

Abstract
Background
Foodborne outbreaks of Salmonella remain a pressing public health concern. We recently detected a large outbreak of Salmonella enterica serovar Enteritidis phage type 14b affecting more than 30 patients in our hospital. This outbreak was linked to community, national and European-wide cases. Hospital patients with Salmonella are at high risk, and require a rapid response. We initially investigated this outbreak by whole-genome sequencing using a novel rapid protocol on the Illumina MiSeq; we then integrated these data with whole-genome data from surveillance sequencing, thereby placing the outbreak in a national context. Additionally, we investigated the potential of a newly released sequencing technology, the MinION from Oxford Nanopore Technologies, in the management of a hospital outbreak of Salmonella.
Results
We demonstrate that rapid MiSeq sequencing can reduce the time to answer compared to the standard sequencing protocol with no impact on the results. We show, for the first time, that the MinION can acquire clinically relevant information in real time and within minutes of a DNA library being loaded. MinION sequencing permits confident assignment to species level within 20 min. Using a novel streaming phylogenetic placement method samples can be assigned to a serotype in 40 min and determined to be part of the outbreak in less than 2 h.
Conclusions
Both approaches yielded reliable and actionable clinical information on the Salmonella outbreak in less than half a day. The rapid availability of such information may facilitate more informed epidemiological investigations and influence infection control practices.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0677-2) contains supplementary material, which is available to authorized users.
Background
Outbreaks of Salmonella from contaminated food are frequently reported in the community, with 1.2 million cases estimated to occur in the US each year [1]. In a population-based study in the UK in 2008–2009, there were >38,600 estimated cases of salmonellosis and 11,300 patients presenting to a primary care physician [2]. Hospital outbreaks of Salmonella may result from patient-to-patient spread and can be lethal in vulnerable patients [3–5]. An example is the hospital outbreak at Stanley Royd Hospital in the UK which led to the deaths of 19 patients and a public inquiry [2, 6]. We recently detected a cluster of more than 30 cases of Salmonella enterica serovar Enteritidis (S. Enteritidis) over a 3-week period at one of three hospital sites in our hospital organisation and from the community. This was against a typical background incidence of five to eight cases per month of all S. enterica isolates in the area served by our hospital. Initially a small number of seemingly unrelated, presumed community-acquired cases were detected on different wards but subsequently a larger number of long-term inpatients on two adjoining wards were affected suggesting the possibility of spread within the hospital. Simultaneously, an increase in community isolates was also detected. At first, it was unclear whether hospital cases were reflecting multiple imports from a community outbreak or spread within the hospital or both. Due to the explosive nature of the outbreak, coupled with uncertainty about the source, a rapid response was required to ensure that infection control measures were appropriately targeted. Outbreak investigations are aided by rapid availability of whole-genome sequencing (WGS) data, as this provides the greatest level of discrimination between isolates when compared to traditional typing methods such as phage typing, multilocus variable number tandem repeat analysis (MLVA) and pulsed-field gel electrophoresis (PFGE) [3–5, 7]. The Illumina MiSeq sequencing platform has emerged as the gold standard for WGS investigations of outbreaks, but results may not be available for as long as 3 working days, depending on the protocol used [8–10]. A number of studies have evaluated the utility of WGS for typing S. enterica isolates; however, to the authors’ knowledge, this is the first use of prospective typing of this organism during an outbreak. Rapid availability of accurate typing results is critical to effective outbreak control. We therefore devised a novel rapid draft sequencing protocol on the MiSeq generating results in under 6 h following library preparation. At the time of the outbreak we were testing a portable, handheld, ‘USB stick’, whole-genome sequencer, the MinION (Oxford Nanopore Technologies, UK), as part of their early access programme. We wished to see what role this technology might play in the management of future outbreaks.
Our initial goals when performing sequencing prospectively were: (1) to determine if cases in the hospital were from the same strain as those circulating in the community, and to discriminate outbreak cases from normal background S. enterica strains; (2) to determine whether there was evidence of a super-shedder patient or specific breakdown in infection control practices; (3) to help link cases to a primary source (for example, person or food) and to compare to previous outbreak strains; and (4) to integrate these results with national surveillance data.
Results and discussion
Epidemiological investigation
In total, 43 isolates of S. Enteritidis were identified in the study period (1 to 24 June) from inpatients, community samples from general practitioners and from environmental isolates. Hospitalised cases were only identified at one hospital site in the group of three hospitals. The same hospital food is distributed to all three hospital sites from a single, central kitchen processing unit where hot food is twice-cooked to standards that would kill salmonellae. All microbiological testing of hospital food was negative for Salmonella. The environmental swabs from affected wards were all negative apart from one isolate of S. Enteritidis recovered from the outside door seal of a food regeneration trolley. This proved to be of the outbreak type. Four separate colony picks were sequenced from this culture. Isolates from staff were sent to the reference laboratory by another laboratory in a different city 14 miles away. These were detected in faecal samples submitted by general practitioners and were found to belong to staff at our hospital working on the affected wards. The first 16 samples, of which six cases had onset dates compatible with community acquisition, were available for sequencing on 10 June and 13 samples sequenced successfully. These were subsequently shown to be distinct from other isolates recently sequenced by national surveillance and were identical to each other, apart from three cases that each had one SNP difference (Fig. 1, Panel a).
Fig. 1.
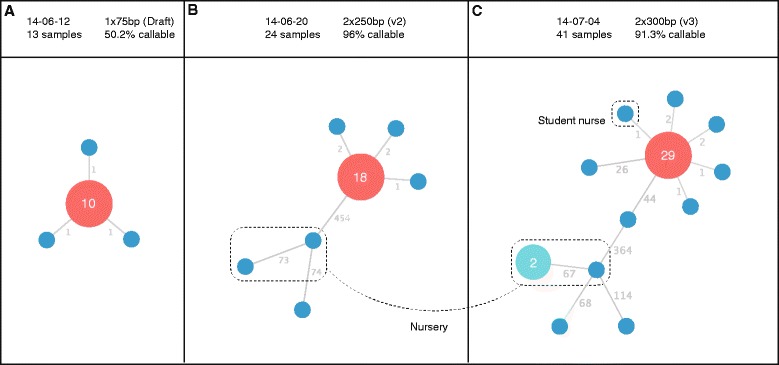
Phylogenetic reconstruction of the outbreak, as sequencing data were generated at three time points during the outbreak (corresponding to Panels a, b and c). The red node demonstrates the most frequently occurring SNP type. Node labels show the number of isolates of this type. Blue nodes are singletons. Edge labels show the number of SNPs between isolates. The turquoise node shows a cluster of two identical isolates from cases associated with a nursery. The header bar shows the date of the sequencing, the Illumina sequencing protocol used, the number of samples in the dataset and the total percentage of the S. Enteritidis reference genome which could be used for SNP calling (core genome). Subtle differences in the number of SNPs between nodes on the tree may be seen, for example in the unrelated nursery isolates (6 SNPs different between Panel a and Panel b). These differences are attributed to differences in core genome size, that is, the number of positions in the reference genome used to generate the results. The precise core genome size figures are shown as a percentage in the panel headings
Sequencing of isolates from two early patient cases on 3 and 4 June showed them to be identical. As both patients had been hospitalised for longer than the Salmonella incubation period this was strongly suggestive of hospital acquisition. There were nine other cases on or prior to 4 June, which together with the typing data helped to inform further infection control actions. All symptomatic patients were isolated and the two wards were closed. Deep cleaning was undertaken with vaporised hydrogen peroxide sterilisation. Four isolates from the later part of the outbreak were identified by SNP typing to be unrelated to the outbreak type. Two of the four isolates were from young children who had recently returned from separate holidays in Egypt. These isolates were different to each other but one was identical to another isolate from a child of similar age who had not travelled abroad, which prompted further epidemiological investigation. It emerged that the two children attended the same nursery in a town just outside the city in which the hospital outbreak had been detected, strongly suggesting transmission had occurred within the nursery (Fig. 1).
