

Abstract
Background
Sesame is an important high-quality oil seed crop. The sesame genome was de novo sequenced and assembled in 2014 (version 1.0); however, the number of anchored pseudomolecules was higher than the chromosome number (2n = 2x = 26) due to the lack of a high-density genetic map with 13 linkage groups.
Results
We resequenced a permanent population consisting of 430 recombinant inbred lines and constructed a genetic map to improve the sesame genome assembly. We successfully anchored 327 scaffolds onto 13 pseudomolecules. The new genome assembly (version 2.0) included 97.5 % of the scaffolds greater than 150 kb in size present in assembly version 1.0 and increased the total pseudomolecule length from 233.7 to 258.4 Mb with 94.3 % of the genome assembled and 97.2 % of the predicted gene models anchored. Based on the new genome assembly, a bin map including 1,522 bins spanning 1090.99 cM was generated and used to identified 41 quantitative trait loci (QTLs) for sesame plant height and 9 for seed coat color. The plant height-related QTLs explained 3–24 % the phenotypic variation (mean value, 8 %), and 29 of them were detected in at least two field trials. Two major loci (qPH-8.2 and qPH-3.3) that contributed 23 and 18 % of the plant height were located in 350 and 928-kb spaces on Chr8 and Chr3, respectively. qPH-3.3, is predicted to be responsible for the semi-dwarf sesame plant phenotype and contains 102 candidate genes. This is the first report of a sesame semi-dwarf locus and provides an interesting opportunity for a plant architecture study of the sesame. For the sesame seed coat color, the QTLs of the color spaces L*, a*, and b* were detected with contribution rates of 3–46 %. qSCb-4.1 contributed approximately 39 % of the b* value and was located on Chr4 in a 199.9-kb space. A list of 32 candidate genes for the locus, including a predicted black seed coat-related gene, was determined by screening the newly anchored genome.
Conclusions
This study offers a high-density genetic map and an improved assembly of the sesame genome. The number of linkage groups and pseudomolecules in this assembly equals the number of sesame chromosomes for the first time. The map and updated genome assembly are expected to serve as a platform for future comparative genomics and genetic studies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2316-4) contains supplementary material, which is available to authorized users.
Background
The sesame (Sesamum indicum) is a member of the family Pedaliaceae and order Lamiales [1] and is a diploid species with 13 pairs of chromosomes (2n = 2x = 26). In our previous study, the sesame genome was estimated to be approximately 357 Mb in size based on de novo sequencing of Zhongzhi No. 13. The assembled genome (version 1.0) was predicted to contain 27,148 protein-coding genes. The version 1.0 assembly anchored 150 scaffolds into 16 pseudomolecules that harbored 85.3 % of the assembled genome and 91.7 % of the predicted genes [2, 3]. We attempted to anchor additional scaffolds and contigs to improve the sesame genome assembly and equalize the number of sesame chromosomes. To achieve this goal, a high-quality and dense genetic map is essential. Although two sesame genetic maps have been published [4, 5], they contain more than 13 linkage groups. The numbers of breeding lines and temporary populations usually contribute to the construction of genetic maps [6].
Genetic maps are useful for discovering, dissecting, and manipulating the genes responsible for simple and complex traits in crop plants [7]. A high-quality genetic map not only improves genome assembly but also provides a foundation for mapping the genes or quantitative trait loci (QTLs) that underlie agronomic traits of important oil crops such as the sesame. The sesame seed is useful because of its high oil content and quality. The sesame antioxidative furofuran lignans such as sesamin, are the focus of research in medicine and pharmacology due to their potent pharmacological properties including the ability to decrease blood lipid [8] and cholesterol [9] levels. Moreover, the diploid characteristic, high oil content, small genome size, and smaller number of lipid-related genes distinguish sesame from other polyploid oil crops such as soybean, peanut, and rapeseed and make it a valuable plant model to study oil biosynthesis and various traits [2]. Despite the importance and long history of cultivation of the sesame, few gene loci have been fine-mapped due to a lack of genetic and genomic studies and gene cloning and functional analyses.
