

Abstract
Background
Cystic fibrosis (CF) is an inherited, rare autosomal recessive disease that results in chronically debilitating morbidities and high premature mortality. We evaluated how ivacaftor treatment affected CF symptoms, functioning, and well-being, as measured by the Cystic Fibrosis Questionnaire-Revised (CFQ-R), a widely-used patient-reported outcome (PRO) measure.
Methods
STRIVE, a double-blind, placebo-controlled randomized trial, evaluated ivacaftor (150 mg) in CF patients aged 12+ with the G551D-CFTR mutation for 48 weeks. Treatment effect analysis used a mixed-effects repeated measures model. Treatment benefit analyses applied the cumulative distribution function and a categorical analysis of change scores (“improvement,” “no change,” or “decline”). Content-based interpretation examined treatment effect on specific item responses.
Results
Data from 152 patients with a baseline CFQ-R assessment were analyzed. The treatment effect analysis favored treatment with ivacaftor over placebo on the Body Image, Eating, Health Perceptions, Physical Functioning, Respiratory, Social Functioning, Treatment Burden, and Vitality scales. Findings were supported by the analysis of categorical change. On all CFQ-R scales, the percentage of patients who improved was greater for ivacaftor. In the content-based analysis, the treatment benefit was characterized by better scores across a broad range of domains.
Conclusions
Results illustrate broad benefits of ivacaftor treatment across many domains: respiratory symptoms, physical and social functioning, health perceptions, and vitality, as measured by the CFQ-R. The breadth of improvements reflects the systemic mechanism of action of ivacaftor compared to other therapies. Findings support the patient-reported value of ivacaftor treatment in this patient population.
Trial Registration
ClinicalTrials.gov NCT00909532
Electronic supplementary material
The online version of this article (doi:10.1186/s12955-015-0293-6) contains supplementary material, which is available to authorized users.
Keywords: Cystic fibrosis, Ivacaftor, Patient-reported outcomes, CFQ-R, Health-related quality of life
Background
Cystic Fibrosis (CF) is a rare, autosomal recessive disease affecting multiple organs, including the lungs, pancreas, sweat glands, and intestinal, biliary and reproductive tracts [1]. CF is caused by defects in the CF transmembrane conductance regulatory (CFTR) protein resulting from mutations in the CFTR gene. Some CFTR mutations produce CFTR protein channels that have defective gating (reduced channel open probability), resulting in little to no net chloride ion transport [2–4]. Patients with CF who have at least one CFTR mutation of this type are at high risk for early lung function decline and progression of other disease manifestations. The most prevalent CFTR mutation that predominantly affects CFTR channel open probability is G551D, which is found in approximately 4 % of patients with CF in the United States [5].
Ivacaftor (Kalydeco®) was approved by the FDA in January 2012 for the treatment of CF in patients 6 years of age and older who have the G551D mutation on at least one CFTR allele. The approval was expanded in the US in February 2014 and December 2014 to include 9 additional mutations [6]. All of the currently indicated mutations affect CFTR channel open probability as their primary molecular dysfunction.
The Phase 3 clinical program for ivacaftor was designed to assess the efficacy and safety of ivacaftor treatment in patients with at least one of the specified CFTR mutations. Treatment-related changes in patient-reported outcomes (PROs) were evaluated and have been previously reported for children (ENVISION study), and adolescents and adults (STRIVE study) [7, 8] with at least one G551D-CFTR mutation.
Developers of drugs are expected to provide direct evidence of the treatment benefit as experienced by patients. Providers, regulators, and payers seek information about how patients feel and function in daily life, with evidence from pivotal trials implementing well-defined and reliable assessments. To that end, we expand on data previously reported that show the impact of CF and its treatment on how patients feel and function in everyday life.
The objective of this study was to evaluate the treatment effects of ivacaftor on patient-reported symptoms and health-related quality of life (HRQoL) as reported by patients with CF 12 years of age and older with the G551D-CFTR mutation participating in the STRIVE study. The specific aim was to interpret treatment benefit using innovative analyses of PRO data covering a broad range of HRQoL domains and CF symptoms, specifically: 1) mean changes in Cystic Fibrosis Questionnaire-Revised (CFQ-R) scale scores from baseline through 48 weeks of treatment; 2) patterns of treatment response using empirically-defined categories of change; and 3) examination of treatment using responses to salient items of the CFQ-R that characterized disease impact.