The earliest date of onset was 25 May and the last 8 July. A total of 37 cases with the outbreak strain were identified of which six had no connection with the hospital, eight were staff members and three were asymptomatic carriers identified by screening patients on outbreak wards. Comparison of the outbreak genome sequence with the Public Health England database of strains from the whole of England and Wales suggested a very close relationship to six isolates from London, Bedford and Northampton. Further epidemiological investigation of these cases found no link to Birmingham or a foodstuff. The outbreak strain was PT 14b and multi-locus variable number tandem repeat (MLVA) type 2-11-9-7-4-3-2-8-9, an uncommon type for PT 14b strains.
Rapid draft sequencing on the Illumina MiSeq
As an initial response to the outbreak, isolates from 16 patients were sequenced overnight on the Illumina MiSeq on 12 June 2014 in order to generate results for an infection control meeting the following day (results shown in Fig. 1 Panel a). To enable this, we devised a new draft sequencing protocol that reduced the run time of the MiSeq instrument to 6 h (contrasted with standard protocols which can take up to 55 h to complete). This was achieved by reducing the read length, cycle time and number of tiles imaged. Of the 16 isolates, 13 had a mean coverage depth of greater than 4× (mean 8×) and could be used for further analysis. Due to the lower coverage of strains, 50.2 % of the core genome was used to generate these results. Despite this, the results generated within 6 h were sufficient to conclude that the initial set of isolates were all part of the same outbreak (10/13 isolates were identical when analysing the core genome of S. Enteritidis, three other isolates each differed by 1 SNP). Later on, when standard protocol MiSeq (paired 250 or 300 bp) data were available as well as HiSeq data from PHE surveillance, we were able to compare these results to that of draft sequencing. We could then conclude that although genome coverage was lower, the rapid draft sequencing method was concordant with both slower methods (Fig. 1). The sequencing quality using the draft protocol was lower (median Q score 36 compared with 38 using the V2 and V3 protocols at cycle 75) (Fig. 2).
Fig. 2.
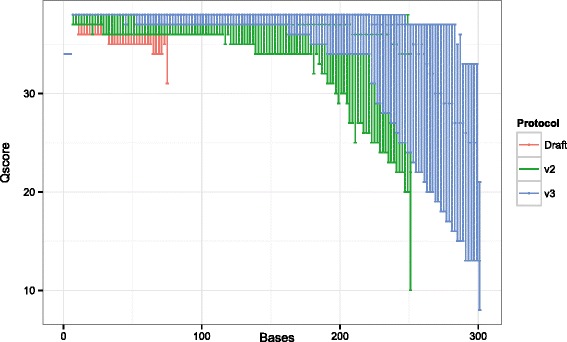
Phred-scaled quality scores (−10 log10 P) for Illumina sequencing demonstrating the impact of read length on read quality scores with the three Illumina MiSeq sequencing modalities used in this study. Red scores indicate results from the draft 1 × 75 base sequencing protocol, which shows minimally worse quality drop-off than running V2 (green, 2 × 250 base) or V3 chemistry (blue, 2 × 300) under standard conditions
Retrospective evaluation of real-time nanopore sequencing
Two samples, one belonging to the outbreak and one unrelated were sequenced on the newly-available MinION from Oxford Nanopore Technologies. During the outbreak, we used an early version of the chemistry termed R6. However, results from this sequencing did not produce sufficient numbers of high-quality two-direction (2D) reads to be of use. In July 2014 R6 chemistry was replaced by R7, which we were able to evaluate retrospectively. The MinION is characterised by very long reads, which have a high error rate compared to the Illumina platform.
Nanopore sequencing results
In order to evaluate the potential benefits of real-time sequencing to enhance infection control procedures we analysed read sets as they would have become available in real time, that is, at 10 min intervals after the run had been initiated. The two samples were run on separate flow cells. The number of reads generated in the first 170 min were 2,865 (first flowcell) and 3,447 (second flowcell) with mean read lengths of 6,340 and 4,664 bp, respectively for each sequence library. The mean read accuracy, determined by counting all differences from the reference genome, was 72 % (first flowcell) and 73 % (second flowcell).
Real-time strain identification from nanopore reads
We found that in the two samples tested we could unambiguously identify the bacterial species S. enterica in less than 30 min (Fig. 3). Additionally, chromosomally encoded phage sequences were detectable and distinguishable between outbreak and non-outbreak strains within 50 min.
Fig. 3.
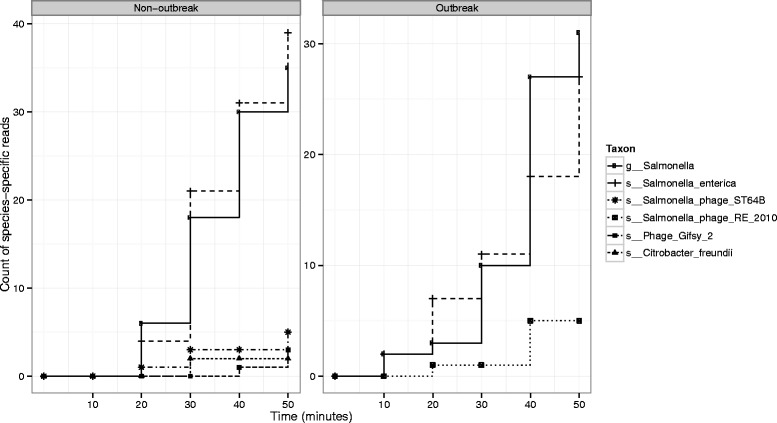
Streaming taxonomic assignments from the Oxford Nanopore MinION are shown for an isolate belonging to the outbreak and an isolate not belonging to the outbreak. Assignments to Salmonella and Salmonella enterica are found within 10 min of starting sequencing for the outbreak strain and within 20 min for the non-outbreak strain. Phage-specific sequences are detected and are distinct between the non-outbreak and outbreak strain. The non-outbreak strain harbours Salmonella phage ST64B and Gifsy-2, whereas the outbreak strain harbours Salmonella phage RE2010
Genotyping from low coverage, error-prone data using phylogenetic placement
Genotyping accuracy improves as more sequencing data are available and a consensus sequence is formed (Table 1). Our genotyping protocol gets increasingly more precise as more reads are added, however recall stays relatively constant. Despite this, a phylogenetic placement method confidently assigned both the outbreak and non-outbreak strains to a clade of S. enterica containing the Gallinarum, Pullorum and Enteritidiis serovars very early on in the sequencing process. By 40 min it was possible to determine that the likely serovar was Enteritidis (Fig. 4). Once assigned to a serovar, further analysis could be restricted to a reference tree of S. Enteritidis strains. It was possible then to show that the outbreak strain unambiguously belonged to the main hospital outbreak cluster within 100 min of starting sequencing (Fig. 5). The non-outbreak strain was assignable to a clade containing several closely related strains (with a mixture of phage types, none of them PT 14b) within 120 min.