Plant architecture is crucial for the grain yield and is determined by the plant height (PH) and other traits [10]. The “green revolution” was characterized by breeding semi-dwarf or dwarf wheat and rice cultivars to achieve an increased harvest index and improved adaption to the irrigated and fertile environments usually inhabited by taller plants [11–13]. Sesame has a low yield capacity compared to other crop plants due to its high PH, low harvest index, and susceptibility to biotic and abiotic stressors [14]. Mutant sesame lines with a determinate growth habit have been generated to reduce the sesame PH. Unfortunately, this characteristic was linked to short fruiting zone length and low yield [14, 15] phenotypes; thus, new dwarf or semi-dwarf sesame genetic resources must be explored.
Another important sesame trait is the seed coat color which varies widely from white to black. Black sesame seeds are favored as food and medication in Asia, whereas white sesame seeds are primarily used to produce oil. Some studies have shown that white sesame seeds typically have higher sesamin or sesamolin content [16], whereas black sesame seeds usually have higher ash and carbohydrate content and lower protein, oil, and moisture ratios [17].
Single nucleotide polymorphisms (SNPs) are the most abundant small-scale form of genetic variation in humans and plants and SNP markers have become increasingly important tools for molecular genetic analyses [18, 19]. Restriction-site associated DNA sequencing (RAD-seq) using next generation sequencing platforms reduces the representation of the genome and allows oversequencing of the nucleotides adjacent to restriction sites; thus, it is a relatively cost-effective and precise method for the detection of SNPs [20]. In this study, we constructed a recombinant inbred line (RIL) population consisting of 430 lines using a newly discovered semi-dwarf sesame strain. A new high-density sesame genetic map with 13 linkage groups was prepared using RAD-seq which enabled us to update the sesame genome assembly to a new level and to finely map important agronomic traits such as the PH and seed coat color.
Results and discussion
Construction of a new sesame genetic map
We RAD sequenced the 430 RILs and the two parental lines and generated more than 1.7 × 108 paired-end reads. Each line produced a mean of 76.5 Mb of high-quality sequence data. All reads were aligned with the Zhongzi No. 13 scaffold sequence which has a total effective genome length of 274 Mb based on analysis using BWA software [21], and 90.97 % of the reads were mapped onto reference sequences across the various lines. Approximately 5.4 % of the reference genome was tagged by RAD-Seq, with a mean value of 4.53-fold coverage.
The tags from the two parents Zhongzhi No. 13 and ZZM2748 were subjected to comparative analysis to detect SNPs. After filtering out poor SNPs and those with a significantly distorted segregation ratio (P < 0.01), 11,924 SNPs were retained for genotyping of the RIL population. Due to the limitation of markernumbers that can be analyzed in JoinMap software, a portion of the SNPs was selected to construct the primary genetic map. Based on the primary genetic map, we rearranged the scaffolds from the previous genome assembly and generated a new sesame genome sequence. Then, a bin map was constructed by joining the consecutive intervals on the genome that lacked a recombination event within the population [22, 23]. The genetic map included 1,522 bins that could be grouped into 13 sesame linkage groups (SLGs) with a total length of 1090.99 cM (Fig. 1, Additional file 1: Table S1). This is the first sesame genetic map in which the number of linkage groups equals the number of chromosome pairs in a sesame cultivar. The lengths of the 13 linkage groups ranged from 57.76 to 125.32 cM (mean value, 83.92 cM). Each linkage group had 84 (SLG12) to 168 (SLG3) bins resulting in a mean interval distance of 0.72 cM between adjacent bins (Additional file 1: Table S2). The bin distance was similar to that generated by RAD-Seq (0.69 cM) [5], but was shorter than that maps constructed using randomly selective amplification markers mainly (1.86 cM) [24] or the specific length amplified fragment sequencing (SLAF-seq) technology (1.20 cM) [4]. Furthermore, more than 99 % of the interval distances between adjacent bins were shorter than 6 cM (Additional file 2: Figure S1), indicating the high density of the new sesame genetic map.
Fig. 1.

