluronic gel. Human saphenous veins were collected from discarded unused portions in coronary artery bypass surgeries, and were cultured in vitro for 7days in the presence of vehicle or IC86340.
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Results
PDE1C is Highly Expressed in Synthetic/Proliferative SMCs in vitro
To identify the PDE isoforms differentially expressed in synthetic versus contractile SMCs, we performed preliminary discovery screening among all PDE genes in contractile SMCs (freshly isolated medial layers) and synthetic SMCs (cultured SMCs) via qRT-PCR. We found that PDE1C expression is selectively associated with synthetic SMCs (data not shown). To confirm this finding, we analyzed PDE1C expression in isolated contractile and synthetic SMCs from rat aortas (Fig. 1A), human aortas (Fig. 1B), and human saphenous veins (Fig. 1C). As expected, SMC markers such as smooth muscle myosin heavy chain (SM-MHC) and calponin are drastically decreased in synthetic SMCs compared to contractile medial SMCs (Fig. 1A-C, middle and right panels), providing validation of a phenotype change. Importantly, we observed a marked increase of PDE1C in all three types of growing synthetic SMCs compared to corresponding contractile SMCs from different species and different vascular beds (Fig. 1A-C, right panels). Consistently, PDE1C protein levels were also increased in synthetic SMCs (Supplemental Fig. S1). Our results corroborate previous findings demonstrating that PDE1C was highly expressed in human aortic proliferating SMCs but is not detected in quiescent SMCs isolated from the tunica media 19, 20.
Figure 1. PDE1C expression is drastically upregulated in synthetic SMCs.
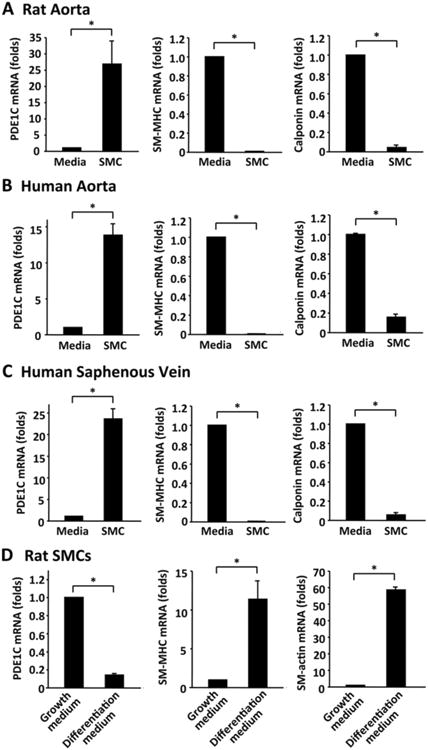
qRT-PCR results showing mRNA levels of PDE1C, SM-MHC and calponin mRNA in contractile SMCs (freshly isolated medial layers) and corresponding synthetic SMCs (cultured SMCs) from rat aortas (A), human aortas (B), and human saphenous veins (C). Contractile SMCs are freshly isolated medial tissues procured by removing endothelial cells and pealing off adventitial layers. Synthetic SMCs are cultured growing SMCs isolated from the corresponding vessel with the explant method. (D) Cultured rat aortic SMCs were in differentiation medium (medium 231 supplemented with Smooth Muscle Growth S (SMGS), from Cascade Biologics) or growth medium (medium 231 supplemented with Smooth Muscle Differentiation Supplement (SMDS), from Cascade Biologics) for 2 days. SM-MHC and calponin are used as contractile SMC markers. Values are mean ± SD of at least four repeats. *P < 0.05.
To further demonstrate the phenotype-dependent expression of PDE1C, we used in vitro models of SMC phenotype modulation via differentiation- and growth-medium as previously described 21. We found that PDE1C was downregulated about 80% when cells were grown in the differentiation-medium compared to growth-medium (Fig. 1D, left panel). Concurrent phenotype modulation was verified by a drastic increase in the SMC contractile marker protein SM-MHC and calponin in the differentiation-medium (Fig. 1D, middle and right panel). This observation is consistent with the previous finding that PDE1C was significantly down-regulated in SMCs cultured on dishes coated with fibrillar/polymer type I collagen (eliciting a contractile-like phenotype) compared to SMCs cultured on non-coated plastic dishes (exhibiting a synthetic phenotype) 20.
PDE1C is Highly Induced in SMC-like cells in Rodent and Human Disease Vessels
To demonstrate the induction of PDE1C in vivo, we first analyzed PDE1C via Immunohistochemical staining in neointimal lesions from different mouse injury models, including carotid artery ligation, femoral artery wire injury, and vein bypass grafting (Fig. 2A-C). The adjacent sections were immunostained with anti-PDE1C and anti-smooth muscle alpha actin (SM-α-actin, a SMC marker) (Fig. 2A-C, middle panels). Carotid artery intima thickening was induced by complete ligation of left carotid arteries for two weeks and contralateral right carotid arteries were used as control vessels. As shown in Figure 2A, PDE1C expression was very low or almost undetectable in the normal control right carotid artery (inset), but was significantly increased in the neointimal and medial areas of the ligated left carotid artery (left and right panels). We also examined PDE1C expression in a mouse model of femoral artery with wire injury 22. As shown in Figure 2B, PDE1C staining was significantly increased in the neointimal and medial areas of the injured left femoral artery (right and left panels) compared to the non-injured right femoral artery (inset). Consistently, PDE1C mRNA was also quantitatively higher in injured compared to uninjured femoral arteries (Supplemental Figure S2A). Moreover, we examined PDE1C in venous neointimal lesions from a mouse model of vein bypass graft with vena cava-to-carotid artery isografting 23. As shown in Figure 2C, we observed a marked induction of PDE1C expression in the graft neointimal lesions (right and left panels) compared to non-grafted veins (insets). The majority of neointimal cells are stained with SM-α-actin and PDE1C, suggesting that PDE1C is induced in the SMC-like cells during vascular pathologic states. The immunofluorescent double-staining of PDE1C and SM-α-actin further indicates that PDE1C is highly induced in SMC-like cells in diseased mouse carotid artery (Supplemental Fig. S2B).
Figure 2. PDE1C expression is markedly induced in injured vessels of different mouse models as well as human disease vessels.
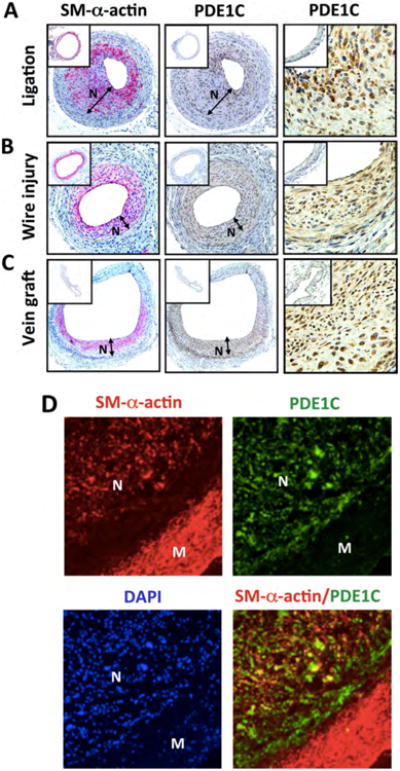
(A-C) Representative immunohistochemistry images showing SM-α-actin and PDE1C staining in injured and uninjured control (inset) vessels from different mouse injury models, including (A) left carotid artery with partial ligation for 14 days in FVB mice or right carotid artery without ligation, (B) left femoral artery with wire injury for 28 days in C57BL/6 mice or right femoral artery without injury, and (C) vena cava subjected to isografting, wherein vena cava was obtained from a donor C57BL/6 mouse and grafted between two ends of the right common carotid artery in a recipient C57BL/6 mouse for 4 weeks. Red: SM-α-actin, Brown: PDE1C, blue: nuclear counterstaining with Hematoxylin. (D) Representative immunofluorescent double-staining images of human coronary artery with neointimal lesions. Red: SM-α-actin, Green: PDE1C, Blue: nuclei stained with DAPI.
To support the results obtained from murine injury models, cross-sections from human coronary arteries with neointimal lesions were immunofluorescently double-stained with PDE1C and SM-α-actin. We observed that PDE1C expression was low in medial SMCs, but was highly elevated in the neointimal cells and largely overlapped with SM-α-actin positive cells (Fig. 2D). These in vivo observations are in line with the findings from cultured SMCs in vitro (Fig. 1). Taken together, our results suggest that PDE1C expression is associated with synthetic SMCs.
PDE1C Ablation Attenuates Neointimal Hyperplasia Following Vascular Injury
To determine the causative role of PDE1C in neointimal hyperplasia and pathological vascular remodeling, we used global PDE1C knockout (PDE1C-/-) mice with backcrossing to C57BL/6 mice for at least 9 generations. This knockout line has normal growth rates and feeding patterns, as well as normal nursing and mating behaviors 24. There are no apparent morphological and histological abnormalities in tissues examined, including aorta, brain, fat, heart, intestine, kidney, liver, lung, ovary, pancreas, skeletal muscle, spleen, and testes (data not shown). We examined the effects of PDE1C depletion on carotid remodeling induced by complete carotid artery ligation, a procedure known to induce intimal hyperplasia caused by blood flow cessation 18, 25. As shown in Figure 3A, there is no obvious change in the appearance of unligated right carotid artery (RCA) between PDE1C+/+ and PDE1C-/- mice (insets). Ligation of the left carotid artery (LCA) for 2 weeks developed more significant vascular wall thickening in PDE1C+/+ mice compared to PDE1C-/- mice (Fig, 3A). Morphometric analyses revealed that the ligation injury caused a marked increase in neointimal and medial thickening in PDE1C+/+ mice (Fig. 3B). However, these changes were largely attenuated in PDE1C-/- mice. Because SMC proliferation contributes to neointima hyperplasia, we conducted immunostaining of Ki67 (a marker of cellular proliferation). SMCs were counterstained with SM-α-actin. There was a significant increase in the number of Ki67-positive SMCs in ligated LCA compared to control RCA in PDE1C+/+ mice, which was significantly decreased in PDE1C-/- mice (Supplemental Fig. S3A). Because reactive oxidative stress (ROS) plays a crucial role in the development of vascular diseases, we measured lipid oxidation by immunostaining of 4-Hydroxy-2-Nonenal (4-HNE). We showed that 4-HNE staining intensity per area is significantly decreased in the media-intima of carotid arteries from PDE1C-/- mice (Supplemental Figure S3B).
Figure 3. PDE1C deficiency or PDE1 inhibition attenuates pathological vascular remodeling.