Table 1.
Streaming alignment statistics from nanopore data
| Flowcell | Time (m) | Reads | Bases | Positions | Missing bases | Covered (%) | True positive | True negative | False positive | False negative | Recall | Precision | Accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Outbreak | 60 | 920 | 5635627 | 7091 | 6463 | 8.86 | 10 | 617 | 0 | 2 | 0.83 | 1.00 | 0.09 |
| Outbreak | 120 | 2037 | 12853716 | 7091 | 4815 | 32.10 | 26 | 2237 | 7 | 7 | 0.79 | 0.79 | 0.32 |
| Outbreak | 180 | 3040 | 19297035 | 7091 | 3580 | 49.51 | 48 | 3436 | 13 | 15 | 0.76 | 0.79 | 0.49 |
| Outbreak | 240 | 3933 | 24900526 | 7091 | 2703 | 61.88 | 62 | 4291 | 17 | 19 | 0.77 | 0.78 | 0.61 |
| Outbreak | 300 | 4525 | 28614437 | 7091 | 2236 | 68.47 | 70 | 4736 | 25 | 25 | 0.74 | 0.74 | 0.68 |
| Outbreak | 360 | 5654 | 35848389 | 7091 | 1499 | 78.86 | 82 | 5454 | 26 | 31 | 0.73 | 0.76 | 0.78 |
| Outbreak | 420 | 6680 | 42498530 | 7091 | 1029 | 85.49 | 87 | 5914 | 25 | 37 | 0.70 | 0.78 | 0.85 |
| Outbreak | 480 | 7516 | 47950926 | 7091 | 749 | 89.44 | 94 | 6185 | 30 | 34 | 0.73 | 0.76 | 0.89 |
| Outbreak | 540 | 7913 | 50372188 | 7091 | 630 | 91.12 | 96 | 6300 | 29 | 37 | 0.72 | 0.77 | 0.90 |
| Outbreak | 600 | 8807 | 56254898 | 7091 | 463 | 93.47 | 103 | 6470 | 20 | 36 | 0.74 | 0.84 | 0.93 |
| Outbreak | 660 | 9666 | 61989423 | 7091 | 337 | 95.25 | 107 | 6588 | 22 | 38 | 0.74 | 0.83 | 0.94 |
| Outbreak | 720 | 10472 | 67171497 | 7091 | 267 | 96.23 | 111 | 6659 | 16 | 39 | 0.74 | 0.87 | 0.95 |
| Outbreak | 780 | 10833 | 69363106 | 7091 | 243 | 96.57 | 112 | 6686 | 16 | 35 | 0.76 | 0.88 | 0.96 |
| Outbreak | 840 | 11708 | 74625788 | 7091 | 191 | 97.31 | 117 | 6737 | 13 | 34 | 0.77 | 0.90 | 0.97 |
| Outbreak | 900 | 12479 | 79551399 | 7091 | 141 | 98.01 | 121 | 6780 | 16 | 34 | 0.78 | 0.88 | 0.97 |
| Outbreak | 960 | 13198 | 84228957 | 7091 | 120 | 98.31 | 124 | 6797 | 16 | 35 | 0.78 | 0.89 | 0.98 |
| Outbreak | 1020 | 13579 | 86600020 | 7091 | 107 | 98.49 | 125 | 6808 | 16 | 36 | 0.78 | 0.89 | 0.98 |
| Outbreak | 1080 | 14359 | 91437571 | 7091 | 90 | 98.73 | 126 | 6823 | 17 | 36 | 0.78 | 0.88 | 0.98 |
| Outbreak | 1140 | 15168 | 96646434 | 7091 | 74 | 98.96 | 124 | 6842 | 15 | 37 | 0.77 | 0.89 | 0.98 |
| Outbreak | 1200 | 15835 | 100970757 | 7091 | 70 | 99.01 | 123 | 6851 | 12 | 36 | 0.77 | 0.91 | 0.98 |
| Outbreak | 1260 | 16205 | 103367082 | 7091 | 63 | 99.11 | 124 | 6857 | 11 | 37 | 0.77 | 0.92 | 0.98 |
| Outbreak | 1320 | 16632 | 106040214 | 7091 | 60 | 99.15 | 125 | 6859 | 12 | 36 | 0.78 | 0.91 | 0.98 |
| Outbreak | 1380 | 17184 | 109618605 | 7091 | 56 | 99.21 | 125 | 6863 | 11 | 37 | 0.77 | 0.92 | 0.99 |
| Outbreak | 1440 | 17332 | 110500445 | 7091 | 55 | 99.22 | 124 | 6865 | 11 | 37 | 0.77 | 0.92 | 0.99 |
| Non-outbreak | 60 | 1268 | 5382184 | 7091 | 6372 | 10.14 | 1 | 717 | 2 | 0 | 1.00 | 0.33 | 0.10 |
| Non-outbreak | 120 | 2554 | 11567191 | 7091 | 4791 | 32.44 | 2 | 2284 | 15 | 0 | 1.00 | 0.12 | 0.32 |
| Non-outbreak | 180 | 3626 | 17058822 | 7091 | 3451 | 51.33 | 4 | 3607 | 29 | 1 | 0.80 | 0.12 | 0.51 |
| Non-outbreak | 240 | 4612 | 22004574 | 7091 | 2500 | 64.74 | 11 | 4545 | 32 | 4 | 0.73 | 0.26 | 0.64 |
| Non-outbreak | 300 | 5483 | 26582592 | 7091 | 1760 | 75.18 | 13 | 5281 | 35 | 3 | 0.81 | 0.27 | 0.75 |