High density genetic map of the sesame genome and the mapped QTLs. Positions of the mapped QTLs for plant height (PH), capsule zone length (CZL), height of the first capsule-bearing node (HFC), internode length (IL), node number (NN), and tip length without the capsule (TL). The seed coat color space L*, a*, and b* values are indicated with colored rectangles centered at the peak of each QTL
Update of sesame genome assembly
The previous de novo sesame assembly provided a total genome assembly of 274 Mb with a scaffold N50 of 2.1 Mb and 200 scaffolds greater than 150 kb in size. The assembly anchored 150 larger scaffolds into 16 pseudomolecules based on a genetic map that included 406 markers, which harbored 85.3 % of the total genome assembly and 91.7 % of the predicted genes (version 1.0) [2, 3]. Using the genetic map constructed in this study, we anchored another 172 scaffolds or contigs in a new sesame genome assembly (version 2.0) and linked together six smaller linkage groups in version 1.0 (LG12 and LG16, LG13 and LG14, and LG9 and LG15).
Despite the addition of 172 new scaffolds to the new version of the genome assembly, their orders were relatively consistent with the orders of the 150 scaffolds of version 1.0. In total, 20 changes occurred during the re-anchoring, including 10 scaffolds that were adjusted in the same pseudomolecules, 4 scaffolds that were relocated to other pseudomolecules, and 6 scaffolds that were split into two parts and relocated separately (Fig. 2; Additional file 1: Table S3 and S4). The inconsistency might be due to a single misassembly in which contig sequences were erroneously joined into these scaffolds through paired-end links during the initial assembly as reported for Cucumis melo [25, 26].
Fig. 2.
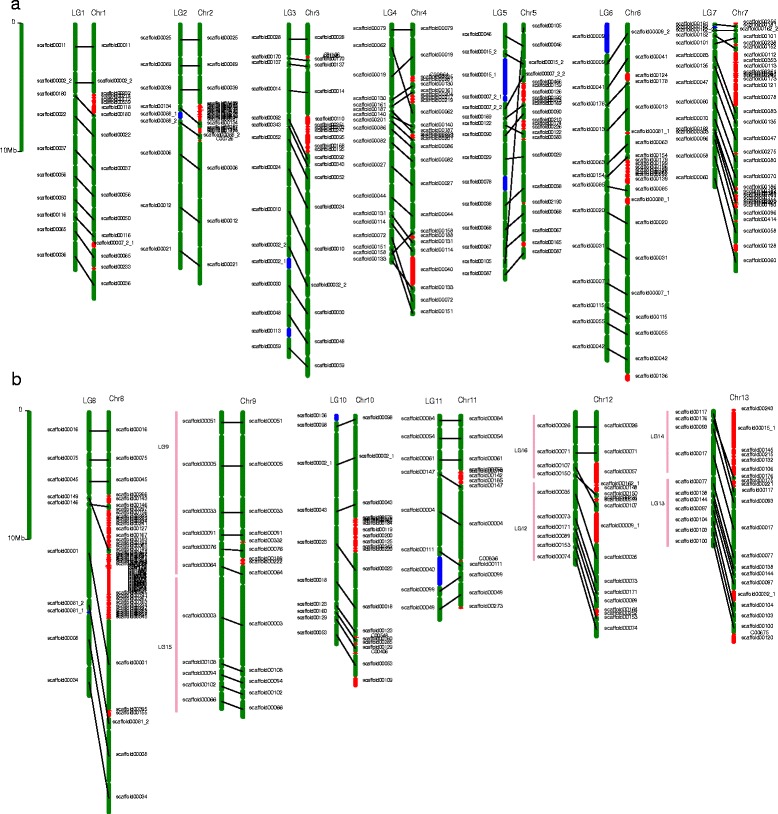
Anchored scaffolds in the current and previously assembled sesame genomes. Green scaffolds indicate anchored scaffolds that are consistent in the current (version 2.0) and previously (version 1.0) assembled genomes. Their relative positions are marked with lines. The newly anchored scaffolds in assembly version 2.0 are highlighted in red. The split scaffolds and relocated scaffolds are indicated in blue. The positions of the six smaller linkage groups in version 1.0 (LG9, LG12, LG13, LG14, LG15 and LG16) were indicated with pink lines
As a result, 327 scaffolds,(ncluding the split parts) were anchored to 13 pseudomolecules (Table 1), which were defined as pseudochromosomes and designated as Chr1 to Chr13 (Figs. 2 and 3). The lengths of the 327 anchored scaffolds ranged from 325 bp to 7.0 Mb (mean value, 784.7 kb; Additional file 2: Figure S2), 97.5 % of the scaffolds with lengths greater than 150 kb in version 1.0 were included in the new assembly version. The total length of the assembled pseudomolecules in the new version increased the assembly from 233.7 Mb to 258.4 Mb, representing 94.3 % of the genome assembly, and 97.2 % of the predicted gene models were anchored successfully (Fig. 3).