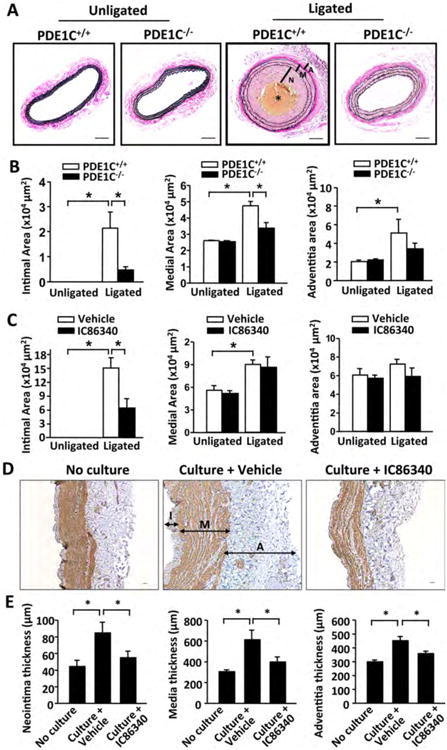
(A-B) Effect of PDE1C deficiency on carotid artery intima/media thickening. A, representative Verhoeff-van Gieson (VVG) staining images of carotid artery cross-sections from PDE1C+/+ and PDE1C-/- mice subjected to left common carotid artery ligation for 14 days. The right common carotid arteries were used as unligated controls (insets). Bar = 50 μm. Blue-black indicates elastic fibers. The middle yellowish part (indicated by asterik) is the blood clot. B, quantitative data of morphometric analyses of neointimal and medial areas measured using ImagePro software. N, neointima; M, media; A: adventitia. Values are mean ± SEM (n=7 for each group). C, Quantitative morphometric data showing the effect of IC86340 on carotid artery intima/media thickening. Values are mean ± SEM (n=8 for each group). (D-E) Effects of PDE1 inhibitor IC86340 on human saphenous vein remodeling. D, Representative images of HSV sections with immunostaining for SM-α-actin (brown). E, quantification of intimal, medial, and adventitial thickness. HSV explants were either no culture or subjected to ex vivo culture for 7 days in the presence of vehicle or IC86340 (30 μM). Bar = 50 μm. Values are mean ± SEM (n=6). I: intima; M: media; A: adventitia. *P < 0.05.
In addition, we tested the effect of a pan-PDE1 inhibitor IC86340 on carotid artery remodeling in FVB mice. IC86340 inhibits all PDE1 isozymes although it is more potent for PDE1C inhibition. IC86340 or vehicle was applied perivascularly through pluronic gel to carotid arteries 26. As shown in Figure 3C, ligation of the artery induced a drastic neointima formation in FVB mice, which was significantly reduced by application of IC86340. The moderate change of media thickness was independent of IC86340. Ki67-positive SMC numbers or 4-HNE staining intensity were also reduced by IC86340 in ligated LCA compared to control RCA (Supplemental Fig. S3C and D). These data indicate that PDE1C plays a critical role in neointimal hyperplasia in response to flow-induced vascular injury.
PDE1 Inhibition Attenuates Human Saphenous Vein Remodeling Ex Vivo
To determine the role of PDE1 in human vascular remodeling, we utilized human saphenous vein (HSV) samples. HSV is used for bypassing stenotic coronary arteries, but late vein graft failure occurs due to remodeling of the vessel wall and the development of stenosis 27, 28. When cultured ex vivo, HSV spontaneously undergoes remodeling, which predominantly involves SMC growth, migration, and extracellular matrix 29. As shown in Figure 3D and E, after HSV were cultured ex vivo for 7 days, the thickness of intimal, medial and adventitial layers was markedly increased compared to the same vessels without culture. PDE1 inhibitor IC86340 significantly reduced HSV remodeling in all three layers. This suggests an important role for PDE1 in human vascular remodeling disorders.
PDE1C Deficiency or PDE1 Inhibitor Antagonizes SMC Proliferation and Migration
Proliferation and migration of SMCs are critical steps in neointimal formation after vascular injury. To examine the role of PDE1C in SMC proliferation, we performed SRB assay (a well-established colorimetric cell viability and proliferation assay) in primary cultured mouse SMCs isolated from PDE1C+/+ and PDE1C-/- mice. As shown in Figure 4A and B, under serum starvation (SF), the rate of cell growth was similar in PDE1C+/+ and PDE1C-/- cells. Serum stimulation (10% FBS) or platelet derived growth factor BB (PDGF-BB) markedly increased cell growth in PDE1C+/+ cells, which is reduced more than 70% in PDE1C-/- cells. We also examined the effect of IC86340 on rat aortic SMCs growth and found that IC86340 significantly reduced rat SMC growth (Fig. 4C). These observations indicate that blocking PDE1C function attenuates SMC proliferation.
Figure 4. PDE1C deficiency or PDE1 inhibition reduces SMC proliferation and migration in vitro and/or ex vivo.
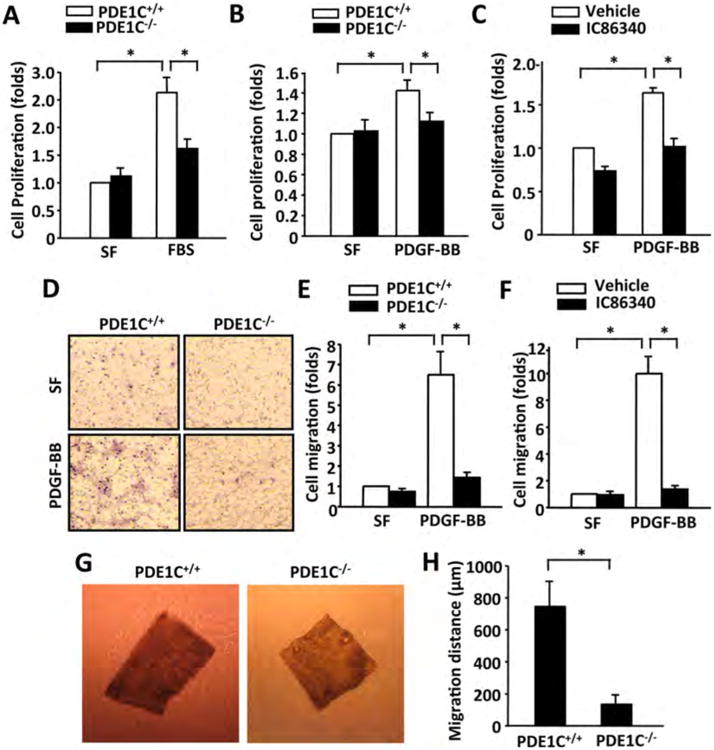
(A-B) PDE1C deficiency inhibited SMC proliferation measured by SRB assay. Mouse SMCs isolated from thoracic aortas of PDE1C+/+ and PDE1C-/- mice were subjected to serum starvation (in serum free medium, SF), followed by the same or addition of 10% FBS stimulation or 50 ng/ml PDGF-BB for 2 days. Values are mean ± SD of (n=3). (C) PDE1 inhibitor IC86340 reduced SMC proliferation. Rat aortic SMCs were subjected to serum starvation, followed by with or without 50 ng/ml PDGF-BB in the presence of vehicle or 15 μmol/L IC86340. Values are mean ± SD of (n=3). (D-E) PDE1C deficiency inhibited SMC migration measured by modified Boyden chamber assay. D, representative images showing transmigrated cells from PDE1C+/+ and PDE1C-/- mice. E, quantitative data of SMC migration. SMC from PDE1C+/+ and PDE1C-/- SMC mice was placed in the upper microchemotaxis chamber, and 25 ng/ml of PDGF-BB was added in the lower polycarbonate filter chamber. The transmigrated cells on the filter membrane were fixed, stained with hematoxylin, photographed, and quantified. Values are mean ± SD of (n=3). (F) PDE1 inhibitor IC86340 reduced SMC migration. Rat aortic SMCs were pretreated with 30 μmol/L IC86340 for 24 h, and then subjected to the Boyden chamber assay. Values are mean ± SD (n=3). (G and H) PDE1C deficiency impaired smooth muscle explant out growth. G, representative images showing outgrowth of SMCs of aortic medial explants from PDE1C+/+ and PDE1C-/- mice in 3D collagen I gel ex vivo. Medial explants of thoracic aortas from PDE1C+/+ and PDE1C-/- mice were embedded in 3D collagen type I gel, and cultured for 10 days. H, quantitative data of migration distance (the distance between the leading front SMCs and the explant tissue). Values are mean ± SD (n=3). *P < 0.05.
We next examined the effects of PDE1C ablation or inhibition on SMC migration by a modified Boyden chamber assay (Fig. 4D-F). We found that PDGF-BB markedly increased the migration of PDE1C+/+ cells (Fig. 4D-E). However, the migratory capacity was almost completely diminished in PDE1C-/- SMCs. Similar observations were obtained in cells treated with IC86340 (Fig. 4F). To further explore the role of PDE1C in SMC migration, we performed an ex vivo 3D-collagen gel migration assay with mouse aortic medial explants. SMC migration was accessed by the migration distances of cells from explants. As shown in Figure 4G and H, when mouse aortic medial explants from PDE1C+/+ mice were cultured in a 3D-collagen matrix for 10 days, SMCs migrated out from aortic explants. In contrast, SMC migration was significantly suppressed in aortic explants from PDE1C-/- mice. These data indicate that PDE1C is critical for SMC migration.
To determine whether PDE1C induction is important in SMC phenotype transition, we measured contractile SMC markers in low passage mouse aortic SMCs isolated from PDE1C+/+ and PDE1C-/- mice. As shown in the supplemental Figure S4A, SM-MHC, calponin, and SM-α-actin mRNA levels were significantly higher in PDE1C-/- than PDE1C+/+ cells. Consistently, SM-MHC and SM-α-actin protein detected by immunostaining were also elevated in PDE1C-/- cells (Supplemental Fig. S4B). These observations suggest that PDE1C induction facilitates SMC phenotype transition.
PDE1C Regulates PDGFRβ Protein Levels
Synthetic SMCs acquire the capacity to proliferate and migrate according to growth factor receptor expression. PDGF signaling, particularly through PDGF receptor beta (PDGFRβ), plays crucial roles in SMC proliferation, migration, and neointimal formation following vascular injury 30-32. Therefore, we first examined the role of PDE1C in regulating PDGFRβ in SMCs. Interestingly, we found that IC86340 dose-dependently reduced PDGFRβ protein levels in rat SMCs (Fig. 5A). IC86340 inhibits both PDE1A and PDE1C isoforms in SMCs. In order to identify the specific PDE1 isoform involved in IC86340-induced reduction of PDGFRβ protein expression, we used adenoviruses expressing PDE1A shRNA or PDE1C shRNA. When high dose adenoviruses were transfected into cells, PDE1A shRNA and PDE1C shRNA specifically decreased PDE1A and PDE1C expression levels by 60-70%, respectively (Supplemental Fig. S5A). As shown in Figure 5B, PDE1C knockdown by its shRNA, but not PDE1A, reduced PDGFRβ protein similar to IC86340, suggesting that the effect of IC86340 on PDGFRβ is primarily through inhibition of PDE1C. To determine whether PDE1C regulates PDGFRβ gene expression, we analyzed PDGFRβ mRNA levels. In an unexpected finding, IC86340 or PDE1C shRNA did not alter PDGFRβ mRNA levels (Supplemental Fig. S5B and C). Instead, IC86340 reduced both endogenous PDGFRβ and exogenously expressed Flag-PDGFRβ protein levels (Supplemental Fig. S5D and F). This further supports the role of PDE1C in PDGFRβ protein regulation. In addition to PDGFRβ, IC86340 or PDE1C shRNA also reduced the protein levels of PDGFR alpha (PDGFRα) and EGF receptor (Supplemental Fig. S5D and E) but did not change their mRNA levels (Supplemental Fig. S5B and C). Together, these results suggest that PDE1C likely regulates multiple growth factor receptors. Due to the robustness of the response, the remainder of investigations focused on PDGFRβ.