| Non-outbreak | 360 | 6198 | 30340527 | 7091 | 1330 | 81.24 | 15 | 5705 | 40 | 2 | 0.88 | 0.27 | 0.81 |
| Non-outbreak | 420 | 6877 | 34040490 | 7091 | 985 | 86.11 | 16 | 6054 | 35 | 2 | 0.89 | 0.31 | 0.86 |
| Non-outbreak | 480 | 7522 | 37471113 | 7091 | 727 | 89.75 | 18 | 6306 | 37 | 4 | 0.82 | 0.33 | 0.89 |
| Non-outbreak | 540 | 8306 | 41387560 | 7091 | 552 | 92.22 | 18 | 6483 | 34 | 5 | 0.78 | 0.35 | 0.92 |
| Non-outbreak | 600 | 9032 | 45052523 | 7091 | 395 | 94.43 | 20 | 6643 | 28 | 6 | 0.77 | 0.42 | 0.94 |
| Non-outbreak | 660 | 9682 | 48325820 | 7091 | 304 | 95.71 | 20 | 6735 | 27 | 6 | 0.77 | 0.43 | 0.95 |
| Non-outbreak | 720 | 10262 | 51312827 | 7091 | 262 | 96.31 | 20 | 6783 | 21 | 6 | 0.77 | 0.49 | 0.96 |
| Non-outbreak | 780 | 10845 | 54417219 | 7091 | 202 | 97.15 | 21 | 6845 | 18 | 6 | 0.78 | 0.54 | 0.97 |
| Non-outbreak | 840 | 11346 | 57135819 | 7091 | 178 | 97.49 | 22 | 6870 | 16 | 6 | 0.79 | 0.58 | 0.97 |
| Non-outbreak | 900 | 11793 | 59514439 | 7091 | 145 | 97.96 | 23 | 6898 | 18 | 8 | 0.74 | 0.56 | 0.98 |
| Non-outbreak | 960 | 12192 | 61590631 | 7091 | 111 | 98.43 | 22 | 6932 | 19 | 8 | 0.73 | 0.54 | 0.98 |
| Non-outbreak | 1020 | 12571 | 63597395 | 7091 | 99 | 98.60 | 22 | 6944 | 19 | 8 | 0.73 | 0.54 | 0.98 |
| Non-outbreak | 1080 | 12926 | 65415215 | 7091 | 87 | 98.77 | 21 | 6959 | 18 | 7 | 0.75 | 0.54 | 0.98 |
| Non-outbreak | 1140 | 13263 | 67138579 | 7091 | 71 | 99.00 | 22 | 6976 | 15 | 8 | 0.73 | 0.59 | 0.99 |
| Non-outbreak | 1200 | 13594 | 68911549 | 7091 | 62 | 99.13 | 22 | 6985 | 15 | 8 | 0.73 | 0.59 | 0.99 |
| Non-outbreak | 1260 | 13881 | 70408443 | 7091 | 59 | 99.17 | 22 | 6992 | 11 | 8 | 0.73 | 0.67 | 0.99 |
| Non-outbreak | 1320 | 14186 | 72080944 | 7091 | 53 | 99.25 | 23 | 7001 | 8 | 7 | 0.77 | 0.74 | 0.99 |
| Non-outbreak | 1380 | 14471 | 73573256 | 7091 | 44 | 99.38 | 23 | 7008 | 10 | 7 | 0.77 | 0.70 | 0.99 |
| Non-outbreak | 1440 | 14683 | 74801565 | 7091 | 40 | 99.44 | 23 | 7012 | 10 | 7 | 0.77 | 0.70 | 0.99 |
The columns show (from left to right): (1) the sample analysed; (2) the cumulative results at this time period (min); (3) the total number of two-direction reads; (4) the total number of nucleotide bases; (5) the total size of the alignment; (6) the number of bases in the alignment missing from the dataset; (7) the percentage of bases in the alignment that can be called; (8) the count of true positives; (9) the count of true negatives; (10) the count of false positives; (11) the count of false negatives; (12) the recall, that is, sensitivity, calculated as TP/(TP + FN); (13) the precision, calculated as TP/(TP + FP); (14) the accuracy, calculated as (TP + TN)/(P + N)
Fig. 4.
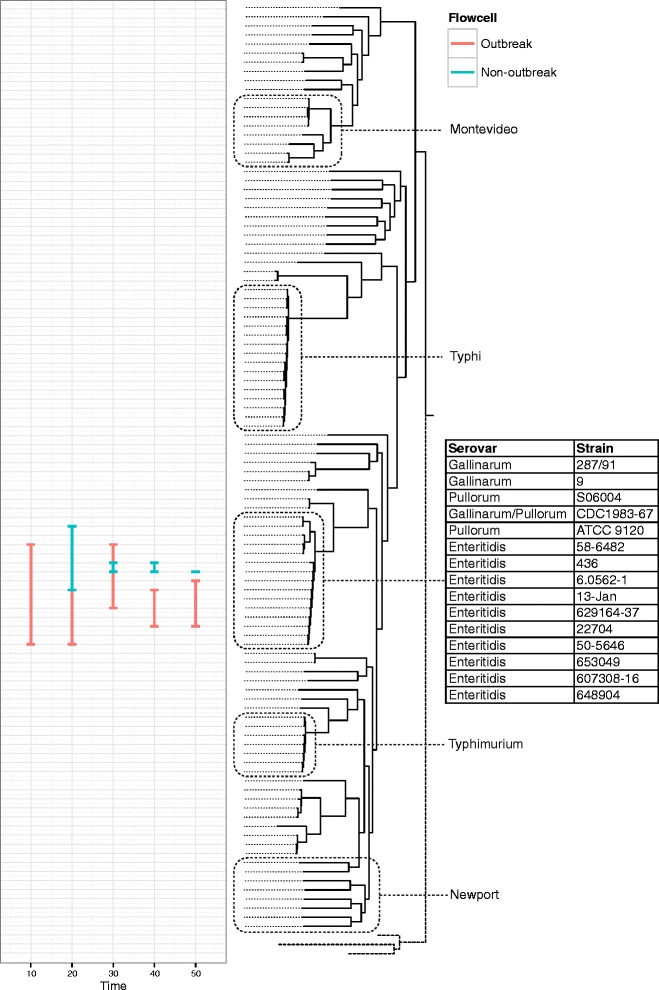
Results of streaming phylogenetic placement from the Oxford Nanopore MinION on a reference tree of representative published Salmonella enterica sequences. Common serovars of Salmonella are highlighted. Both outbreak and non-outbreak strains are unambiguously identified as Salmonella enterica serovar Enteritidis by their position on the phylogenetic tree within 50 min. The line demonstrates the potential range of placements reported by pplacer. The red placements indicate the positions of the outbreak isolate and the blue placements indicate the positions of the non-outbreak isolate
Fig. 5.