Methods
Data source
STRIVE was a randomized, double-blind, placebo-controlled study evaluating ivacaftor, a CFTR potentiator, in subjects 12 years of age or older with CF and at least one G551D-CFTR mutation. Patients were randomly assigned in a 1:1 ratio to receive 150 mg of ivacaftor or placebo every 12 h for 48 weeks. Throughout the study, all patients continued to take their pre-study medications (with the exception of hypertonic saline). The primary endpoint was the estimated mean absolute change from baseline through week 24 in the percent predicted forced expiratory volume in 1 s (FEV1 % predicted). Secondary endpoints included the change from baseline through week 48 in the FEV1 % predicted, time to first pulmonary exacerbation, change in body weight, change in concentration of sweat chloride, as well as patient-reported scores from the CFQ-R Respiratory Symptoms Scale [9–11].
The study included a 2-week screening period, a 2-week run-in period, and a 48-week treatment period. Efficacy and safety findings from this study have been previously reported [9].
Cystic fibrosis questionnaire-revised
The CFQ-R was the first disease-specific PRO instrument developed for use by patients with CF and their caregivers [12]. The original CFQ was developed from qualitative and quantitative studies which included a conceptual framework, interviews with patients, parents, and health care professionals, cognitive testing, and psychometric evaluation [10, 12–14]. Modifications were subsequently made to the instrument leading to the current revised version, which has been identified as a widely used PRO measure for CF [15–17].
Three versions of the CFQ-R are available: a self-completed Teen/Adult version for patients with CF aged 14 and older, a Child version for children aged 6–13 years (self-report for ages 12–13 and interviewer-administered for ages 6–11), and a proxy-completed Parent/Caregiver version. The Teen/Adult and Parent/Caregiver versions include the following scales: Body Image, Digestive Symptoms, Eating Problems, Emotional Functioning, Health Perceptions, Physical Functioning, Respiratory Symptoms, Role Functioning, Social Functioning, Treatment Burden, Vitality, and Weight. The Child version does not include Health Perceptions, Role Functioning, Vitality, and Weight. Items included in each scale of the CFQ-R are summed and standardized to a 0–100 scale, with higher scores indicating better outcomes or fewer symptoms from the patient perspective.
In the STRIVE study, the CFQ-R was administered at the beginning of the run-in period, baseline, day 15, week 8, and every 8 weeks thereafter, up to week 48. Patients aged 14 and older completed the Teen/Adult version while those aged 12 and 13 at baseline completed the Child version. Parents of children aged 12 and 13 at baseline also completed the Parent/Caregiver version. This analysis included pooled data from the two self-completed Teen/Adult and Child versions (data from the Parent/Caregiver version were not included).
Analyses
Treatment-related changes in CFQ-R domains
For each scale of the CFQ-R, mean changes from baseline through week 48 were evaluated by treatment group using a mixed-effects model for repeated measures [18] with absolute change from baseline as the dependent variable, fixed effects for study visit and treatment group, and adjustment for continuous baseline values of age, FEV1 % predicted, and baseline scale score, using an unstructured covariance matrix. Treatment effect was calculated as the difference in mean change from baseline between the ivacaftor and placebo groups. A p-value of <0.05 was employed to assess statistical significance.
In addition to evaluating treatment benefit as mean change from baseline, the current analyses also included evaluating treatment group differences in terms of the percentage of patients reporting various levels of change across the different CFQ-R domains. First, to evaluate treatment group differences across the entire range of observed change, we used a method based on the cumulative distribution function (CDF). The CDF of change scores presents the proportion of patients who experienced an improvement or decrement at or below a specific value [19]. When presented separately by treatment group, CDF plots illustrate the separation between groups at each threshold of change. For evaluating treatment response, CDFs are most easily interpreted when the y-axis is reversed to show the proportion of patients with a score change > |X|, an approach known as cumulative response curves (CRCs) [20]. We evaluated the CRCs for change from baseline to week 48 by CFQ-R scale, by treatment group. Statistical significance was assessed using the two-sample Kolmogorov-Smirnov test.
In addition to the CRC analysis, we used a distribution-based methodology [21] to establish threshold values that help interpret the magnitude of change observed in HRQoL scores. One-half standard deviation (SD) of the change from beginning to end of the 2-week run-in period (2 weeks prior to study drug initiation) was used as a threshold value [22] to identify minimal important change. The direction and magnitude of score changes were used to analyze treatment response. Positive or negative changes from baseline to week 48 exceeding (in absolute value) the threshold were considered “improvements” or “declines”, respectively. Change scores smaller (in absolute value) than the threshold value were considered “no change”. The Chi-square test was used to assess statistical significance of treatment group differences.