Figure 5. PDE1C regulates PDGFRβ protein levels via a cAMP-PKA dependent mechanism.
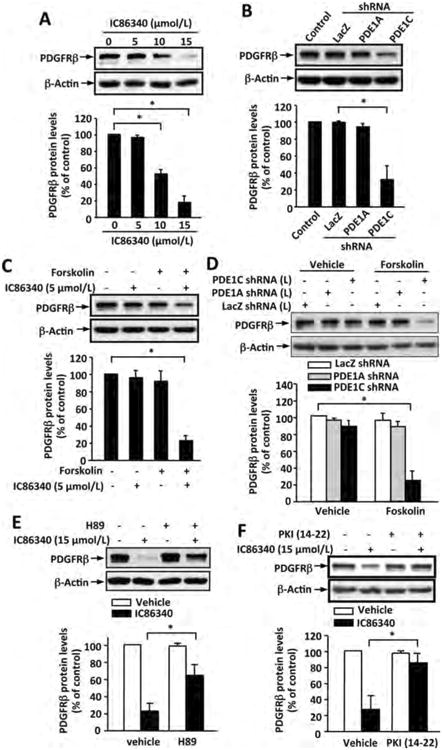
(A) PDE1 inhibitor IC86340 dose-dependently decreased PDGFRβ protein levels. Rat aortic SMCs were treated with indicated doses of IC86340 for 24 h. (B) Knockdown of PDE1C but not PDE1A decreased PDGFRβ protein. Rat aortic SMCs were transfected with adenoviral vectors expressing a high dose of adenovirus encoding LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days. (C) Forskolin enhanced the effect of IC86340 on PDGFRβ protein reduction. Rat aortic SMCs were treated with a low dose of IC86340 (5 μmol/L) with or without 10 μmol/L forskolin for 24 h in DMEM containing 0.1% FBS. (D) Forskolin augmented PDE1C knockdown-induced PDGFRβ protein reduction. Rat aortic SMCs were transfected with a low dose of adenovirus encoding LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, followed with or without 10 μmol/L forskolin treatment for 24 h in DMEM containing 0.1% FBS. (E) PKA inhibitor H89 blocked IC86340-induced PDGFRβ protein reduction. Rat aortic SMCs were treated with a high dose of IC86340 (15 μmol/L) with or without of 5 μmol/L H89 for 24 h in DMEM containing 0.1% FBS. (F) Specific PKA inhibitor PKI (14-22) blocked IC86340-induced PDGFRβ protein reduction. Rat aortic SMCs were treated with a high dose of IC86340 (15 μmol/L) with or without 5 μmol/L PKI (14-22) for 24 h in DMEM containing 0.1% FBS. PDGFRβ protein levels and b-actin equal loading were analyzed by immunoblotting with the anti-PDGFRβ and β-actin antibody, respectively. The blots were analyzed by densitometry. Fold changes normalized to the left lane. Values are mean ± SD (n= 3). *p<0.05.
To further determine the PDGF-mediated cellular signaling response, we pretreated cells with IC86340 for 24 hours to down-regulate PDGFR proteins. After washing out IC86340, cells were stimulated with PDGF-BB for 5 and 30 min. We found that PDGF-BB-mediated Erk1/2 and Akt activation was significantly attenuated in IC86340-pretreated cells (Supplemental Fig. S5G). We have previously found that treating SMCs with IC86340 up to 30 min did not affect PDGF-BB stimulated Erk1/2 and Akt (data not shown), suggesting that PDE1C does not directly regulate PDGFR activation.
PDE1C Modulates a tmAC-cAMP-PKA Signaling Critical for Regulating PDGFRβ Protein Level
PDE1C is able to hydrolyze both cAMP and cGMP with high affinity in vitro 33. The most common cAMP and cGMP effector molecules include cAMP dependent protein kinase (PKA), exchange protein activated by cAMP (Epac), and cGMP-dependent protein kinase (PKG). We, therefore, examined the role of cAMP/PKA, cAMP/Epac, and cGMP/PKG in PDE1C-mediated regulation of PDGFRβ protein expression. First, we tested forskolin, a transmembrane adenylate cyclase (tmAC) activator that activates tmAC to produce cAMP. We found that 10 μmol/L forskolin or low dose IC86340 (5 μmol/L) alone had minimal effect on PDGFRβ protein expression (Fig. 5C). However, forskolin and IC86340 (5 μmol/L) together elicited a synergistic effect on reducing PDGFRβ protein (Fig. 5C). At a low dose, PDE1C shRNA alone had little appreciable effect on PDGFRβ protein expression, but forskolin combined with a low dose of PDE1C shRNA also showed a synergistic PDGFRβ protein reduction (Fig. 5D). An inhibitor of tmAC, 2,5-ddA, abrogated the enhanced effects of forskolin/IC86340 or forskolin/PDE1C shRNA on PDGFRβ protein reduction (Supplemental Fig. S6A and B). These results suggest that PDE1C is coupled to a tmAC/cAMP signaling, which is important for the regulation of PDGFRβ protein levels.
To examine whether PKA was involved in IC86340-induced PDGFRβ protein reduction, two different PKA inhibitors H89 and PKI (14-22) were utilized. As shown in Figure 5E and F, both H89 and PKI (14-22) largely suppressed IC86340-induced PDGFRβ protein reduction. Consistently, H89 also blocked the enhanced effects of forskolin/IC86340 or forskolin/PDE1C shRNA on PDGFRβ protein reduction (Supplemental Fig. S6C and D). Additionally, we examined the role of Epac I using Epac I siRNA. As shown in supplemental Figure S7A, knocking down Epac I expression by 90% did not affect IC86340-induced PDGFRβ protein reduction, suggesting that Epac I does not play a major role in this pathway. Therefore, these results suggest that PDE1C regulates a tmAC/cAMP/PKA signaling pathway that mediates a reduction of PDGFRβ protein.
We next examined the role of cGMP/PKG pathway in regulating PDGFRβ protein. We found that knocking down PKG1 via its siRNA did not alter IC86340-induced PDGFRβ protein reduction (Supplemental Fig. S7B). Moreover, elevating intracellular cGMP concentration by SNAP (a nitrite oxide donor), YC-1 (a soluble guanylyl cyclase activator), or CNP (a membrane guanylyl cyclase activator) did not enhance the effect of IC86340 on PDGFRβ protein reduction (Supplemental Fig. S7C-E). These results suggest that sGC/cGMP/PKG pathway is unlikely to be involved in PDE1C regulation of PDGFRβ protein.
PDE1C Regulates Lysosome-dependent PDGFRβ Protein Degradation
The fact that Inhibiting PDE1C decreases both endogenous and exogenous PDGFRβ protein but not mRNA prompted us to hypothesize that PDE1C regulates PDGFRβ degradation. Both proteasome and lysosomes have been implicated in PDGFRβ degradation 34-36. Therefore, we first tested the proteasome inhibitor MG132 and found that inhibiting proteasome function did not block the effect of IC86340 on PDGFRβ reduction (Data not shown). This suggests that IC86340-induced PDGFRβ protein reduction is not mediated by proteasome degradation.
We next tested the role of lysosomes using vacuolar-type H (+)-ATPase (V-ATPase) inhibitor bafilomycin A1 and lysosome pH neutralizer NH4Cl to inhibit lysosomal function. We found that both bafilomycin A1 and NH4Cl significantly blocked IC86340 and PDE1C shRNA-induced PDGFRβ protein reduction (Fig. 6A and B). Furthermore, we observed that bafilomycin A1 and NH4Cl also blocked the enhanced effect of forskolin/IC86340 on PDGFRβ protein degradation (Supplemental Fig. S7F). These results suggest that PDE1C-regulated PDGFRβ protein degradation occurs through a lysosome-dependent mechanism.
Figure 6. Role of lysosomes in PDE1C-mediated regulation PDGFRβ protein degradation.
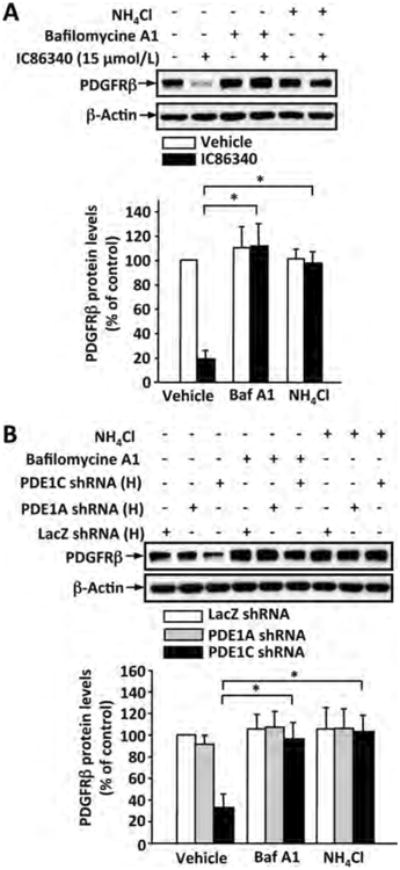
(A) Lysosome inhibitors blocked IC86340-induced PDGFRβ protein reduction. Rat aortic SMCs were pretreated with lysosome inhibitor NH4Cl (20 mmol/L) or Bafilomycine A (50 nmol/L) for 0.5 h, followed by treatment with 15 μmol/L IC86340 for additional 24 h in DMEM containing 0.1% FBS. (B) Lysosome inhibitors blocked PDE1C knockdown-induced PDGFRβ protein reduction. Rat aortic SMCs were transfected with a high dose of adenovirus encoding LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, and treated with 20 mmol/L NH4Cl or 50 nmol/L Bafilomycine A for 24 h in DMEM containing 0.1% FBS. Protein levels of PDGFRβ and β-actin equal loading were determined by immunoblotting and analyzed by densitometry. Fold changes normalized to the left lane. Values are mean ± SD (n= 3). *p<0.05.