Results of streaming phylogenetic placement from the Oxford Nanopore MinION on a reference tree of Salmonella enterica serovar Enteritidis isolates collected by Public Health England during routine surveillance. The left-most panel demonstrates the confident placing of the outbreak isolate in the outbreak clade within 100 min, and the confident placing of the non-outbreak isolate into a clade containing multiple serotypes of Salmonella within 120 min. The red placements indicate the positions of the outbreak isolate and the blue placements indicate the positions of the non-outbreak isolate. The right-most panel shows a phylogenetic reconstruction of isolates from the outbreak and their source, set in context of a national outbreak of phage type 14b. Uncertainty in the phylogenetic placement technique is demonstrated early on in sequence data collection due to the low accuracy of the variant calls collected. As more data are collected, the number of possible phylogenetic placements reduces and the confidence values increase (not shown)
The availability of definitive typing data so early on in this outbreak enabled us to identify transmission between hospital wards and take rapid action to control spread. The appearance of cases in unrelated wards was puzzling initially, but WGS confirmed that the hospital SNP type was the same as that circulating in the community. This reassured the infection control team that there was not hospital-wide spread via some unknown vector. Preliminary food sample testing results were only available one day later. The finding of the outbreak strain on the door seal of the food trolley with the subsequent confirmation of cases in staff members supported the hypothesis that some local spread had occurred via the environment. Person-to-person spread may also have occurred. Towards the end of the outbreak the ability to rapidly identify cases not involved prevented much wastage of effort and resources. Remarkably we identified transmission of another strain of S. Enteriditis probably acquired in Egypt in a childcare group at a distant site because of the resolution of the typing information directing epidemiological investigations. Recent outbreaks of PT 14b strains in the UK have previously been associated with Spanish eggs, although the antibiotic resistance profile of the outbreak described here is different [11, 12]. Contemporaneously, outbreaks of S. Enteritidis PT 14b associated with consumption of eggs were reported in France, Austria and Germany, triggering an urgent outbreak investigation by the ECDC and EFSA [12]. Strains associated with this outbreak were of MLVA type 2-12-7-3-2 (using the 5-locus scheme), varying by a single locus from the isolates identified in this study. In these cases S. Enteritidis was isolated from eggs originating from a producer in Germany [12]. There is no definitive link between the outbreak reported in this study and the consumption of German eggs. However, the MLVA type in the European outbreak was also detected in the UK and eggs from the German producer are distributed for sale in the UK. Further whole-genome sequencing of European isolates is now being undertaken and may help determine whether the two outbreaks are linked to a common source.
This study illustrates a substantial future benefit from extremely rapid definitive WGS typing. The epidemiology of non-typhoidal Salmonella has changed significantly in the UK over the last decade and to a lesser extent in the rest of Europe [2, 13]. While non-typhoidal Salmonella rates have fallen overall, particularly in the UK following chicken flock vaccination, the proportion of disease caused by S. Enteritidis associated with travel has risen greatly. The ability to both identify serovars via deduced multi-locus sequence typing (MLST) and specific strains within a day of bacterial colonies being available will enable outbreaks to be investigated at a stage where accurate travel/food histories and possible person-to-person transmission can be elucidated and control measures introduced. We show that our method of rapid draft sequencing on the MiSeq is able to generate reliable results, despite generating reduced genome coverage. We anticipate this method will be of value to research groups needing to generate results in the timescale of a single working day, a considerable reduction compared to the standard protocols on this instrument.
The availability of national and international databases of sequencing data of food-borne pathogens marks an exciting step forward for epidemiological investigations. Surveillance by WGS has been pioneered by the US Food and Drug Administration, with results published online on the National Center for Biotechnology Information’s GenomeTrakr service, an advantage of the portable, digital nature of genome data [8, 14]. In the UK, since 1 April 2014, Public Health England has been routinely sequencing all Salmonella enterica strains reported by hospitals and general practitioners to the Salmonella Reference Service, Colindale. Through integration with this dataset, we determined that the outbreak strains formed a distinct cluster, although this cluster varied by only a single core SNP from cases observed elsewhere in the UK.
We evaluated two sequencing methodologies in this study, both capable of providing rapid whole-genome sequencing information. The MinION senses individual DNA strands as they move through a protein nanopore. A unique property of this technology is that sequence data are available in real time, and analysis can be performed on a continuous stream of long reads. We wished to evaluate the potential impact of a real-time approach for analysis of clinical bacterial isolates. We exploited this feature to perform rapid identification and typing of genomic DNA prepared from a pure colony isolate. Given the high error rate reads generated in this study we employed a database of taxon-defining genes from microbial species to make bacterial and bacteriophage identifications [15]. This approach is tolerant of low-coverage, high-error reads making it useful for real-time analysis of nanopore sequences. However, due to the higher error rate of this platform, a de novo SNP calling approach as utilised with MiSeq data would not produce informative results within the short time scales of interest here. Other studies have investigated the error rate and mode of this instrument in greater detail [15, 16, 17]. We show that despite the high error rate, effective genotyping is possible using phylogenetic placement techniques. Phylogenetic placement has been used to good effect in metagenomics studies where only low-coverage data are available, for example in the diagnosis of infectious diseases from ancient DNA samples, directly from sputum and from the hospital environment [18–20]. Using this approach, and a simple heuristic algorithm to call the most likely genotype it was possible to reliably place streaming nanopore data onto a reference phylogeny despite the high read error rate. Other studies have shown that genotyping accuracy can reach 99 % when very high coverage (>120×) is available. This would permit a de novo genotyping approach which did not rely on phylogenetic placement, as is more typical in studies employing traditional high-throughput sequencing [14].
Both the draft sequencing protocol presented for the MiSeq and the real-time evaluation of nanopore sequencing demonstrate that these approaches have utility for generating data of use in outbreak investigations in less than one day (Fig. 6). It is not our intention here to perform direct comparisons between the instruments in this study, particularly as they are quite different in their mode of operation.
Fig. 6.