Table 1.
Comparison of the anchored sesame genome versions 1.0 and 2.0
| Version 2.0 | V2.0_scaff_number | V2.0_size (Mb) | Version 1.0 | V1.0_scaff_number | V1.0_size (Mb) |
|---|---|---|---|---|---|
| Chr1 | 17 | 20.26 | LG1 | 10 | 18.58 |
| Chr2 | 25 | 18.42 | LG2 | 8 | 18.50 |
| Chr3 | 22 | 25.85 | LG3 | 14 | 24.93 |
| Chr4 | 27 | 20.58 | LG4 | 18 | 17.36 |
| Chr5 | 23 | 16.58 | LG5 | 13 | 18.90 |
| Chr6 | 24 | 25.97 | LG6 | 13 | 25.29 |
| Chr7 | 34 | 16.76 | LG7 | 14 | 11.73 |
| Chr8 | 65 | 26.18 | LG8 | 9 | 21.52 |
| Chr9 | 14 | 22.85 | LG9 & LG15 | 11 | 22.46 |
| Chr10 | 22 | 19.49 | LG10 | 10 | 17.25 |
| Chr11 | 14 | 14.05 | LG11 | 9 | 15.45 |
| Chr12 | 18 | 16.28 | LG12 & LG16 | 10 | 11.34 |
| Chr13 | 22 | 16.47 | LG13 & LG14 | 11 | 9.93 |
| Total | 327 | 259.73 | 150 | 233.22 |
Fig. 3.
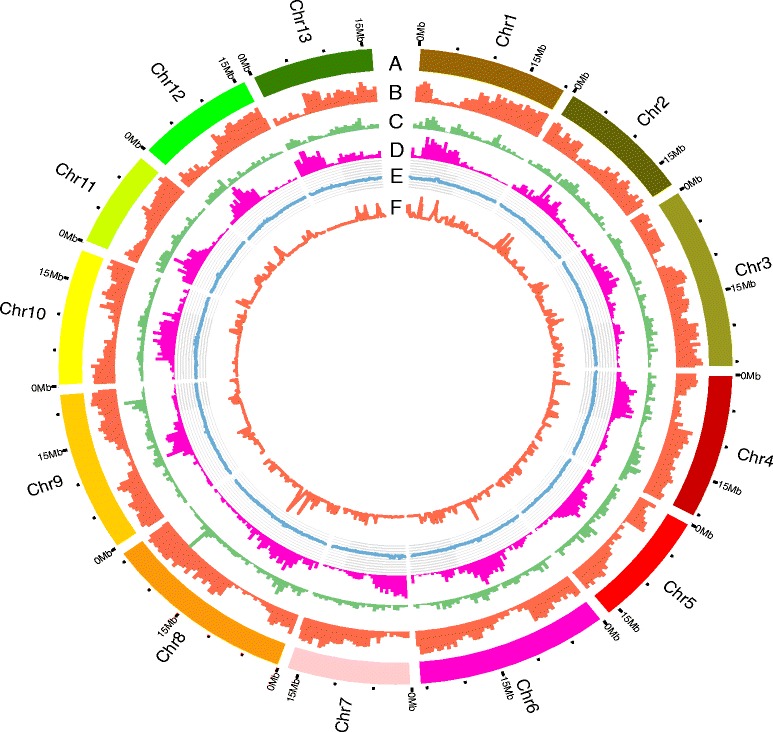
Distributions of basic elements of the sesame genome in the current assembly. a Pseudomolecules. b Gene density (mRNA); the frequency of sites within gene regions per 500 kb ranged from 0.04 to 0.61. c DNA transposon element (TE) density; the frequency of sites within the DNA TE regions per 500 kb ranged from 0 to 0.22. d Retrotransposon element density; the frequency of sites per 500 kb within the retrotransposon element regions ranged from 0 to 0.71. e GC content; the ratio of GC sites per 100 kb ranged from 0.32 to 0.40. f SNP density; the frequency of sites within SNP regions per 100 kb ranged from 1 × 10−5 to 1 × 10−3. Circos software (http://circos.ca) was used to construct the diagram
There were 5 scaffolds (GenBank accession nos. KI866576.1, KI866583.1, KI866588.1, KI866606.1, and KI866610.1) with lengths greater than 150 kb that were still missing in the 13 newly assembled pseudomolecules. These sequences may represent conserved regions with few mutation sites or lower frequencies of exchange during chromosome recombination. Other high-density genetic maps based on new mapping populations or fluorescence in situ hybridization (FISH) may be required to anchor the remaining scaffold and to make the map more perfect. The