Sensitivity analyses were conducted using the standard error of measurement (SEM). The SEM reflects the measurement precision and the amount of random variation from repeated assessments. One SEM has been proposed as a measure of minimal clinically important difference (MCID) and was used to identify “improvement”, “no change”, or “decline” [23], with the Chi-square test for assessment of group differences. The SEM was evaluated as , where SD∆ is the SD of change scores from the run-in period and the ICC is the intraclass correlation coefficient, a measure of reliability related to the repeated administration of the same test under the assumption that substantial change in the concept of interest has not occurred.
Content-based interpretation of treatment effects
These analyses examined the impact of treatment on responses to specific “sentinel” CFQ-R items empirically selected to represent each scale. Polyserial or Spearman [24] correlations between change in each item and change in its scale were calculated. The item with the strongest association with its scale change score was identified as the sentinel item. Response options for each sentinel item were collapsed into “none” vs. “any” impairment. The change in percent of patients with no impairment was evaluated by treatment group at baseline and week 48.
Ethics
The STRIVE clinical trial (“A Phase 3, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of VX-770 in Subjects with Cystic Fibrosis and the G551D Mutation”) protocol was reviewed and approved by the institutional review board at each participating center, and each subject provided written informed consent or written or oral assent.
Results and discussion
Characteristics of study sample
The STRIVE study randomized 167 subjects to two treatment arms. Of these, 161 subjects subsequently received at least one dose (intention-to-treat analysis). This study sample contained the 152 patients who completed a baseline CFQ-R assessment. The two treatment groups were similar in terms of gender (53.0 % female) and mean age (25.7 years). Most patients were 18 years of age or older (77.6 %). At baseline, the treatment groups also were similar in average FEV1 % predicted (64.6 %), mean sweat chloride (100.4 mmol/L), height (167 cm), body weight (78.9 kg), and body mass index (BMI) (21.9 kg/m2).
Treatment-related changes in CFQ-R scales
Figure 1 shows the observed mean and standard error of the change from baseline at each study visit, by scale and treatment group. Mean change on the Body Image, Eating Problems, Health Perceptions, Physical Functioning, Respiratory Symptoms, Social Functioning, Treatment Burden, and Vitality scales was consistently higher for ivacaftor vs. placebo. Favorable effects of ivacaftor were observed within the first 2 months of treatment initiation.
Fig. 1.
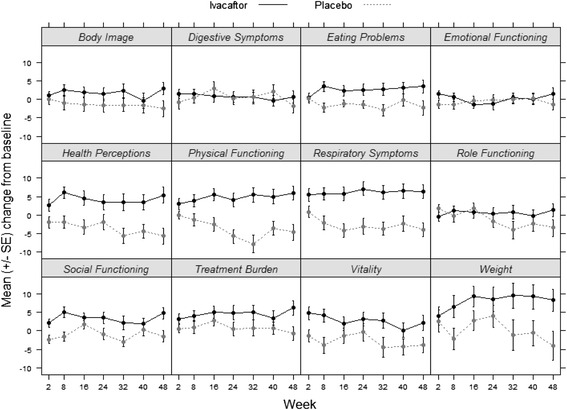
Change from baseline in CFQ-R scores for each visit by treatment group (observed data). SE = standard error; means and standard error are unadjusted
Table 1 presents baseline score, mean change from baseline through week 48, and treatment effects (difference between treatment groups in mean change) by CFQ-R scale and treatment group. For seven of the twelve CFQ-R scales, the mean post-baseline change was statistically significant, after adjustment for age, FEV1 % predicted, and baseline CFQ-R scale score. The largest treatment effect was found on the Respiratory Symptoms scale with a mean improvement of 5.9 points observed among patients receiving ivacaftor, exceeding the MCID value of 4 points, vs. a mean decline of 2.7 points among those receiving placebo (treatment effect of 8.6 points, P < 0.001) [12].
Table 1.