PDE1C Regulates Endosome-Mediated Internalization of PDGFRβ
It is well known that the internalized receptors within endosomes are either recycled to the plasma membrane or trafficked to late endosome/lysosome for degradation 37. Therefore, we determined the role of endocytosis in IC86340-induced PDGFRβ protein degradation through inhibiting the function of dynamin, a protein that is essential for clathrin-dependent coated vesicle formation and receptor endocytosis. As shown in Figure 7A, IC86340-induced PDGFRβ protein reduction was almost completely abolished by dynasore (a cell-permeable inhibitor of dynamin) in SMCs. We then further examined the effect of IC86340 on PDGFRβ internalization by first labeling SMC surface proteins with biotin and then detecting intracellular biotin-labeled PDGFRβ through streptavidin-immunoprecipitation. As shown in Figure 7B, the levels of internalized PDGFRβ were increased ≈ 3 fold by IC86340 compared to vehicle control. We also examined whether IC86340 induced endosome localization of PDGFRβ through double immunostaining of PDGFRβ and EEA-1, an early endosome marker protein. As shown in Figure 7C-E, there was a substantial amount of PDGFRβ localized on the plasma membrane in the absence of IC86340. However treatment with IC86340 caused a significant reduction of PDGFRβ protein on the membrane, and increase of PDGFRβ co-localization with EEA1. Moreover, we found that the effect of IC86340 on suppressing PDGF-BB-induced cell proliferation is largely abolished in the presence of dynasore, a dynamin inhibitor for blocking the endocytic pathway (Fig. 7F). Together, these results together suggest that PDE1C inhibition promotes PDGFRβ endocytosis and degradation, subsequently attenuating SMC growth.
Figure 7. PDE1 inhibitor IC86340 induces PDGFRβ protein degradation via endocytosis.
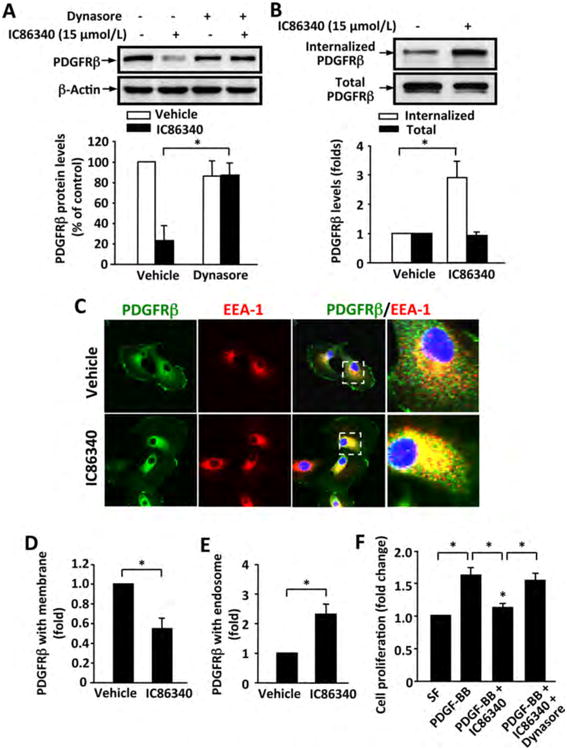
(A) Dynamin inhibitor dynasore blocked IC86340-induced PDGFRβ protein reduction. Rat aortic SMCs were treated with 15 μmol/L IC86340 with or without 2.5 μmol/L dynasore for 24 h in DMEM containing 0.1% FBS. (B) IC86340 induced PDGFRβ internalization. Rat aortic SMC membrane proteins were labeled with biotin and stimulated with 15 μmol/L IC86340 for 12 h in DMEM containing 0.1% FBS. Cell lysates were immunoprecipitated with streptavidin beads, and the recovered internalized biotinylated PDGFRβ were immunoblotted with PDGFRβ antibody. (C-E) IC86340 increased PDGFRβ co-localization with endosomes. Rat aortic SMCs were treated with 15 μmol/L IC86340 for 12 h in DMEM containing 0.1% FBS. PDGFRβ and early endosome marker EEA-1 were determined by immunostaining. C, representative images showing the localization of PDGFRβ in plasma membrane and endosomes. Selected boxes were enlarged for better view. D, quantitative data of PDGFRβ on cell membrane. E, quantitative data of PDGFRβ in endosomes. (F) Dynasore abrogated the inhibitory effect of IC86340 on SMC growth. Rat aortic SMCs were treated with IC86340, dynasore or a combination of IC86340 and dynasore, and stimulated with 50 ng/ml PDGF-BB for 48 h. Cell proliferation was measured by SRB assay. Values are mean ± SD of (n=3). *P < 0.05.
PDE1C Associates with PDGFRβ
Based on the facts that PDE1C couples to tmAC/cAMP and PDE1C regulates membrane PDGFRβ internalization, we hypothesized that PDE1C could be located on the plasma membrane and associated with PDGFRβ. To prove this hypothesis, we first performed immunofluorescent double-staining of PDE1C and PDGFRβ. As shown in Figure 8A, in actively growing SMCs, PDE1C was largely detected in perinuclear areas and the cell membrane. PDE1C is co-localized with PDGFRβ, particularly on the cell membrane. To further confirm the co-localization of PDE1C and PDGFRβ, we exogenously expressed EGFP-PDE1C and Flag-tagged PDGFRβ in SMCs via electroporation. Consistently, EGFP-PDE1C was localized in both plasma membrane and perinuclear areas, similar to endogenous PDE1C (Fig. 8B). Flag-PDGFRβ and GFP-PDE1C was largely co-localized on the cell membrane (Fig. 8B).
Figure 8. PDE1C is associated with PDGFRβ.
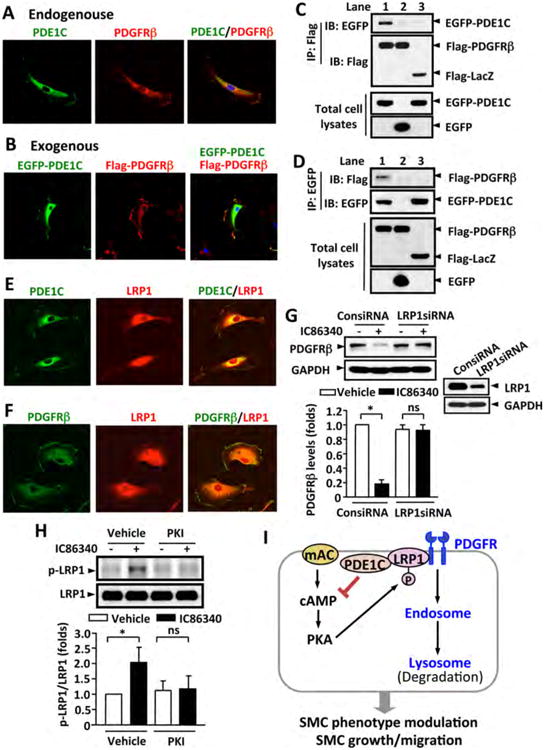
(A) Representative images showing endogenous PDE1C and PDGFRβ co-localized on cytoplasmic membrane. Rat aortic SMCs were immunostained with anti-PDE1C and anti-PDGFRβ antibodies. (B) Representative images showing exogenous PDE1C and PDGFRβ co-localization on plasma membrane. Rat aortic SMCs were co-transfected with Flag-PDGFRβ and EGFP-PDE1C by electroporation, and exogenous Flag-PDGFRβ and EGFP-PDE1C were detected by immunostaining with anti-flag and anti-GFP antibodies, respectively. Cells were visualized using confocal microscopy. (C-D) Co-immunoprecipitation (IP) revealed that PDE1C and PDGFRβ associate together. HEK293A cells were co-transfected with Flag-PDGFRβ and EGFP-PDE1C (lane 1), Flag-PDGFRβ and EGFP (lane 2), or Flag-LacZ and EGFP-PDE1C (lane 3), C, IP with anti-Flag antibody and IB with anti-EGFP or anti-Flag antibody. D, IP with anti-EGFP antibody and IB with anti-Flag or anti-EGFP antibody. The expression of Flag-tagged and EGFP proteins in total cell lysates were immunoblotted with anti-Flag or anti-EGFP antibody, respectively. (E-F) Representative images showing the co-localization of PDE1C and LRP1 (E) or PDGFRβ and LRP1 (F) by immunofluorescent staining. Rat aortic SMCs were immunostained with anti-PDE1C or anti-PDGFRβ together with anti-LRP1 antibodies. (G) Knockdown of LRP-1 attenuated IC86340-induced PDGFRβ degradation. Rat aortic SMCs were transfected with 50 nmol/L control siRNA (ConsiRNA) or LRP1 siRNA (LRP1siRNA) for 2 days, followed by treatment with 15 μmol/L IC86340 in DMEM supplied with 0.1% FBS for 24 h. The protein levels were determined by western blot. (H) PDE1 inhibitor IC86340 stimulates LRP1 phosphorylation in a PKA-dependent manner. Rat aortic SMCs were treated with vehicle or 15 μmol/L IC86340 for 12 h in the presence of vehicle or 5 μ mol/L PKI (14-22) peptide. The phosphorylation of LRP1 was detected by immunoblotting with a phospho-PKA substrate antibody after immunoprecipitation with LRP1 antibody. Values are mean ± SD of (n=3-4). *P < 0.05; ns: no significant difference. (I) Proposed model: a tmAC-derived cAMP-PKA signaling is critical in promoting PDGFRβ internalization and endocytosis in a LRP1-dependent manner.
In addition, we examined if EGFP-PDE1C and Flag-PDGFRβ is able to be co-immunoprecipitated (Fig. 8C and D). We found that in the cells expressing EGFP-PDE1C and Flag-PDGFRβ (Lane 1), immunoprecipitation (IP) of PDGFRβ using an anti-Flag antibody pulled down EGFP-PDE1C (Fig. 8C) and IP of PDE1C using an anti-EGFP antibody pulled down Flag-PDGFRβ (Fig. 8D). However, we failed to detect the interaction between Flag-PDGFRβ and EGFP (Lane 2), or EGFP-PDE1C and Flag-LacZ (Lane 3). These results strongly suggest an association between PDGFRβ and PDE1C.
Moreover, we examined PDE1C and PDGFRβ expression and localization in neointimal lesions in vivo (Supplemental Fig. S8). Similar to PDE1C, PDGFRβ expression level was very low in the normal mouse carotid artery (Fig. S8A) as well as the medial layer of human coronary arteries (Fig. S8B), but was markedly induced in the neointimal lesions. Most importantly, PDGFRβ and PDE1C proteins were highly co-localized in pathologic SMCs, supporting the association of PDE1C with PDGFRβ in vivo.