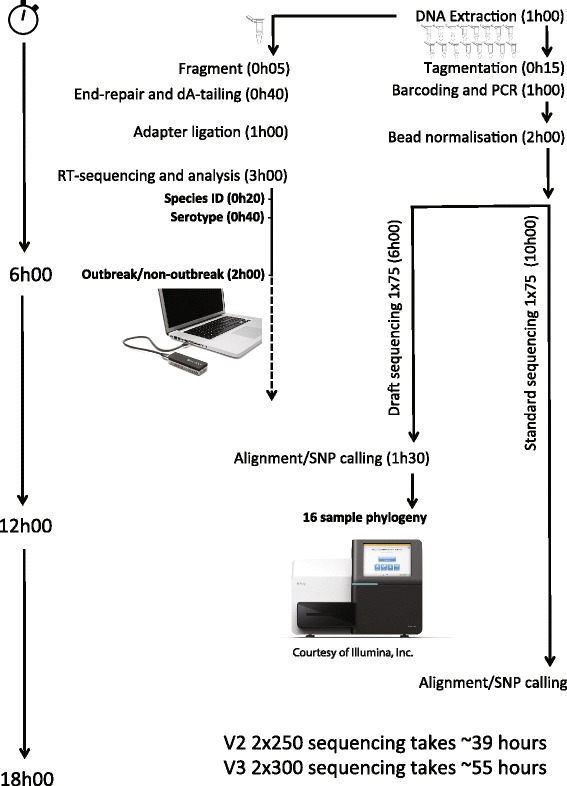
A schematic showing time-to-result for the sequencing analysis strategies used in this study, starting from DNA input
The MiSeq is typically run in a factory-style ‘batch’ mode, where many bacterial samples (up to 100 on a MiSeq, or potentially many hundreds on the larger HiSeq instrument) are run simultaneously, and processed in serial at the end of the instrument operation. This approach reduces the cost of sequencing by taking advantage of the very high output offered from these instruments (>1 terabase for the HiSeq in High Output Mode). The precipitous drop in the cost of sequencing bases has meant that for bacterial applications the cost of library preparation is rapidly becoming the most expensive component. However, batch methods, particularly with the very highest output modes result in a flexibility trade-off; such an approach means that data cannot be analysed until at least the barcode identifiers have been read (usually not until after halfway through the run).
This is in contrast to the real-time sequencing approach of the MinION whereby individual samples are loaded, and results are generated and analysed in real time until the results are sufficient to address the clinical question. Such an approach has appealing properties for applications such as infectious disease diagnostics. A second attribute of the MinION that is notable is its extreme portability, comparable in size to a USB flash drive and requiring only a basic laptop to draw power from and connect to. This suggests that it may, in principle, be possible in the future to move sequencing closer to the sample, and particularly when coupled with a culture-independent approach.
However, at present the instrument depends on access to a basic molecular biology laboratory infrastructure, including access to freezer, and basic laboratory equipment such as heater blocks and pipettes. The existing library preparation method, although relatively quick, is quite labour-intensive for each sample. Presently there is no method for multiplexing large numbers of bacterial genomes (as with the MiSeq instrument), nor would the throughput be amenable to this. Therefore, it seems likely for large-scale surveillance efforts this platform is not the obvious choice, for reasons of labour and cost. Instead, we envisage that development of rapid library preparation assays will be necessary in order to see this platform become usable in a clinical microbiology laboratory or patient setting in the manner described here.
Furthermore, the need for culture enrichment remains a significant bottleneck for rapid identification of bacteria and this also applies to other studies employing whole-genome sequencing. Culturing of Salmonella takes between 24 h (presumptive diagnosis) and 48 h (pure culture for sequencing). Our approach, which relies on sequencing single colonies from each sample, is a limitation of this and similar studies. However, sequencing of four individual colonies from the food trolley demonstrated very limited heterogeneity with three isolates being identical to the majority of other cases in the outbreak, and one showing two SNP differences. A culture-free approach for bacterial diagnostics has been recently proposed and this would permit detection of mixed infections as well as cutting down the time to result significantly, for example in the case of direct sequencing of Shiga-toxin producing E. coli from stool samples and M. tuberculosis directly from sputum [21, 22]. However, sequencing mixed communities reduces the genomic coverage of the pathogenic target of interest, and so for such an approach to be successful it is likely to rely on generating greater throughput than currently achievable on the MinION. Enrichment for the target organism, most easily attained through traditional microbiology culture, is therefore still a required stage.
Conclusion
The combination of rapid prospective sequencing during an outbreak and detailed characterisation of cases occurring on a national scale has potential implications for the future of outbreak investigation [23]. We describe a novel protocol for draft sequencing on the MiSeq that is sufficiently quick to determine whether an outbreak is occurring. For this vision to become a reality, further work is needed to enable sharing of data between hospitals and community practitioners with public health laboratories. Larger scale integration with national genome databases represents the first implementation of a new paradigm for the investigation of outbreaks. The use of rapid, draft sequencing can delineate the context of an outbreak very quickly even at lower than usual genome coverage.
Materials and methods
Sample and bacterial culture collection
Faeces samples from patients with diarrhoea were submitted for culture and plated on XLD medium. Presumptive S. enterica isolates were confirmed using biochemical tests and O- and H-antigen agglutination sera and all those identified as S. Enteritidis were retained for molecular typing. Environmental swabs were taken from the affected wards within 24 h of the ward clusters being identified and were processed as above.
Genomic DNA extraction
Genomic DNA was prepared from nutrient agar slopes incubated for 4–18 h at 37 °C. Cells were harvested using 100 μL of sterile PBS added to the surface and a sterile loop used to emulsify bacteria into a suspension that was then pipetted into a sterile Eppendorf tube. This suspension was used to harvest DNA with the ‘Invisorb spin cell mini kit’ (Invitek, Germany) according to the manufacturer’s instructions. The quantity of DNA in each sample was determined using a Qubit 2.0 fluorometer and dsDNA HS assay (Life Technologies, Paisley, UK).
Library preparation for Illumina MiSeq sequencing
Sequence-ready libraries were generated from 1 ng DNA per sample using the Nextera XT library preparation kit (Illumina, Great Chesterford, UK) according to the manufacturer’s instructions.
Rapid draft sequencing on the Illumina MiSeq
In order to provide results for an emergency infection control meeting the next morning, we adapted the standard sequencing protocol on the Illumina MiSeq to rapidly generate sufficient data to analyse 16 strains. We utilised a standard V3 600-cycle reagent kit. By modifying the recipe files on the instrument we reduced the chemistry time by 40 s per cycle and the number of tiles imaged by 50 %. This resulted in a cycle time of approximately 3 min per cycle and allowed 75 base single-read sequencing with dual barcoding to complete within 6 h. We chose 75 base reads as a trade-off between expected genome coverage and available time in order to have results available sufficiently quickly for analysis. The sequencing protocol can be downloaded from [24].
Standard sequencing on Illumina MiSeq and HiSeq
Later in the outbreak isolates were sequenced using the Illumina MiSeq with standard V3 protocol at the University of Birmingham, UK, also prepared with Nextera XT reagents. In addition, some outbreak isolates were sequenced on the Illumina HiSeq 2500 with TruSeq V3 reagents as part of the Public Health England (PHE) WGS sequencing pipeline at Colindale, UK (Additional file 1: Table S1).