CFQ-R adjusted change scores from baseline through week 48 by treatment group
| CFQ-R Scale | Placebo | Ivacaftor | ||||
|---|---|---|---|---|---|---|
| (N = 70b) | (N = 80b) | |||||
| Baseline | Change | Baseline | Change | Treatment effect | p-valuea | |
| Body Image | 80.3 | −1.2 | 81.0 | 1.5 | 2.7 | 0.086 |
| Digestive Symptoms | 85.4 | 0.4 | 85.2 | 0.8 | 0.5 | 0.732 |
| Eating Problems | 91.9 | −1.1 | 91.8 | 2.2 | 3.3 | 0.002 |
| Emotional Functioning | 83.6 | −1.4 | 86.0 | 0.7 | 2.1 | 0.096 |
| Health Perceptions | 71.7 | −3.6 | 72.1 | 4.0 | 7.6 | <0.001 |
| Physical Functioning | 80.2 | −1.7 | 76.1 | 2.7 | 4.4 | 0.006 |
| Respiratory Symptoms | 68.5c | −2.7 | 70.2 | 5.9 | 8.6 | <0.001 |
| Role Functioning | 85.9 | 0.1 | 86.3 | −0.5 | −0.6 | 0.651 |
| Social Functioning | 71.9 | −1.0 | 72.1 | 3.3 | 4.3 | 0.003 |
| Treatment Burden | 65.7 | 1.0 | 64.5 | 4.3 | 3.3 | 0.042 |
| Vitality | 64.7 | −2.8 | 64.3 | 2.7 | 5.5 | 0.002 |
| Weight | 78.1 | 1.7 | 79.0 | 6.9 | 5.3 | 0.053 |
Health Perceptions, Role Functioning, Vitality and Weight are not included in the Child self-report version of the CFQ-R; results for these scales are based on the Teen/Adult version only (N = 64 for placebo and N = 76 for Ivacaftor)
a P-value for overall post-baseline treatment effect, estimated using a mixed-effect model for repeated measures with fixed effects for study visit, treatment group, and adjustment for continuous baseline values of age, percent predicted FEV1 and domain score
bAnalysis sample includes patients with a baseline assessment and at least one post-baseline assessment
c n = 71
Figure 2 presents CRCs of change scores from baseline to week 48, by treatment group. For nearly all CFQ-R scales, lower cumulative change scores (worse outcomes) were seen for the placebo group, indicated by the dashed curve for the placebo group appearing to the left of the solid curve for ivacaftor. These findings suggest that the proportion of subjects who exceeded a particular level of change (better outcomes) was higher in the ivacaftor than placebo group. Differences between treatment groups (favoring ivacaftor) were greatest for Respiratory Symptoms (p < 0.001), Physical Functioning (p = 0.002), Health Perceptions (p = 0.019), and Vitality (p = 0.030). Using illustrative values of 5 and 10 points, the percentage of patients with change scores greater than 5 on the Respiratory Symptoms scale was 59 % for ivacaftor vs. 27 % in the placebo group. For changes greater than 10 points, percentages were 47 and 11 % for ivacaftor and placebo, respectively. For change scores of 5 and 10 points on Physical Functioning, percentages of ivacaftor-treated patients with improvement were 36 and 23 %, respectively, compared to 13 and 8 % in the placebo group.
Fig. 2.
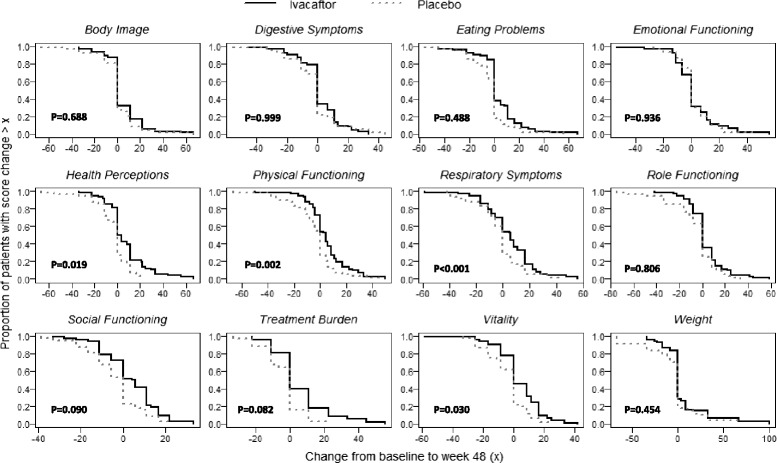
Cumulative response curves for CFQ-R scales by treatment group. P-values from the Kolmogorov-Smirnov two-sample test for equality of cumulative distribution functions
Figure 3 presents the percentage of patients who experienced “improvement”, “no change”, or “decline” after 48 weeks of treatment with either ivacaftor or placebo, according to our change threshold of 0.5 SD. Differences favoring ivacaftor were observed for Respiratory Symptoms (P < 0.001), Social Functioning (P = 0.026), Vitality (P = 0.006), Treatment Burden (P = 0.016), Health Perceptions (P = 0.003), Physical Functioning (P < 0.001), Eating Problems (P = 0.015), and Weight (P = 0.015).
Fig. 3.