LRP1 is critical for PDE1-mediated regulation of PDGFRβ protein
The LDL receptor-related protein 1 (LRP1) is a large endocytic receptor that modulates the endocytosis and trafficking of a number of membrane receptors and extracellular macromolecules 38, 39. It has been shown that LRP1 forms a complex with PDGFRβ, which alters PDGFRβ subcellular trafficking 35, 40-42. PKA-dependent phosphorylation of LRP1 has been shown to be critical for LRP1-mediated endocytosis 43. Therefore, we examined the relationship between PDE1C and LRP1 and the role of LRP1 in PDE1 inhibition-induced PDGFβ reduction. Interestingly, immunofluorescent double-staining revealed that LRP1 was co-localized with PDE1C and PDGFRβ in cell membrane as well as in some intracellular structures (Fig. 8E and F). In addition, LRP1 and PDE1C or LRP1 and PDGFRβ co-expression and co-localization were found in mouse or human neointimal lesions in vivo (Fig. S9). Moreover, siRNA mediated depletion of LRP1 largely blocked the effect of IC86340 on PDGFRβ reduction (Fig. 8G). IC86340 significantly increased LRP1 phosphorylation detected by the phospho-antibody recognizing PKA substrates, which was attenuated upon PKA inhibition by the PKI peptide treatment (Fig. 8H). These observations indicate that LRP1 is required for PDE1-mediated regulation of PDGFRβ protein, likely via modulating PKA-dependent phosphorylation of LRP1.
Discussion
Experimental evidence has strongly supported the conclusion that PDE1C is a synthetic SMC specific enzyme. Previous studies by Rybalkin et al showed that PDE1C was highly expressed in proliferating human aortic SMCs but not detectable in quiescent human aortas 19, 20. In the current study, we more comprehensively demonstrated that PDE1C is specifically expressed in synthetic SMCs from multiple species and different vascular beds, not only under in vitro culture conditions but also in vivo in vascular lesions from a number of different small animal injury models as well as in diseased human vessels. More importantly, we provided in vitro, ex vivo, and in vivo evidence demonstrating that PDE1C is critical for SMC growth, migration, and neointima formation. Furthermore, we defined a novel mechanism by which PDE1C negatively regulates PDGFRβ endocytosis and degradation in an LRP1-dependent manner. PDGF signaling has multiple actions in SMCs, including phenotypic modulation, cell proliferation, migration, and ECM metabolism, all of which contribute to pathological vascular remodeling 30-32. PDE1C, through modulating PDGF signaling, is thus capable of serving as an important multifunctional regulator in synthetic SMCs. As shown in the proposed model (Fig. 8I), our experimental evidence suggests that a tmAC-derived cAMP-PKA signaling is critical in promoting PDGFRβ internalization and endocytosis. PDE1C upregulation antagonizes the tm-AC-cAMP-PKA signaling and thus suppresses PDGFRβ degradation, which facilitates SMC phenotype modulation and accelerates SMC growth/migration. PKA-dependent phosphorylation of LRP1 might be important in PDE1C-cAMP regulation of PDGFRβ protein degradation. Taken together, these experimental results strongly implicate that the induction of PDE1C in SMCs is responsible for the pathogenesis of synthetic function, contributing to vascular hyperplasia. PDEs have been proven to be worth targets for drug development. Thus our findings may have great therapeutic impact as it may lead to the development of novel therapeutic strategies using PDE1 inhibitors (ideally PDE1C-selective inhibitors) in treating a number of vascular hyperplasic disorders. Currently, a pan PDE1 inhibitor is under development for treating schizophrenia through targeting the PDE1B isozyme in the brain 44.
Receptor tyrosine kinases (RTKs) are subjected to endocytosis. Internalized RTKs have a number of different fates: sustained signaling within early endosomes, being recycled to the plasma membrane, or trafficked to late endosomes/lysosomes for degradation 37, 45. Receptor endocytosis and subsequent lysosome degradation is one of the important mechanisms to prevent sustained RTK activation on the plasma membrane as well as in early endocytic vesicles 46, 47. Deregulation of the endocytic pathway and impairment of the degradation system has been found in cell transformation and tumorgenesis 45-47. Therefore, it is believed that targeting RTK endocytosis and degradation may represent a promising perspective in cancer therapy 37, 48. In this study, we identified PDE1C as a novel regulator of PDGFRβ endocytosis/degradation in vascular SMCs. The role of PDE1C may not be only restricted to PDGFRβ because we found that PDE1C also regulates other RTKs, such as PDGFRα and EGFR (Fig. Supplemental Fig. S5). A previous study showed that in SMCs, human cytomegalovirus decreased both PDGFRα and PDGFRβ protein, accompanied by increased localization of these receptor proteins in endosomes and lysosomes 49. PDE1C is also important for the growth of human malignant melanoma cell lines 50. Therefore, our findings in SMCs may also be applicable to tumor cells and suggest that PDE1C inhibition may represent a novel strategy to target RTK degradation in vascular diseases as well as cancer therapy.
LRP1 is a multifunctional scavenger and signaling receptor. It plays diverse roles in a variety of biological processes, including lipoprotein metabolism, clearance of plasma proteins, protease degradation, as well as receptor trafficking and signaling 38, 39. Our current study suggests that LRP1 is important in PDE1C/cAMP-mediated regulation of PDGFRβ stability and availability. Previous studies have also shown that LRP1 depletion in SMCs resulted in elevated PDGFRβ level and activation, increased SMC proliferation and migration, and accelerated atherosclerosis and aortic aneurysm in SMC-specific LRP1 knockout mice 51-53. Blockade of PDGFR signaling with imatinib (a tyrosine kinase inhibitor) prevented atherosclerosis progression in LRP1 knockout mice 51. These lines of experimental evidence strongly suggest an important role of LRP1 in negatively modulating PDGF signaling and SMC pathogenesis in vascular diseases. The molecular mechanism by which cAMP regulates LRP1-medited PDGFRβ endocytosis/degradation has not been fully characterized. It has been shown that LRP1 interacts with stimulatory heterotrimeric G-protein (Gsα) that leads to cAMP production and PKA activation 54. LRP1-mediated endocytosis of urokinase-type plasminogen activator receptor (uPAR) is regulated by PKA 55. In addition, PKA-dependent phosphorylation of Serine 76 of LRP1 cytoplasmic tail is critical in receptor endocytosis 43. These observations suggest a potential role of cAMP/PKA in directly modulating LRP1 function, likely through PKA phosphorylation of LRP1. In the current study, we have shown that an mtAC-PDE1C controlled cAMP/PKA signaling regulates LRP1 phosphorylation and subsequent PDGFRβ endocytosis/degradation. Future studies are necessary to determine the specific phosphorylation site and the role of LRP1 phosphorylation in PDE1C-mediated regulation of PDGFRβ endocytosis and SMC proliferation/migration.
It has long been believed that vascular medial SMCs change from quiescent/contractile to active/synthetic phenotype, thereby contributing to neointimal hyperplasia 56. However, there is also evidence supporting the possible transdifferentiation of adventitial fibroblasts 57; the differentiation of progenitor cells/stem cells 58, 59; or endothelial-to-mesenchymal transition 60, 61 to these SMC-α-actin positive, synthetic SMC-like cells in the neointimal lesions. Thus PDE1C-positive cells in neointimal lesions might nave multiple origins. Regardless of the origins, synthetic SMC-like cells are able to proliferate, migrate, and secrete ECM proteases and proteins. In addition, they produce pro-inflammatory molecules, providing an inflammatory microenvironment for leukocyte penetration, accumulation and activation. Therefore, developing novel strategies, impeding the phenotype transition from the contractile to synthetic state will be of great interest. Thus, PDE1C may represent a novel therapeutic target in combating SMC phenotype modulation under disease states. Besides those SMC-like cells, inflammatory cells also contribute to neointimal hyperplasia. We failed to detect PDE1C in mouse peritoneal macrophages and found that PDE1C deficiency does not alter LPS-stimulated cytokine expression in macrophages nor circulating inflammatory molecule levels in mice (our unpublished observations). This is also consistent with the previous findings that the PDE1B isozyme represents the major PDE1 activity in macrophages 62, 63. Together, these suggest that PDE1C does not regulate macrophage function and systemic inflammation in murine animals. Nevertheless, the specific contribution of SMC-origin PDE1C needs to be further determined using SMC-specific PDE1C knockout mice in the future.
Supplementary Material
Figure S1. PDE1C protein level is increased in synthetic SMCs. Western blotting results showing protein levels of PDE1C and calponin in contractile SMCs (freshly isolated medial layers) and corresponding synthetic SMCs (cultured SMCs) from rat aortas (A), human aortas (B), and human saphenous veins (C). Contractile SMCs are freshly isolated medial tissues procured by removing endothelial cells and pealing off adventitial layers. Synthetic SMCs are cultured growing SMCs isolated from the corresponding vessel with the explant method. (D) Cultured rat aortic SMCs were in differentiation medium (medium 231 supplemented with Smooth Muscle Growth S (SMGS), from Cascade Biologics) or growth medium (medium 231 supplemented with Smooth Muscle Differentiation Supplement (SMDS), from Cascade Biologics) for 2 days. SM-MHC and calponin are used as contractile SMC markers. Values are mean ± SD of triplicate experiments. *P < 0.05.
Supplemental Figure S2. (A) PDE1C is elevated in femoral arteries after wire injury. qRT-PCR showing the PDE1C mRNA levels of in uninjured (right) and injured (left) femoral arteries. C57BL/6J mice were subjected to femoral artery wire injury for 28 days, total RNA were isolated from right uninjured and left injured femoral arteries. The levels of PDE1C mRNA were assessed by qPCR. Values are means ± SD of triplicate (three arteries are pooled together). *P < 0.05. (B) PDE1C are induced in SMC-like cells. Representative images of carotid artery sections subjected to immunofluorescent double staining with SM-α-actin (red) and PDE1C (green), Nuclei were stained wit DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel. The merged images (right panels) show that PDE1C-positive cells are largely overlapped with SM-α-actin-positive cells in the neointimal region. N: neointima.
Supplemental Figure S3. (A and C) PDE1C deficiency or PDE1 inhibition attenuates SMC proliferation in response to vascular injury. Top panels: representative images of carotid artery sections immunostained with SM-α-actin (pink) or Ki67 (brown). Bottom panel: quantitative data of Ki67 positive cells in intima and media. A, PDE1C+/+ and PDE1C-/- mice or were subjected to left common carotid artery ligation for 14 days. C, FVB mice were subjected to left common carotid artery ligation for 14 days in the presence of vehicle or 30 μmol/L IC86340 applied perivascularly via pluronic gel. SM-α-actin and Ki67 immunostaining were performed in cross-sections of carotid arteries. Ki-67-positive cells in the intima and the media were calculated. Values are means ± SEM (n=3). *P < 0.05. N: neointima; M: media. (B-D) PDE1C deficiency suppresses ROS production in carotid arteries after vascular injury. Top panels: Representative images of carotid arteries immunostained with 4-HNE (an oxidative stress marker). C, PDE1C+/+ and PDE1C-/- mice were subjected to left common carotid artery ligation for 14 days. D, FVB mice were subjected to left common carotid artery ligation for 14 days in the presence of vehicle or 30 μmol/L IC86340 applied perivascularly via pluronic gel. 4-HNE immunostaining were performed in cross-sections of carotid arteries. Quantification were performed using Image Pro Plus software. Values are means ± SEM (n=3). *P < 0.05.