Phylogenetic reconstruction from draft sequencing
Before being mapped against the reference genome S. Enteritidis P125109 (PRJNA59247) with BWA-MEM (version 0.7.5), 75 base single-read data generated by draft sequencing on the Illumina MiSeq was adapter and quality trimmed with Trimmomatic [25, 26]. Single nucleotide polymorphisms (SNPs) were called using samtools mpileup (version 0.1.18) and VarScan (version 2.3.6), specifying a minimum read depth of 2 [27, 28]. Filtered SNPs (those positions with an allele frequency of >80 % to call a variant or <20 % to call the reference base in all samples) were extracted to make a concatenated FASTA alignment. FastTree (version 2.1.7) was used to generate an approximate maximum likelihood phylogenetic tree [29]. Phyloviz was used to produce minimum spanning tree reconstructions [30]. Functional annotation of these variants was performed using snpEff (version 3.1) [31].
Phylogenetic reconstruction from PHE surveillance sequencing
Before being mapped against the reference genome S. Enteritidis AM933172 (PRJEA30687) with BWA-MEM, 100 base pair paired-end data generated on the Illumina HiSeq 2500 was adapter and quality trimmed [26]. SNPs were called using GATK [32]. High quality SNPs (>10-fold coverage, >30 mapping quality, 90 % consensus) were selected and uploaded into SNPdatabase (SNPdb). This is an in-house PostgreSQL database containing genome position and variant base for each SNP and low quality/missing positions for all S. Enteritidis eBURST group 4 (EBG 4) isolates sequenced by PHE. SNPs in the core genome of the strain set being analysed were extracted from an in-house SNPdb and FastTree was used to derive approximate maximum likelihood phylogenetic trees. Annotation data came from the in-house PHE GastroDataWarehouse (GDW).
Due to the clinical interest in these cases, strains with below standard sequencing depth (30×) were analysed and this had no impact on the analysis outcome, with identical tree topologies detected in all cases.
Real-time sequencing on the MinION
An outbreak and a non-outbreak isolate, as determined by earlier MiSeq sequencing, were chosen for sequencing on the MinION (Oxford Nanopore Technologies, Oxford, UK) to assess its suitability for future outbreak investigations. High-molecular weight input DNA (1 μg) was fragmented using a Covaris G-Tube (Covaris, Woburn, USA) at 5,000 rpm in a centrifuge. Fragmented DNA was end-repaired using the NEB repair module (New England Biolabs, Ipswich, USA), then cleaned-up using SPRI beads with a ratio of 1:1 beads to reaction mixture. End-repaired DNA was then A-tailed using the NEB A-tailing module. Following this a sequence-ready library was generated using the gDNA sequencing kit and protocol provided as part of the MinION access program (MAP). The diluted library (150 μL) was loaded into the MinION flowcell via the sample loading port. A 72-h sequencing protocol was initiated using the MinION control software, MinKNOW (version 0.45.2.6). Read event data were base-called by the software Metrichor (version 0.16.37960) using workflow 1.0.2_R7. The FASTA sequences and strand translocation times were extracted for further analysis using the poretools FASTA extraction function [33]. All sequence data are deposited into the Short Read Archive (SRA) with study reference ERP006904 (MinION data) and ERP007194 (Illumina data).
Species identification from nanopore reads
Identification of bacterial and viral species present in each sample was carried out using an alignment method to the MetaPhlAn 2 database of taxon-defining marker genes [33]. First, the database was extracted into FASTA format using the fastacmd utility supplied with NCBI BLAST. Alignment of nanopore reads was performed using the LAST package (version 475), invoking lastal with custom settings as per Quick et al. [15], using a gap creation penalty and extension of 1 and a mismatch penalty of 2 (match score 1), corresponding to command line arguments -a1 -b1 -q2.
Subspecies level classification from nanopore reads
Serovars of S. enterica can often be assigned by phylogenetic methods. A phylogenetic reference tree was created from the available draft or complete Salmonella enterica genomes in RefSeq. From each of the sequences 600,000 simulated paired-end reads were generated using wgsim (version 0.3.1) [34]. These were mapped against the reference genome S. Typhimurium LT2 (PRJNA57799) with BWA-MEM (version 0.7.5) [26]. samtools mpileup (version 0.1.18) and VarScan (version 2.3.6) were used to call variants [27, 28]. Variant filtering was done using filter_non_discriminatory_variants.py [35] in order to remove non-discriminatory positions, as well as heterozygous positions and regions of putative recombination. Variant alleles for each sample were concatenated into a fasta file using vcf2phyloviz.py [36]. This file was de-duplicated using the mogrify command in seqmagick (version 0.6.0) to remove identical sequences which can affect placements. A phylogenetic reconstruction was created using FastTree (version 2.1.7) following a generalised time reversible model, after which taxtastic (version 0.5.1) was used to build the reference package [37].
To determine the subspecies level classification from the nanopore sequencing data, the reads were mapped against the reference genome with lastal with settings -a1 -b1 -q2. For each read, the highest scoring alignment was taken before being converted into BAM format using samtools. Using samtools mpileup and the script get_alleles_from_pileup.py the alignment was interrogated at all coordinates used for the reference tree. Aligned bases at these coordinates were counted and the dominant allele was used if at least two concordant bases were in the alignment. Alleles were concatenated into an alignment. Gap characters were used to represent uncertain positions not meeting the above criteria. The phylogenetic placement utility, pplacer, was used to place the sequence onto the reference tree producing a file containing the most likely position and logML probability for this placement. Placements with a likelihood value of greater than −500 were excluded [37]. This placement process was repeated for the read dataset available at each timepoint (10 min apart). New reads generated during each 10 min time interval were mapped to the reference, converted to a BAM file and merged with the BAM file generated at the previous time period.
S. enterica outbreak reconstruction
As with the subspecies level classification, phylogenetic placement can be used as a method for classifying samples in or out of an ongoing outbreak and in a national and international context. In order to do this, we leveraged the routine surveillance sequencing of S. enterica by PHE using 575 S. Enteritidis genomes of phage type 14b. Using the method described above a phylogenetic reference tree was created for these genomes (448 remained after de-duplication) before the nanopore sequences were placed onto the tree to predict whether or not they belonged to the outbreak cluster.
Acknowledgements
Thank you to the staff of Heartlands Hospital for assistance in this investigation. We are grateful to Paul Keim of Northern Arizona University for introducing us to phylogenetic placement techniques. We thank Torsten Seemann of Monash University, Australia for critical reading of the manuscript. We are grateful to the staff of Oxford Nanopore Technologies for admission to the MinION Early Access Programme and instrument and software technical support. NJL is funded by a Medical Research Council Special Training Fellowship in Biomedical Informatics. JQ is funded by the NIHR Surgical Reconstruction and Microbiology Research Centre. The Cloud Infrastructure for Microbial Bioinformatics (CLIMB) cyberinfrastructure was used for the data analysis presented in this manuscript. The authors would like to thank the anonymous reviewers of this paper for their constructive feedback during the peer-review process.
Additional file
Isolate identifiers sequenced in this study using rapid draft MiSeq sequencing, standard MiSeq sequencing and HiSeq sequencing during routine PHE surveillance.
Footnotes
Joshua Quick and Philip Ashton contributed equally to this work.