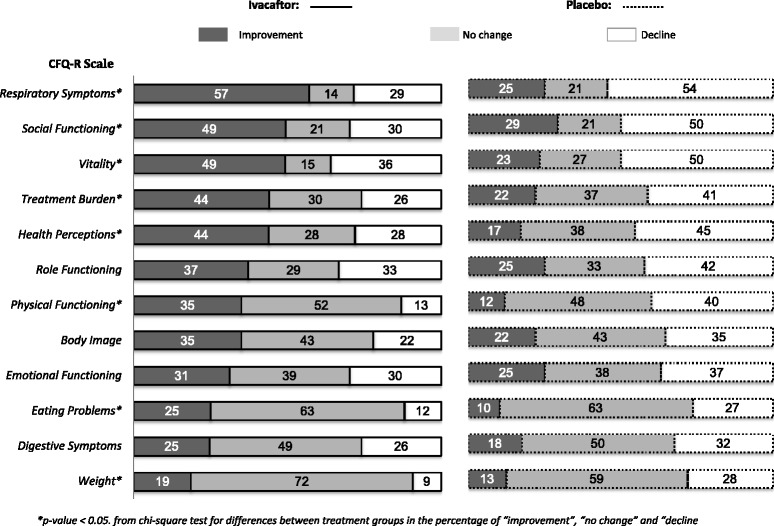
Analysis of categorical change from baseline to week 48 by CFQ-R scale and treatment group. *p-value < 0.05. from chi-square test for differences between treatment groups in the percentage of “improvement”, “no change” and “decline” patients
Results were similar under the 1 SEM criterion (see Additional file 1: Tables S1 and Additional file 2: Table S2).
Content-based interpretation of treatment effects
Table 2 shows the sentinel items by CFQ-R scale and the baseline percentage of patients reporting “no impairment”.
Table 2.
Percentage of patients with “no impairment” in representative CFQ-R items at baseline and 48 weeks
| Representative items | % No Impairment at baseline (pooled) | |||
|---|---|---|---|---|
| CFQ-R scale | Teen-adult version/Child version | Response optiona | Corr.b with domain change score | |
| Body image | Look different from others | Very false/Not at all true | 0.82 | 60.0 |
| Digestive symptoms | Problems with gas/stomach hurt | Never | 0.85c | 48.1 |
| Eating problems | Force myself to eat/pushed to eat | Very false/Never | 0.82c | 77.7 |
| Emotional functioning | Felt sad/worried | Never | 0.79 | 71.1 |
| Health perceptions | Feel healthy | Very true | 0.86 | 32.5 |
| Physical functioning | Walking as fast as others | No difficulty | 0.85 | 65.9 |
| Respiratory symptoms | Coughing | Not at all/Never | 0.85 | 3.7 |
| Role functioning | Running errands out of the house | Never | 0.85 | 41.0 |
| Social functioning | Comfortable going out/got together with friends | Very true | 0.66 | 59.2 |
| Treatment burden | Tx makes life more difficult/Tx bothered you | Not at all/Not at all true | 0.78 | 25.1 |
| Vitality | Felt exhausted/grouchy | Never | 0.81 | 47.4 |
| Weightd | Trouble gaining weight | Not at all | e | 62.0 |
aIdentified to represent patient report of no impairment
bPolyserial correlation unless otherwise noted
cSpearman correlation
dSingle item scale
eNot applicable for single item scales
Pooled = ivacaftor and placebo
The proportion of patients treated with ivacaftor who showed “no impairment” generally increased between baseline and week 48 and decreased among patients treated with placebo (data not shown). The items with at least 10 % separation between ivacaftor and placebo at week 48 were: “feel healthy” (17.5 % separation), “walking as fast as others” (15.1 %), “coughing” (14.1 %), “going out with friends” (14.1 %), “trouble gaining weight” (14.0 %), “force myself to eat” (13.5 %), and “treatment makes life more difficult” (12.5 %).
Figure 4 presents the change in percentage with “no impairment” from baseline to week 48 by treatment group. As before, the percentage with “no impairment” increased with ivacaftor and decreased with placebo between baseline and week 48 for most items, eg, “walking as fast as others”(+13.7 vs. −10.7 %, respectively), “feel healthy” (+10.2 vs. −4.5 %), “running errands out of the house” (+18.0 vs. −1.2 %), “going out with friends” (+11.2 vs. −0.7 %), [never feeling] “bothered by treatment” (+15.7 vs. −1.0 %).
Fig. 4.