mean sesame whole-genome recombination rate (cM/Mb) was 4.25; the rate ranged from 3.45 for Chr11 to 5.38 for Chr10. Comparison of the physical and genetic distances between markers revealed that most of the SLGs contained a low recombination region located approximately 5 Mb from the proximal end or center (Additional file 2: Figure S3).
QTLs for traits related to sesame PH
The sesame plant height is generally determined by the height of the first capsule-bearing node (HFC), capsule zone length (CZL), and tip length without the capsule (TL) traits. The CZL is also related to the node number (NN) and internode length (IL) traits. The mature Zhongzhi No. 13 plants are normally greater than 1.8 m in height, whereas ZZM2748 is a semi-dwarf cultivar which grows to less than 0.9 m. Additionally, the two parents exhibited obvious differences in other traits related to PH (Additional file 2: Figure S4). Among the 430 progeny lines, the values for PH, HFC, CZL, TL, IL and NN were 0.65–1.96 (m), 0.15–1.16 (m), 0.22–1.28 (m), 0–15.8 (cm), 1.5–8.2 (cm) and 4.3–52.6, respectively (Additional file 2: Figures S5 and S6). PH, HFC, and IL fluctuated less than the other traits based on the coefficient of variation (CV) across the three field trials, and higher broad-sense heritabilities were also observed (Additional file 1: Table S5). Positive and negative transgressive segregation was observed in the RIL population suggesting that a germplasm could be created with the expected PH, CZL, TL, NN, and IL traits independent of environmental effects; however, the negative heterobeltiosis of HFC appeared to depend largely on the environment. PH was correlated with HFC, CZL, TL, NN, and IL (P < 0.01), and strong correlations were observed between PH and HFC and between CZL and NN (Fig. 4a). Interestingly, the correlation between NN and IL was negative across the three trial sites. This result supported the hypothesis that dwarf sesame cultivars could be bred without the loss of yield because reducing the internode length did not appear to affect the capsule node number.
Fig. 4.