Supplemental Figure S4. (A) Real-time RT-PCR showing the mRNA expression of SM-MHC, calponin, and SM-α-actin in low-passage aortic SMCs isolated for PDE1C+/+ and PDE1C-/- mice. (B) Immanofluorescence staining of SM-MHC or SM-α-actin in PDE1C+/+ and PDE1C-/- SMCs. Green: SM-MHC; Red: SM-α-actin; Blue: nuclei.
Supplemental Figure S5. Role of PDE1C in regulating PDGFRβ levels. (A) PDE1A and PDE1C mRNA levels in rat SMCs treated with a high dose or a low dose of adenovirus encoding LacZ shRNA, PDE1A shRNA or PDE1C shRNA. (B-C) PDE1 inhibitor IC86340 or PDE1C shRNA does not affect mRNA levels of multiple growth factor receptors including PDGFRβ, PDGFRα, and EGFR. Levels of mRNA were determined by RT-PCR. (D-E) PDE1 inhibitor IC86340 and PDE1C shRNA reduced the protein levels of multiple growth factor receptors. The protein levels of PDGFRβ, PDGFRα, and EGFR were assessed by immunoblotting. Rat aortic SMCs were treated with indicated doses of PDE1 inhibitor IC86340 for 24 h or transduced with adenovirus encoding shRNA against LacZ, PDE1A,or PDE1C shRNA. (F) PDE1 inhibitor IC86340 reduced exogenously expressed PDGFRβ protein but not PDE1C protein. Rat aortic SMCs were transfected with Flag-PDGFRβ or EGFP-PDE1C via electropolation for 2 days and then treated with 15 μmol/L IC86340 for 24 h. PDGFRβ and PDE1C were detected by anti-Flag and anti-GFP antibodies, respectively. (G) PDGF-stimulated Erk1/2 and AKT activation was attenuated when PDGFR was downregulated. Rat aortic SMCs were treated with 15 μmol/L IC86340 in DMEM with 0.1% FBS for 24 h, washed, and stimulated with 10 ng/ml PDGF-BB for 5 or 30 min. Phospho-Erk1/2, Erk 1, phospho-AKT, and AKT were measured by immunoblotting.
Supplemental Figure S6. Role of tmAC-cAMP signaling in PDE1C-mediated regulation of PDGFRβ. (A) The tmAC inhibitor blocked the synergistic effect of IC86340 and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340, 10 μmol/L forskolin, or both in the presence or absence of tmAC inhibitor 2′,5′-dideoxyadenosine (2′ 5′-ddA,10 μmol/L) for 24 h in DMEM containing 0.1% FBS. (B) The tmAC inhibitor blocked the synergistic effect of PDE1C shRNA and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were transfected with a low dose of adenovirus expressing LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, followed by treatment with 10 μmol/L forskolin in the presence or absence of 10 μmol/L 2′,5′-ddA for 24 h in DMEM containing 0.1% FBS. (C) PKA inhibitor H89 blocked the synergistic effect of IC86340 and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340, or 10 μmol/L forskolin, or both in the presence or absence of 5 μmol/L H89 for 24 h in DMEM containing 0.1% FBS. (D) PKA inhibition by H89 blocked the synergistic effect of PDE1C shRNA and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were transfected with a low dose of adenovirus expressing LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, and then treated with 10 μmol/L forskolin in the presence of absence of 5 μmol/L H89 for 24 h in DMEM containing 0.1% FBS. Percentile changes normalized to the left lane. Values are mean ± SD (n= 3). *p<0.05.
Supplemental Figure S7. (A) Epac was not involved in PDE1 inhibition-induced PDGFRβ protein reduction. Rat aortic SMCs were transfected with 50 nmol/L control siRNA or Epac siRNA for 3 days, and then treated with 15 μmol/L IC86340 for 24 h in DMEM containing 0.1% FBS. Right panel showed Epac knockdown by specific siRNA using RT-PCR. (B) PKG1 was not involved in PDE1 inhibition-induced the PDGFRβ protein reduction. Rat aortic SMCs were transfected with 50 nmol/L control siRNA or PKG1 siRNA for 3 days, and treated with 15 μmol/L IC86340 for 24 h in DMEM containing 0.1% FBS. (C-E) cGMP and PDE1 inhibition does not have synergistic effect on PDGFRβ. Rat aortic SMCs were treated with either 5 μmol/L IC86340 alone, one of cGMP elevators (100 μmol/L NO donor SNAP, 10 μmol/L sGC activator YC-1, and 100 nmol/L CNP), or both of IC86340 and a cGMP elevator for 24 h in DMEM containing 0.1% FBS. (F) Lysosome inhibitors abrogated enhanced effect of forskolin and IC86340 on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340 and 10 μmol/L forskolin in the presence or absence of 20 mmol/L NH4Cl or 50 nmol/L Bafilomycine A for 24 h in DMEM containing 0.1% FBS. Protein levels of PDGFRβ and β-actin equal loading were determined by immunoblotting. PDGFRβ protein and equal loading β-actin were determined by immnoblotting. Quantitative data show percentile changes normalized to the 1st lane. Values are mean ± SD (n= 3). *p<0.05. ns: no significant difference.
Supplemental Figure S8. PDE1C and PDGFRβ co-localization in neointimal lesions. (A) Representative images of carotid artery sections subjected to co-immunofluorescent staining of PDE1C (green) and PDGFRβ (red), and nuclei are stained with DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel. N: neointima. (B) Reprehensive co-immunofluorescent staining images of human coronary artery with neointimal lesions. Green: PDE1C, Red: PDGFRβ, Blue: nuclei stained with DAPI. The merged images (right panels) show that PDE1C-positive cells are largely overlapped with SM-α-actin-positive cells in the neointimal region. M: media, N: neointima.
Supplemental Figure S9. Co-expression of LRP1 with PDE1C or PDGFRβ in neointimal lesions. (A-B) Representative images of carotid artery sections subjected to co-immunofluorescent staining of LRP1 (green) together with PDE1C (red) or PDGFRβ (red), and nuclei are stained with DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel (inset). N: neointima. (C-D) Reprehensive co-immunofluorescent staining images of human coronary artery with neointimal lesions. Green: LRP1, Red: PDE1C or PDGFRβ, Blue: nuclei stained with DAPI. The merged images (right panels) show that LRP1-postive cells are largely overlapped with PDE1C- or PDGFRβ-positive cells in the neointimal region. M: media, N: neointima.
Acknowledgments
Sources of Funding: This work was supported by NIH grants HL111291 and HL088400 (to C.Y), the American Heart Association (AHA) Grant-in-Aid 12GRNT12080014 (to C.Y.), National Marfan Foundation (to C.Y), and AHA Scientific-Development-Grant 13SDG16560024 (to Y.C).
Non-standard Abbreviations and Acronyms
- CA
coronary artery
- DES
drug-eluting stent
- ECM
extracellular matrix
- Epac
exchange protein activated by cAMP
- GPCR
G-protein coupled receptor
- FRET
fluorescent resonance energy transfer
- IP
immunoprecipitation
- LCA
left carotid artery
- LDL
low-density lipoprotein
- LRP1
LDL receptor-related protein 1
- tmAC
transmembrane adenylyl cyclase
- NO
nitric oxide
- PDE
cyclic nucleotide phosphodiesterase
- PDGFR
platelet-derived growth factor receptor
- PGI2
prostacyclin
- SMC
smooth muscle cell
- PKA
cAMP dependent protein kinase
- PKG
cGMP-dependent protein kinase
- PKI
PKA inhibitor
- RCA
right carotid artery
- RTK
receptor tyrosine kinases
- sGC
soluble guanylate cyclase
- SM-MHC
smooth muscle myosin heavy chain
- V-ATPase
vacuolar-type H (+)-ATPase
Footnotes
Disclosure: None
References
- 1.Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Kearney M, Pieczek A, Haley L, Losordo DW, Andres V, Schainfeld R, Rosenfield K, Isner JM. Histopathology of in-stent restenosis in patients with peripheral artery disease. Circulation. 1997;95(8):1998–2002. doi: 10.1161/01.cir.95.8.1998. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ Res. 2007;100(7):967–978. doi: 10.1161/01.RES.0000261982.76892.09. [DOI] [PubMed] [Google Scholar]
- 4.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 5.Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol (1985) 2001;90(1):358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- 6.Geary RL, Wong JM, Rossini A, Schwartz SM, Adams LD. Expression profiling identifies 147 genes contributing to a unique primate neointimal smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2002;22(12):2010–2016. doi: 10.1161/01.atv.0000038147.93527.35. [DOI] [PubMed] [Google Scholar]
- 7.Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, Caputo RP, Kereiakes DJ, Williams DO, Teirstein PS, Jaeger JL, Kuntz RE. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med. 2003;349(14):1315–1323. doi: 10.1056/NEJMoa035071. [DOI] [PubMed] [Google Scholar]
- 8.Stone GW, Ellis SG, Cox DA, Hermiller J, O'Shaughnessy C, Mann JT, Turco M, Caputo R, Bergin P, Greenberg J, Popma JJ, Russell ME. A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease. N Engl J Med. 2004;350(3):221–231. doi: 10.1056/NEJMoa032441. [DOI] [PubMed] [Google Scholar]
- 9.Pendyala LK, Yin X, Li J, Chen JP, Chronos N, Hou D. The first-generation drug-eluting stents and coronary endothelial dysfunction. JACC Cardiovasc Interv. 2009;2(12):1169–1177. doi: 10.1016/j.jcin.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Koyama H, Bornfeldt KE, Fukumoto S, Nishizawa Y. Molecular pathways of cyclic nucleotide-induced inhibition of arterial smooth muscle cell proliferation. J Cell Physiol. 2001;186(1):1–10. doi: 10.1002/1097-4652(200101)186:1<1::AID-JCP1012>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 11.Klein C. Nitric oxide and the other cyclic nucleotide. Cell Signal. 2002;14(6):493–498. doi: 10.1016/s0898-6568(01)00283-2. [DOI] [PubMed] [Google Scholar]
- 12.Knight WE, Yan C. Cardiac cyclic nucleotide phosphodiesterases: function, regulation, and therapeutic prospects. Horm Metab Res. 2012;44(10):766–775. doi: 10.1055/s-0032-1321870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight W, Yan C. Therapeutic potential of PDE modulation in treating heart disease. Future Med Chem. 2013;5(14):1607–1620. doi: 10.4155/fmc.13.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75(4):725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 15.Chan S, Yan C. PDE1 isozymes, key regulators of pathological vascular remodeling. Curr Opin Pharmacol. 2011;11(6):720–724. doi: 10.1016/j.coph.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lugnier C, Schini VB. Characterization of cyclic nucleotide phosphodiesterases from cultured bovine aortic endothelial cells. Biochem Pharmacol. 1990;39(1):75–84. doi: 10.1016/0006-2952(90)90650-a. [DOI] [PubMed] [Google Scholar]
- 17.Kishi Y, Ashikaga T, Numano F. Phosphodiesterases in vascular endothelial cells. Adv Second Messenger Phosphoprotein Res. 1992;25:201–213. [PubMed] [Google Scholar]
- 18.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17(10):2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 19.Rybalkin SD, Bornfeldt KE, Sonnenburg WK, Rybalkina IG, Kwak KS, Hanson K, Krebs EG, Beavo JA. Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J Clin Invest. 1997;100(10):2611–2621. doi: 10.1172/JCI119805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res. 2002;90(2):151–157. doi: 10.1161/hh0202.104108. [DOI] [PubMed] [Google Scholar]
- 21.Streb JW, Miano JM. Cross-species sequence analysis reveals multiple charged residue-rich domains that regulate nuclear/cytoplasmic partitioning and membrane localization of a kinase anchoring protein 12 (SSeCKS/Gravin) J Biol Chem. 2005;280(30):28007–28014. doi: 10.1074/jbc.M414017200. [DOI] [PubMed] [Google Scholar]