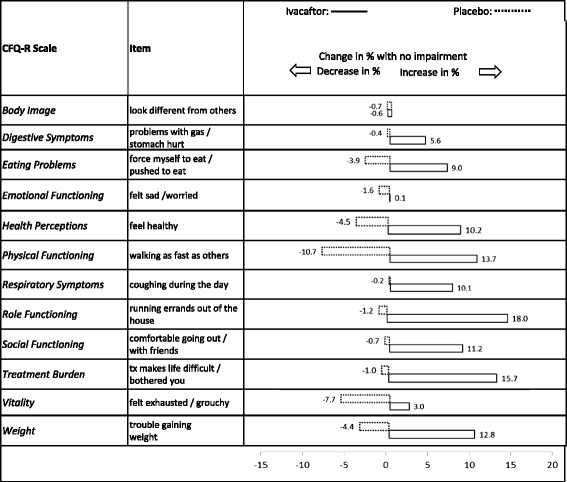
Change in the percentage of patients with no impairment after 48 weeks of treatment
Conclusions
Our analyses provide empirical data describing and quantifying the benefits of ivacaftor treatment in patients aged 12 years and older with CF and the G551D-CFTR mutation.
In the STRIVE study, early and sustained treatment effects from ivacaftor were observed through week 48 on the CFQ-R Respiratory Symptom scale. The magnitude and significance of the improvements in Respiratory Symptoms was consistently observed across the analyses in line with improvements in % predicted FEV1 in the study population. Of interest, sustained improvements were also observed on other scales of the CFQ-R, including Physical Functioning, Social Functioning, Eating Problems, Treatment Burden, Health Perceptions, and Vitality. Trends also were favorable for ivacaftor-treated patients on the Body Image, Digestive Symptoms, Role Functioning, and Emotional Functioning scales, but their magnitude was smaller and failed to reach statistical significance.
These positive changes across multiple CFQ-R scales are supported by findings from categorical change analyses. Using change scores classified into “improvement”, “no change”, or “decline”, the treatment effects favoring ivacaftor over placebo became even more apparent. On all 12 CFQ-R scales, the percentage who improved by week 48 was greater for ivacaftor-treated patients. Further, the percentage who declined was greater for placebo-treated patients, although differences across the three categories of change were not statistically significant for Role Functioning, Body Image, Emotional Functioning, and Digestive Symptoms. For the Respiratory Symptoms and Physical Functioning scales, the ivacaftor group included at least five times more patients with improvements compared to placebo. For the Social Functioning, Vitality, Treatment Burden, and Role Functioning scales, the ivacaftor group included three to five times more patients with improvements compared to placebo. Worsening scores through week 48 were more prevalent among patients on placebo. Of particular interest are the Weight scores, with a three-fold difference between ivacaftor and placebo in the percentage of patients who declined during the study (9 vs. 28 %).
The content-based analysis of sentinel items-showed that ivacaftor led to improvements in a broad range of functional outcomes, including the respiratory, nutritional, physical, social, and treatment-related domains.
Although several trial-based publications have documented treatment benefits using the CFQ-R, they have focused on selected scales: Respiratory Symptoms, Physical Functioning, and Vitality [25–30]. Additional data, particularly from observational studies, would provide needed real-world evidence to complement trial-based data on the patient-reported benefits associated with ivacaftor treatment. In the PERSIST open-label extension study sample, including 75 % (n = 144) of the STRIVE sample, the effect of ivacaftor on the CFQ-R Respiratory Symptoms scale was maintained over an additional 96 weeks of treatment [31]. In an observational study of ivacaftor (US GOAL), clinically meaningful and statistically significant gains (7.4 points, p < 0.0001) were observed in the Respiratory Symptoms scale after 6 months of treatment [32].
Clinicians, patients, and other stakeholders will benefit from understanding the impact of ivacaftor treatment on patients with selected CF mutations using a patient-centered point-of-view. We have presented direct evidence of therapy effects, extending the interpretation of treatment benefit beyond the previously reported clinical markers. These results complement the clinical results, providing evidence of how patients feel and function in daily life. Our findings suggest that ivacaftor treatment led to significant improvements that were substantial, sustained over 48 weeks, and spanned a wide range of symptoms, functioning, and well-being in patients with the G551D-CFTR mutation in the STRIVE study.
Results from the STRIVE study illustrate broad benefits of ivacaftor treatment across highly salient aspects of HRQoL: respiratory symptoms, physical and social functioning, health perceptions, and vitality as measured by the CFQ-R in patients with CF 12 years of age and older with the G551D-CFTR mutation. The breadth of improvements reflects the systemic mechanism of action of ivacaftor compared to other (symptomatic) therapies. Our results are supportive of the patient-reported value of ivacaftor treatment in this patient population.