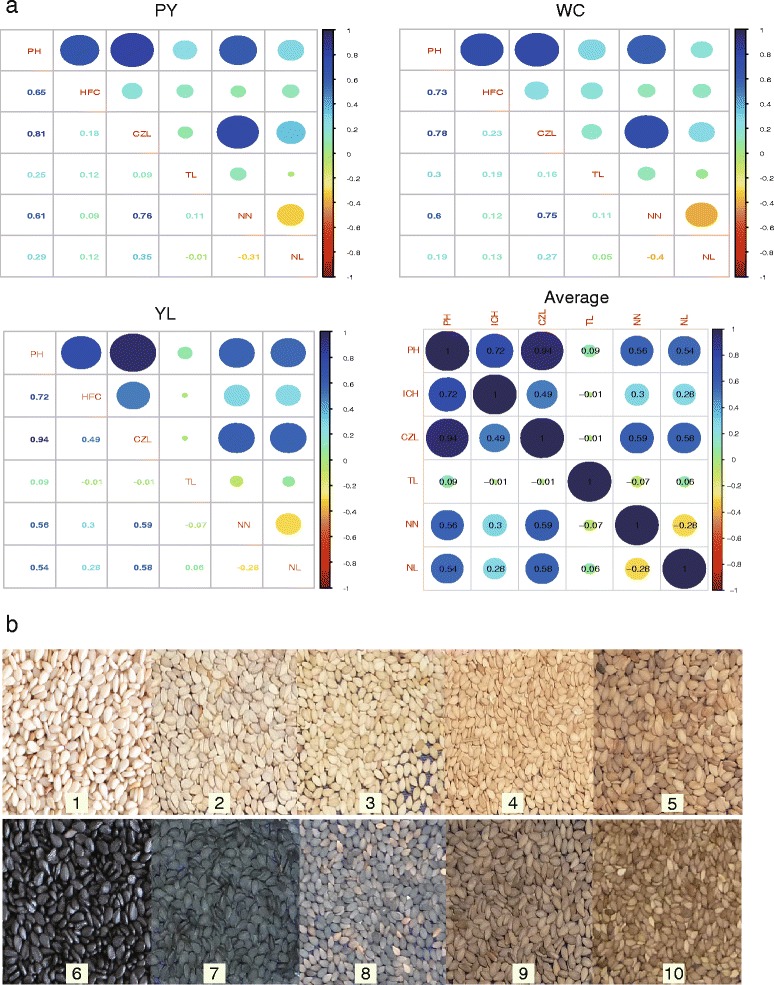
Characteristics of sesame plant height and seed coat color. a Correlations between sesame plant height and related traits across three trial sites. PH, plant height; CZL, capsule zone length; HFC, height of the first capsule-bearing node; IL, internode length; NN, node number; TL, tip length without the capsule; WC, Wuchang field trial location; PY, Pingyu field trial location; YL, Yangluo field trial location. The size and color of the ovals mirrored the correlation coefficient given in the lower triangle or ovals. b Variation in seed coat color in the RIL population. Subpanels 1 and 6 represent the seeds of the parental lines Zhongzhi No. 13 and ZZM2748, and the other panels represent the separation of the seed coat color in the population
Using the bin map, we identified QTLs underlying the sesame PH and the related traits HFC, CZL, TL, NN, and IL. Phenotypic datasets from the three experimental environments were coupled with the genotypic data using Windows QTL Cartographer 2.5 software [27]. The threshold used for the LOD scores to evaluate the statistical significance of the QTL effects was determined using 1,000 permutations. In total, 41 QTLs in nine SLGs were detected for the traits with contribution rates of 3–24 % (mean value, 8 %; Table 2, Fig. 1). Among them, 14, 15, and 12 QTLs were detected in three, two, and one environment(s), respectively. These loci were not distributed randomly because SLG3, SLG4, SLG8, and SLG9 together contained more than 78 % of the QTLs (Fig. 1).
Table 2.
Significant QTLs* associated with sesame plant height and the seed coat color space
| QTL | Linkage group | Position (cM) | Flanking bin_marker | Chr_space (kb) | LOD | Additive effect | R2 | WC | PY | YL |
|---|---|---|---|---|---|---|---|---|---|---|
| qPH-3.1 | SLG3 | 80.91 | SLG3_bin98-SLG3_bin99 | 525.3 | 7.62 | −5.17 | 0.06 | ✓ | ✓ | ✓ |
| qPH-3.2 | SLG3 | 92.41 | SLG3_bin114-SLG3_bin115 | 735.1 | 20.75 | −8.12 | 0.15 | ✓ | ✓ | ✓ |
| qPH-3.3 | SLG3 | 98.01 | SLG3_bin126-SLG3_bin127 | 928.1 | 22.44 | −8.91 | 0.18 | ✓ | ✓ | ✓ |