- 22.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32(11):2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 23.Zou Y, Dietrich H, Hu Y, Metzler B, Wick G, Xu Q. Mouse model of venous bypass graft arteriosclerosis. Am J Pathol. 1998;153(4):1301–1310. doi: 10.1016/S0002-9440(10)65675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cygnar KD, Zhao H. Phosphodiesterase 1C is dispensable for rapid response termination of olfactory sensory neurons. Nat Neurosci. 2009;12(4):454–462. doi: 10.1038/nn.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol. 2003;23(12):2185–2191. doi: 10.1161/01.ATV.0000103120.06092.14. [DOI] [PubMed] [Google Scholar]
- 26.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol. 2009;29(7):1074–1079. doi: 10.1161/ATVBAHA.108.181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cable DG, Caccitolo JA, Caplice N, O'Brien T, Simari RD, Daly RC, Dearani JA, Mullany CJ, Orszulak TA, Schaff HV. The role of gene therapy for intimal hyperplasia of bypass grafts. Circulation. 1999;100(19 Suppl):II392–396. doi: 10.1161/01.cir.100.suppl_2.ii-392. [DOI] [PubMed] [Google Scholar]
- 28.Cai Y, Miller CL, Nagel DJ, Jeon KI, Lim S, Gao P, Knight PA, Yan C. Cyclic nucleotide phosphodiesterase 1 regulates lysosome-dependent type I collagen protein degradation in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2011;31(3):616–623. doi: 10.1161/ATVBAHA.110.212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mekontso-Dessap A, Kirsch M, Guignambert C, Zadigue P, Adnot S, Loisance D, Eddahibi S. Vascular-wall remodeling of 3 human bypass vessels: organ culture and smooth muscle cell properties. J Thorac Cardiovasc Surg. 2006;131(3):651–658. doi: 10.1016/j.jtcvs.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 30.Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, Nishikawa S, Nishikawa SI, Kita T. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103(24):2955–2960. doi: 10.1161/01.cir.103.24.2955. [DOI] [PubMed] [Google Scholar]
- 31.Ishigaki T, Imanaka-Yoshida K, Shimojo N, Matsushima S, Taki W, Yoshida T. Tenascin-C enhances crosstalk signaling of integrin alphavbeta3/PDGFR-beta complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J Cell Physiol. 2011;226(10):2617–2624. doi: 10.1002/jcp.22614. [DOI] [PubMed] [Google Scholar]
- 32.Kohno T, Urao N, Ashino T, Sudhahar V, Inomata H, Yamaoka-Tojo M, McKinney RD, Fukai T, Ushio-Fukai M. IQGAP1 links PDGF receptor-beta signal to focal adhesions involved in vascular smooth muscle cell migration: role in neointimal formation after vascular injury. Am J Physiol Cell Physiol. 2013;305(6):C591–600. doi: 10.1152/ajpcell.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan C, Zhao AZ, Bentley JK, Beavo JA. The calmodulin-dependent phosphodiesterase gene PDE1C encodes several functionally different splice variants in a tissue-specific manner. J Biol Chem. 1996;271(41):25699–25706. doi: 10.1074/jbc.271.41.25699. [DOI] [PubMed] [Google Scholar]
- 34.Baron V, Schwartz M. Cell adhesion regulates ubiquitin-mediated degradation of the platelet-derived growth factor receptor beta. J Biol Chem. 2000;275(50):39318–39323. doi: 10.1074/jbc.M003618200. [DOI] [PubMed] [Google Scholar]
- 35.Takayama Y, May P, Anderson RG, Herz J. Low density lipoprotein receptor-related protein 1 (LRP1) controls endocytosis and c-CBL-mediated ubiquitination of the platelet-derived growth factor receptor beta (PDGFR beta) J Biol Chem. 2005;280(18):18504–18510. doi: 10.1074/jbc.M410265200. [DOI] [PubMed] [Google Scholar]
- 36.Reddi AL, Ying G, Duan L, Chen G, Dimri M, Douillard P, Druker BJ, Naramura M, Band V, Band H. Binding of Cbl to a phospholipase Cgamma1-docking site on platelet-derived growth factor receptor beta provides a dual mechanism of negative regulation. J Biol Chem. 2007;282(40):29336–29347. doi: 10.1074/jbc.M701797200. [DOI] [PubMed] [Google Scholar]
- 37.Hu CT, Wu JR, Wu WS. The role of endosomal signaling triggered by metastatic growth factors in tumor progression. Cell Signal. 2013;25(7):1539–1545. doi: 10.1016/j.cellsig.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88(3):887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dieckmann M, Dietrich MF, Herz J. Lipoprotein receptors--an evolutionarily ancient multifunctional receptor family. Biol Chem. 2010;391(11):1341–1363. doi: 10.1515/BC.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boucher P, Liu P, Gotthardt M, Hiesberger T, Anderson RG, Herz J. Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low Density lipoprotein receptor-related protein in caveolae. J Biol Chem. 2002;277(18):15507–15513. doi: 10.1074/jbc.M200428200. [DOI] [PubMed] [Google Scholar]
- 41.Loukinova E, Ranganathan S, Kuznetsov S, Gorlatova N, Migliorini MM, Loukinov D, Ulery PG, Mikhailenko I, Lawrence DA, Strickland DK. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function betwenn LRP and the PDGF. J Biol Chem. 2002;277(18):15499–15506. doi: 10.1074/jbc.M200427200. [DOI] [PubMed] [Google Scholar]
- 42.Newton CS, Loukinova E, Mikhailenko I, Ranganathan S, Gao Y, Haudenschild C, Strickland DK. Platelet-derived growth factor receptor-beta (PDGFR-beta) activation promotes its association with the low density lipoprotein receptor-related protein (LRP). Evidence for co-receptor function. J Biol Chem. 2005;280(30):27872–27878. doi: 10.1074/jbc.M505410200. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, van Kerkhof P, Marzolo MP, Strous GJ, Bu G. Identification of a major cyclic AMP-dependent protein kinase A phosphorylation site within the cytoplasmic tail of the low-density lipoprotein receptor-related protein: implication for receptor-mediated endocytosis. Mol Cell Biol. 2001;21(4):1185–1195. doi: 10.1128/MCB.21.4.1185-1195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deal watch: Intra-Cellular Therapies and Takeda to develop PDE1 inhibitors for schizophrenia. Nat Rev Drug Discov. 2011;10(5):329. doi: 10.1038/nrd3438. [DOI] [PubMed] [Google Scholar]
- 45.Abella JV, Park M. Breakdown of endocytosis in the oncogenic activation of receptor tyrosine kinases. Am J Physiol Endocrinol Metab. 2009;296(5):E973–984. doi: 10.1152/ajpendo.90857.2008. [DOI] [PubMed] [Google Scholar]
- 46.Kirisits A, Pils D, Krainer M. Epidermal growth factor receptor degradation: an alternative view of oncogenic pathways. Int J Biochem Cell Biol. 2007;39(12):2173–2182. doi: 10.1016/j.biocel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 47.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8(11):835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 48.Mellman I, Yarden Y. Endocytosis and cancer. Cold Spring Harb Perspect Biol. 2013;5(12):a016949. doi: 10.1101/cshperspect.a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gredmark S, Straat K, Homman-Loudiyi M, Kannisto K, Soderberg-Naucler C. Human cytomegalovirus downregulates expression of receptors for platelet-derived growth factor by smooth muscle cells. J Virol. 2007;81(10):5112–5120. doi: 10.1128/JVI.02197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu K, Murata T, Watanabe Y, Sato C, Morita H, Tagawa T. Characterization of phosphodiesterase 1 in human malignant melanoma cell lines. Anticancer Res. 2009;29(4):1119–1122. [PubMed] [Google Scholar]
- 51.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: Role in Vascular Wall Integrity and Protection from Atherosclerosis. Science. 2003;300(5617):329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 52.Boucher P, Gotthardt M. LRP and PDGF signaling: a pathway to atherosclerosis. Trends Cardiovasc Med. 2004;14(2):55–60. doi: 10.1016/j.tcm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Zhou L, Takayama Y, Boucher P, Tallquist MD, Herz J. LRP1 regulates architecture of the vascular wall by controlling PDGFRbeta-dependent phosphatidylinositol 3-kinase activation. PLoS One. 2009;4(9):e6922. doi: 10.1371/journal.pone.0006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goretzki L, Mueller BM. Low-density-lipoprotein-receptor-related protein (LRP) interacts with a GTP-binding protein. Biochem J. 1998;336(Pt 2):381–386. doi: 10.1042/bj3360381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goretzki L, Mueller BM. Receptor-mediated endocytosis of urokinase-type plasminogen activator is regulated by cAMP-dependent protein kinase. J Cell Sci. 1997;110(Pt 12):1395–1402. doi: 10.1242/jcs.110.12.1395. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, Giachelli CM, Parmacek MS, Raines EW, Rusch NJ, Speer MY, Sturek M, Thyberg J, Towler DA, Weiser-Evans MC, Yan C, Miano JM, Owens GK. Smooth muscle cell plasticity: fact or fiction? Circ Res. 2013;112(1):17–22. doi: 10.1161/CIRCRESAHA.112.281048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of Adventitial Fibroblasts to Neointima Formation and Vascular Remodeling: From Innocent Bystander to Active Participant. Circ Res. 2001;89(12):1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 58.Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8(4):403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 59.Torsney E, Xu Q. Resident vascular progenitor cells. J Mol Cell Cardiol. 2010;50(2):304–311. doi: 10.1016/j.yjmcc.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6(227):227ra234. doi: 10.1126/scitranslmed.3006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.James D, Rafii S. Maladapted endothelial cells flip the mesenchymal switch. Sci Transl Med. 2014;6(227):227fs212. doi: 10.1126/scitranslmed.3008623. [DOI] [PubMed] [Google Scholar]
- 62.Bender AT, Ostenson CL, Wang EH, Beavo JA. Selective up-regulation of PDE1B2 upon monocyte-to-macrophage differentiation. Proc Natl Acad Sci U S A. 2005;102(2):497–502. doi: 10.1073/pnas.0408535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bender AT, Beavo JA. PDE1B2 regulates cGMP and a subset of the phenotypic characteristics acquired upon macrophage differentiation from a monocyte. Proc Natl Acad Sci U S A. 2006;103(2):460–465. doi: 10.1073/pnas.0509972102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PDE1C protein level is increased in synthetic SMCs. Western blotting results showing protein levels of PDE1C and calponin in contractile SMCs (freshly isolated medial layers) and corresponding synthetic SMCs (cultured SMCs) from rat aortas (A), human aortas (B), and human saphenous veins (C). Contractile SMCs are freshly isolated medial tissues procured by removing endothelial cells and pealing off adventitial layers. Synthetic SMCs are cultured growing SMCs isolated from the corresponding vessel with the explant method. (D) Cultured rat aortic SMCs were in differentiation medium (medium 231 supplemented with Smooth Muscle Growth S (SMGS), from Cascade Biologics) or growth medium (medium 231 supplemented with Smooth Muscle Differentiation Supplement (SMDS), from Cascade Biologics) for 2 days. SM-MHC and calponin are used as contractile SMC markers. Values are mean ± SD of triplicate experiments. *P < 0.05.