Limitations: The analyses reported here are post-hoc in nature (not pre-specified). Although an MCID has been established for the Respiratory Symptoms scale [12], no such benchmarks for interpretation of change exist for the remaining CFQ-R scales. Further, we used distribution-based methods to identify thresholds of change [21] because use of an anchor-based method was not possible using these data. The STRIVE study included only patients with the G551D-CFTR mutation. As noted, our findings are based on results of a clinical trial and the magnitude of treatment effects of ivacaftor on patient-reported outcomes may differ in actual clinical practice.
Acknowledgements
We extend our deepest appreciation to the adolescents and adults with CF who participated in the STRIVE study. Medical writing assistance was provided by Caroline Jennermann of Optum. Editorial input to earlier versions of the manuscript was provided by Cecile LeCamus MD, Barry Lubarsky PhD, and Jeff Wagener MD. The STRIVE clinical trial was funded and conducted by Vertex Pharmaceuticals Incorporated, Boston, Massachusetts. This post hoc analysis of data from STRIVE was also funded by Vertex Pharmaceuticals; however, the conception and design of these analyses and the interpretation of results was guided by all authors, as described above in “Authors’ Contributions”. Martha Bayliss is the guarantor of this paper.
Additional files
Categorical change from baseline-week 48 by CFQ-R scale and treatment group: 0.5 SD threshold.
Categorical change from baseline-week 48 by CFQ-R scale and treatment group: SEM threshold.
Footnotes
Competing interests
Author AQ has received Investigator-initiated grants from Vertex and Gilead Sciences and consulting income from AbbVie, Vertex, and Gilead. Authors ES, BC, and MV-L are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. Authors RR-B and MB are employed by Optum, which has been contracted by Vertex to carry out research activities related to this presentation; these authors have no conflicts of interest to disclose. Author IS-G as Principal Investigator received grant supports for conduct of clinical trials from Vertex Pharmaceuticals Incorporated. The CFQ-R is free to use for clinical purposes once the electronic copyright form is submitted.
Authors’ contributions
Qualifications for authorship have been met as follows: study conception and design (AQ, ES, MV-L, BC, MB, RR-B); acquisition of data (AQ, ES, MV-L, BC, IS-G); analysis and interpretation of data (AQ, ES, MV-L, BC, MB, R-RB); drafting of manuscript (MV-L, MB, and RR-B); and critical revision (all authors). All authors read and approved the final manuscript.
Contributor Information
Alexandra Quittner, Email: aquittner@miami.edu.
Ellison Suthoff, Email: ellison_suthoff@vrtx.com.
Regina Rendas-Baum, Email: rrbaum@qualitymetric.com.
Martha S. Bayliss, Email: mbayliss@qualitymetric.com
Isabelle Sermet-Gaudelus, Email: isabelle.sermet@nck.aphp.fr.
Brenda Castiglione, Email: brenda_castiglione@vrtx.com.
Montserrat Vera-Llonch, Email: montserrat_vera-llonch@vrtx.com.
References
- 1.Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–70. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 2.Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros. 2012;11:237–45. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Wilschanski M, Dupuis A, Ellis L, Jarvi K, Zielenski J, Tullis E, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med. 2006;174:787–94. doi: 10.1164/rccm.200509-1377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation Patient Registry. 2012 Annual Data Report. Cystic Fibrosis Foundation. 2012. http://www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf.
- 6.KALYDECO® Prescribing Information. Vertex Pharmaceuticals, Inc. 2014. http://pi.vrtx.com/files/uspi_ivacaftor.pdf.