Competing interests
NJL is a member of the MinION Access Programme (MAP) and has received free-of-charge reagents for nanopore sequencing presented in this study. JQ has received travel and accommodation expenses to speak at an Oxford Nanopore-organised symposium. NJL and JQ have ongoing research collaborations with Oxford Nanopore but do not receive financial compensation for this.
Authors’ contributions
NJL, ER and PH conceived the study. CC, SG, JH, SN, K. Neal, K. Nye, TP, EdP, ER, KS, AC, TD and PH performed the microbiological and epidemiological investigation. MW performed DNA extractions. SC and JQ performed sequencing. JQ, PA, TD, ER, PH and NJL analysed data. NJL, TD, PH and MW contributed reagents. NJL, PH and JQ wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Joshua Quick, Email: j.quick@bham.ac.uk.
Philip Ashton, Email: philip.ashton@phe.gov.uk.
Szymon Calus, Email: s.t.calus@bham.ac.uk.
Carole Chatt, Email: carol.chatt@phe.gov.uk.
Savita Gossain, Email: savita.gossain@heartofengland.nhs.uk.
Jeremy Hawker, Email: Jeremy.Hawker@phe.gov.uk.
Satheesh Nair, Email: satheesh.nair@phe.gov.uk.
Keith Neal, Email: Keith.Neal@phe.gov.uk.
Kathy Nye, Email: Kathy.Nye@phe.gov.uk.
Tansy Peters, Email: Tansy.Peters@phe.gov.uk.
Elizabeth De Pinna, Email: Elizabeth.DePinna@phe.gov.uk.
Esther Robinson, Email: E.R.Robinson@warwick.ac.uk.
Keith Struthers, Email: keith.struthers@uhcw.nhs.uk.
Mark Webber, Email: m.a.webber@bham.ac.uk.
Andrew Catto, Email: andrew.catto@heartofengland.nhs.uk.
Timothy J. Dallman, Email: Tim.Dallman@phe.gov.uk
Peter Hawkey, Email: peter.hawkey@heartofengland.nhs.uk.
Nicholas J. Loman, Email: n.j.loman@bham.ac.uk
References
- 1.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, et al. Foodborne illness acquired in the United States–major pathogens. Emerg Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Brien SJ. The “decline and fall” of nontyphoidal salmonella in the United Kingdom. Clin Infect Dis. 2013;56:705–10. doi: 10.1093/cid/cis967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Telzak EE, Budnick LD, Greenberg MS, Blum S, Shayegani M, Benson CE, et al. A nosocomial outbreak of Salmonella enteritidis infection due to the consumption of raw eggs. N Engl J Med. 1990;323:394–7. doi: 10.1056/NEJM199008093230607. [DOI] [PubMed] [Google Scholar]
- 4.Palmer SR, Rowe B. Investigation of outbreaks of salmonella in hospitals. Br Med J (Clin Res Ed) 1983;287:891–3. doi: 10.1136/bmj.287.6396.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mason BW, Williams N, Salmon RL, Lewis A, Price J, Johnston KM, et al. Outbreak of Salmonella indiana associated with egg mayonnaise sandwiches at an acute NHS hospital. Commun Dis Public Health. 2001;4:300–4. [PubMed] [Google Scholar]
- 6.Department of Health and Social Security . Report of the committee of inquiry into an outbreak of food poisoning at Stanley Royd Hospital. London: HMSO; 1986. [Google Scholar]
- 7.Lienau EK, Strain E, Wang C, Zheng J, Ottesen AR, Keys CE, et al. Identification of a salmonellosis outbreak by means of molecular sequencing. N Engl J Med. 2011;364:981–2. doi: 10.1056/NEJMc1100443. [DOI] [PubMed] [Google Scholar]
- 8.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–9. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 9.Reuter S, Ellington MJ, Cartwright EJP, Köser CU, Török ME, Gouliouris T, et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern Med. 2013;173:1397–404. doi: 10.1001/jamainternmed.2013.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open. 2012;2:e001124. doi: 10.1136/bmjopen-2012-001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janmohamed K, Zenner D, Little C, Lane C, Wain J, Charlett A, et al. National outbreak of Salmonella Enteritidis phage type 14b in England, September to December 2009: case–control study. Euro Surveill. 2011;16:19840. [PubMed] [Google Scholar]
- 12.European Centre for Disease Prevention and Control, European Food Safety Authority. Multi-country outbreak of Salmonella Enteritidis infections associated with consumption of eggs from Germany – 25 August 2014. Stockholm and Parma: ECDC/EFSA; 2014. http://www.efsa.europa.eu/en/supporting/pub/646e.htm.
- 13.Schmid H, Baumgartner A. Epidemiology of infections with enteric salmonellae in Switzerland with particular consideration of travelling activities. Swiss Med Wkly. 2013;143:w13842. doi: 10.4414/smw.2013.13842. [DOI] [PubMed] [Google Scholar]
- 14.den Bakker HC, Allard MW, Bopp D, Brown EW, Fontana J, Iqbal Z, et al. Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg Infect Dis. 2014;20:1306–14. doi: 10.3201/eid2008.131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9:811–4. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2015;33:296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 17.Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12:351–6. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kay GL, Sergeant MJ, Giuffra V, Bandiera P, Milanese M, Bramanti B, et al. Recovery of a medieval Brucella melitensis genome using shotgun metagenomics. MBio. 2014;5:e01337–14. doi: 10.1128/mBio.01337-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doughty EL, Sergeant MJ, Adetifa I, Antonio M, Pallen MJ. Culture-independent detection and characterisation of Mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer. Peer J. 2014;2:e585. doi: 10.7717/peerj.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quick J, Cumley N, Wearn CM, Niebel M, Constantinidou C, Thomas CM. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: an observational study using whole-genome sequencing. BMJ Open. 2014;4:e006278. doi: 10.1136/bmjopen-2014-006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA. 2013;309:1502–10. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 22.Chan JZ-M, Sergeant MJ, Lee OY-C, Minnikin DE, Besra GS, Pap I, et al. Metagenomic analysis of tuberculosis in a mummy. N Engl J Med. 2013;369:289–90. doi: 10.1056/NEJMc1302295. [DOI] [PubMed] [Google Scholar]
- 23.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group. Henderson DK, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quick J. MiSeq. Draft Sequencing Protocol. https://github.com/joshquick/miseq-draft-sequencing
- 25.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço JA. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics. 2012;13:87. doi: 10.1186/1471-2105-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loman NJ, Quinlan AR. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics. 2014;30:3399–401. doi: 10.1093/bioinformatics/btu555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. wgsim Github repository. https://github.com/lh3/wgsim.
- 35.Quick J. Github repository. https://github.com/joshquick.
- 36.Loman NJ. Github repository. https://github.com/nickloman/misc-genomics-tools.
- 37.Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]