Supplemental Figure S2. (A) PDE1C is elevated in femoral arteries after wire injury. qRT-PCR showing the PDE1C mRNA levels of in uninjured (right) and injured (left) femoral arteries. C57BL/6J mice were subjected to femoral artery wire injury for 28 days, total RNA were isolated from right uninjured and left injured femoral arteries. The levels of PDE1C mRNA were assessed by qPCR. Values are means ± SD of triplicate (three arteries are pooled together). *P < 0.05. (B) PDE1C are induced in SMC-like cells. Representative images of carotid artery sections subjected to immunofluorescent double staining with SM-α-actin (red) and PDE1C (green), Nuclei were stained wit DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel. The merged images (right panels) show that PDE1C-positive cells are largely overlapped with SM-α-actin-positive cells in the neointimal region. N: neointima.
Supplemental Figure S3. (A and C) PDE1C deficiency or PDE1 inhibition attenuates SMC proliferation in response to vascular injury. Top panels: representative images of carotid artery sections immunostained with SM-α-actin (pink) or Ki67 (brown). Bottom panel: quantitative data of Ki67 positive cells in intima and media. A, PDE1C+/+ and PDE1C-/- mice or were subjected to left common carotid artery ligation for 14 days. C, FVB mice were subjected to left common carotid artery ligation for 14 days in the presence of vehicle or 30 μmol/L IC86340 applied perivascularly via pluronic gel. SM-α-actin and Ki67 immunostaining were performed in cross-sections of carotid arteries. Ki-67-positive cells in the intima and the media were calculated. Values are means ± SEM (n=3). *P < 0.05. N: neointima; M: media. (B-D) PDE1C deficiency suppresses ROS production in carotid arteries after vascular injury. Top panels: Representative images of carotid arteries immunostained with 4-HNE (an oxidative stress marker). C, PDE1C+/+ and PDE1C-/- mice were subjected to left common carotid artery ligation for 14 days. D, FVB mice were subjected to left common carotid artery ligation for 14 days in the presence of vehicle or 30 μmol/L IC86340 applied perivascularly via pluronic gel. 4-HNE immunostaining were performed in cross-sections of carotid arteries. Quantification were performed using Image Pro Plus software. Values are means ± SEM (n=3). *P < 0.05.
Supplemental Figure S4. (A) Real-time RT-PCR showing the mRNA expression of SM-MHC, calponin, and SM-α-actin in low-passage aortic SMCs isolated for PDE1C+/+ and PDE1C-/- mice. (B) Immanofluorescence staining of SM-MHC or SM-α-actin in PDE1C+/+ and PDE1C-/- SMCs. Green: SM-MHC; Red: SM-α-actin; Blue: nuclei.
Supplemental Figure S5. Role of PDE1C in regulating PDGFRβ levels. (A) PDE1A and PDE1C mRNA levels in rat SMCs treated with a high dose or a low dose of adenovirus encoding LacZ shRNA, PDE1A shRNA or PDE1C shRNA. (B-C) PDE1 inhibitor IC86340 or PDE1C shRNA does not affect mRNA levels of multiple growth factor receptors including PDGFRβ, PDGFRα, and EGFR. Levels of mRNA were determined by RT-PCR. (D-E) PDE1 inhibitor IC86340 and PDE1C shRNA reduced the protein levels of multiple growth factor receptors. The protein levels of PDGFRβ, PDGFRα, and EGFR were assessed by immunoblotting. Rat aortic SMCs were treated with indicated doses of PDE1 inhibitor IC86340 for 24 h or transduced with adenovirus encoding shRNA against LacZ, PDE1A,or PDE1C shRNA. (F) PDE1 inhibitor IC86340 reduced exogenously expressed PDGFRβ protein but not PDE1C protein. Rat aortic SMCs were transfected with Flag-PDGFRβ or EGFP-PDE1C via electropolation for 2 days and then treated with 15 μmol/L IC86340 for 24 h. PDGFRβ and PDE1C were detected by anti-Flag and anti-GFP antibodies, respectively. (G) PDGF-stimulated Erk1/2 and AKT activation was attenuated when PDGFR was downregulated. Rat aortic SMCs were treated with 15 μmol/L IC86340 in DMEM with 0.1% FBS for 24 h, washed, and stimulated with 10 ng/ml PDGF-BB for 5 or 30 min. Phospho-Erk1/2, Erk 1, phospho-AKT, and AKT were measured by immunoblotting.
Supplemental Figure S6. Role of tmAC-cAMP signaling in PDE1C-mediated regulation of PDGFRβ. (A) The tmAC inhibitor blocked the synergistic effect of IC86340 and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340, 10 μmol/L forskolin, or both in the presence or absence of tmAC inhibitor 2′,5′-dideoxyadenosine (2′ 5′-ddA,10 μmol/L) for 24 h in DMEM containing 0.1% FBS. (B) The tmAC inhibitor blocked the synergistic effect of PDE1C shRNA and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were transfected with a low dose of adenovirus expressing LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, followed by treatment with 10 μmol/L forskolin in the presence or absence of 10 μmol/L 2′,5′-ddA for 24 h in DMEM containing 0.1% FBS. (C) PKA inhibitor H89 blocked the synergistic effect of IC86340 and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340, or 10 μmol/L forskolin, or both in the presence or absence of 5 μmol/L H89 for 24 h in DMEM containing 0.1% FBS. (D) PKA inhibition by H89 blocked the synergistic effect of PDE1C shRNA and forskolin on PDGFRβ protein reduction. Rat aortic SMCs were transfected with a low dose of adenovirus expressing LacZ-shRNA, PDE1A-shRNA, or PDE1C-shRNA for 3 days, and then treated with 10 μmol/L forskolin in the presence of absence of 5 μmol/L H89 for 24 h in DMEM containing 0.1% FBS. Percentile changes normalized to the left lane. Values are mean ± SD (n= 3). *p<0.05.
Supplemental Figure S7. (A) Epac was not involved in PDE1 inhibition-induced PDGFRβ protein reduction. Rat aortic SMCs were transfected with 50 nmol/L control siRNA or Epac siRNA for 3 days, and then treated with 15 μmol/L IC86340 for 24 h in DMEM containing 0.1% FBS. Right panel showed Epac knockdown by specific siRNA using RT-PCR. (B) PKG1 was not involved in PDE1 inhibition-induced the PDGFRβ protein reduction. Rat aortic SMCs were transfected with 50 nmol/L control siRNA or PKG1 siRNA for 3 days, and treated with 15 μmol/L IC86340 for 24 h in DMEM containing 0.1% FBS. (C-E) cGMP and PDE1 inhibition does not have synergistic effect on PDGFRβ. Rat aortic SMCs were treated with either 5 μmol/L IC86340 alone, one of cGMP elevators (100 μmol/L NO donor SNAP, 10 μmol/L sGC activator YC-1, and 100 nmol/L CNP), or both of IC86340 and a cGMP elevator for 24 h in DMEM containing 0.1% FBS. (F) Lysosome inhibitors abrogated enhanced effect of forskolin and IC86340 on PDGFRβ protein reduction. Rat aortic SMCs were treated with 5 μmol/L IC86340 and 10 μmol/L forskolin in the presence or absence of 20 mmol/L NH4Cl or 50 nmol/L Bafilomycine A for 24 h in DMEM containing 0.1% FBS. Protein levels of PDGFRβ and β-actin equal loading were determined by immunoblotting. PDGFRβ protein and equal loading β-actin were determined by immnoblotting. Quantitative data show percentile changes normalized to the 1st lane. Values are mean ± SD (n= 3). *p<0.05. ns: no significant difference.
Supplemental Figure S8. PDE1C and PDGFRβ co-localization in neointimal lesions. (A) Representative images of carotid artery sections subjected to co-immunofluorescent staining of PDE1C (green) and PDGFRβ (red), and nuclei are stained with DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel. N: neointima. (B) Reprehensive co-immunofluorescent staining images of human coronary artery with neointimal lesions. Green: PDE1C, Red: PDGFRβ, Blue: nuclei stained with DAPI. The merged images (right panels) show that PDE1C-positive cells are largely overlapped with SM-α-actin-positive cells in the neointimal region. M: media, N: neointima.
Supplemental Figure S9. Co-expression of LRP1 with PDE1C or PDGFRβ in neointimal lesions. (A-B) Representative images of carotid artery sections subjected to co-immunofluorescent staining of LRP1 (green) together with PDE1C (red) or PDGFRβ (red), and nuclei are stained with DAPI (blue). FVB mice were subjected to left common carotid artery (LCA) ligation for 14 days. Right common carotid artery (RAC) was used as the control vessel (inset). N: neointima. (C-D) Reprehensive co-immunofluorescent staining images of human coronary artery with neointimal lesions. Green: LRP1, Red: PDE1C or PDGFRβ, Blue: nuclei stained with DAPI. The merged images (right panels) show that LRP1-postive cells are largely overlapped with PDE1C- or PDGFRβ-positive cells in the neointimal region. M: media, N: neointima.