- 7.Quittner AL, Ramsey B, Dong Q, Yen K, Elborn JS. Patient-reported outcomes in Phase 3 trials of ivacaftor in subjects with CF who have the G551D-CFTR mutation. J Cyst Fibros. 2012;11(Suppl 1):S67. doi: 10.1016/S1569-1993(12)60212-4. [DOI] [Google Scholar]
- 8.Quittner AL, Ramsey B, Dong Q, Yen K, Elborn JS. Patient-reported outcomes in Phase 3 trials of ivacaftor in patients with CF who have the G551D-CFTR mutation. Pediatr Pulmonol. 2012;47(Suppl 35):297. [Google Scholar]
- 9.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quittner AL, Sawicki GS, McMullen A, Rasouliyan L, Pasta DL, Yegin A, et al. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Qual Life Res. 2012;21:1267–78. doi: 10.1007/s11136-011-0036-z. [DOI] [PubMed] [Google Scholar]
- 11.Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimally clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest. 2009;135:1610–8. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quittner AL, Buu A, Messer MA, Modi AC, Watrous M. Development and validation of the cystic fibrosis questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest. 2005;128:2347–54. doi: 10.1378/chest.128.4.2347. [DOI] [PubMed] [Google Scholar]
- 13.Henry B, Aussage P, Grosskopf C, Launois R. Constructing a disease-specific quality of life questionnaire for children and adults with cystic fibrosis. Isr J Med Sci. 1996;32(Suppl):S181. [Google Scholar]
- 14.Henry B, Aussage P, Grosskopf C, Goehrs JM. Development of the Cystic Fibrosis Questionnaire (CFQ) for assessing quality of life in pediatric and adult patients. Qual Life Res. 2003;12:63–76. doi: 10.1023/A:1022037320039. [DOI] [PubMed] [Google Scholar]
- 15.Goss CH, Quittner AL. Patient-reported outcomes in cystic fibrosis. Proc Am Thorac Soc. 2007;4:378–86. doi: 10.1513/pats.200703-039BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palermo TM, Long AC, Lewandowski AS, Drotar D, Quittner AL, Walker LS. Evidence-based assessment of health-related quality of life and functional impairment in pediatric psychology. J Pediatr Pscyhol. 2008;33:983–96. doi: 10.1093/jpepsy/jsn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Committee for Medicinal Products for Human Use (CHMP). Guideline on the clinical development of medicinal products for the treatment of cystic fibrosis. European Medicines Agency. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017055.pdf. Accessed 21 Apr 2014.
- 18.Siddiqui O, Hung HM, O’Neill R. MMRM vs. LOCF: a comprehensive comparison based on simulation study and 25 NDA datasets. J Biopharm Stat. 2009;19:227–46. doi: 10.1080/10543400802609797. [DOI] [PubMed] [Google Scholar]
- 19.McLeod LD, Coon CD, Martin SA, Fehnel SE, Hays RD. Interpreting patient-reported outcome results: U.S. FDA guidance and emerging methods. Expert Rev Pharmacoecon Outcomes Res. 2011;11:163–9. doi: 10.1586/erp.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cappelleri JC, Zou KH, Bushmakin AG, Carlsson MO, Symonds T. Cumulative response curves to enhance interpretation of treatment differences on the self-esteem and relationship questionnaire for men with erectile dysfunction. BJU Int. 2013;111:E115–20. doi: 10.1111/j.1464-410X.2012.11489.x. [DOI] [PubMed] [Google Scholar]
- 21.Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. J Clin Epidemiol. 2008;61:102–9. doi: 10.1016/j.jclinepi.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Guyatt GH, Bombardier C, Tugwell PX. Measuring disease-specific quality of life in clinical trials. CMAJ. 1986;134:889–95. [PMC free article] [PubMed] [Google Scholar]
- 23.Crosby RD, Kolotkin RL, Williams GR. Defining clinically meaningful change in health-related quality of life. J Clin Epidemiol. 2003;56:395–407. doi: 10.1016/S0895-4356(03)00044-1. [DOI] [PubMed] [Google Scholar]
- 24.Cox NR. Estimation of the correlation between a continuous and a discrete variable. Biometrics. 1974;30:171–8. doi: 10.2307/2529626. [DOI] [PubMed] [Google Scholar]
- 25.McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB. Inhaled azteronam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med. 2008;178:921–8. doi: 10.1164/rccm.200712-1804OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Retsch-Bogart GZ, Quittner AL, Gibson RL, Oermann CM, McCoy KS, Montgomery AB, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest. 2009;135:1223–32. doi: 10.1378/chest.08-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oermann CM, Retsch-Bogart GZ, Quittner AL, Gibson RL, McCoy KS, Montgomery AB, et al. An 18-month study of the safety and efficacy of repeated courses of inhaled aztreonam lysine in cystic fibrosis. Pediatr Pulmonol. 2010;45:1121–34. doi: 10.1002/ppul.21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–50. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 29.Saiman L, Marshall BC, Mayer-Hamblett N, Burns JL, Quittner AL, Cibene DA, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–56. doi: 10.1001/jama.290.13.1749. [DOI] [PubMed] [Google Scholar]
- 30.Bilton D, Robinson P, Cooper P, Gallagher CG, Kolbe J, Fox H, et al. Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. Eur Respir J. 2011;28:1071–80. doi: 10.1183/09031936.00187510. [DOI] [PubMed] [Google Scholar]
- 31.McKone E, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the G551D-CFTR mutation: Response through 144 weeks of treatment (96 weeks of PERSIST) Pediatr Pulmonol. 2013;48(Suppl 36):287. [Google Scholar]
- 32.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175–84. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]