| qPH-4.1 | SLG4 | 34.11 | SLG4_bin49-SLG4_bin50 | 103.0 | 10.58 | 5.86 | 0.08 | ✓ | ✓ | |
| qPH-4.2 | SLG4 | 42.61 | SLG4_bin56-SLG4_bin57 | 908.3 | 7.16 | 5.09 | 0.06 | ✓ | ✓ | ✓ |
| qPH-8.1 | SLG8 | 65.61 | SLG8_bin107-SLG8_bin108 | 33.3 | 9.41 | 70.07 | 0.09 | ✓ | ✓ | |
| qPH-8.2 | SLG8 | 70.51 | SLG8_bin111-SLG8_bin112 | 360.2 | 29.75 | 10.18 | 0.23 | ✓ | ✓ | ✓ |
| qPH-9.1 | SLG9 | 78.81 | SLG9_bin102-SLG9_bin103 | 63.4 | 12.28 | 6.33 | 0.09 | ✓ | ✓ | |
| qPH-9.2 | SLG9 | 84.71 | SLG9_bin105-SLG9_bin106 | 1255.3 | 12.22 | 6.56 | 0.10 | ✓ | ✓ | ✓ |
| qPH-12.1 | SLG12 | 53.01 | SLG12_bin66-SLG12_bin67 | 94.3 | 3.52 | 41.35 | 0.03 | ✓ | ✓ | ✓ |
| qCZL-3.1 | SLG3 | 99.00 | SLG3_bin126-SLG3_bin127 | 928.1 | 10.43 | −4.88 | 0.09 | ✓ | ||
| qCZL-4.1 | SLG4 | 34.00 | SLG4_bin48-SLG4_bin49 | 127.4 | 7.95 | 4.09 | 0.07 | ✓ | ||
| qCZL-4.2 | SLG4 | 48.50 | SLG4_bin59-SLG4_bin60 | 237.1 | 3.94 | 32.40 | 0.04 | ✓ | ✓ | |
| qCZL-8.1 | SLG8 | 71.50 | SLG8_bin111-SLG8_bin112 | 360.2 | 27.69 | 7.82 | 0.24 | ✓ | ✓ | ✓ |
| qCZL-9.1 | SLG9 | 79.60 | SLG9_bin103-SLG9_bin104 | 725.9 | 7.47 | 4.00 | 0.06 | ✓ | ✓ | |
| qCZL-10.1 | SLG10 | 57.60 | SLG10_bin104-SLG10_bin105 | 176.6 | 2.78 | 2.39 | 0.02 | ✓ | ||
| qCZL-12.1 | SLG12 | 52.20 | SLG12_bin65-SLG12_bin66 | 101.8 | 3.90 | 2.83 | 0.03 | ✓ | ✓ | ✓ |
| qCZL-13.1 | SLG13 | 24.40 | SLG13_bin32-SLG13_bin33 | 110.9 | 6.65 | 42.23 | 0.07 | ✓ | ✓ | |
| qHFC-3.1 | SLG3 | 92.21 | SLG3_bin112-SLG3_bin113 | 324.9 | 9.99 | −34.32 | 0.09 | ✓ | ||
| qHFC-3.2 | SLG3 | 97.01 | SLG3_bin126-SLG3_bin127 | 928.1 | 23.26 | −3.65 | 0.22 | ✓ | ✓ | ✓ |
| qHFC-4.1 | SLG4 | 32.41 | SLG4_bin45-SLG4_bin46 | 359.2 | 6.13 | 1.75 | 0.05 | ✓ | ✓ | ✓ |
| qHFC-8.1 | SLG8 | 65.71 | SLG8_bin107-SLG8_bin108 | 33.3 | 5.59 | 30.10 | 0.05 | ✓ | ✓ | |
| qHFC-8.2 | SLG8 | 69.51 | SLG8_bin111-SLG8_bin112 | 360.2 | 10.91 | 2.37 | 0.09 | ✓ | ✓ | ✓ |
| qHFC-9.1 | SLG9 | 78.81 | SLG9_bin102-SLG9_bin103 | 63.4 | 12.40 | 2.46 | 0.10 | ✓ | ✓ | |
| qHFC-9.2 | SLG9 | 84.71 | SLG9_bin105-SLG9_bin106 | 1255.3 | 12.96 | 2.58 | 0.11 | ✓ | ||
| qHFC-9.3 | SLG9 | 91.01 | SLG9_bin112-SLG9_bin113 | 78.7 | 12.40 | 2.48 | 0.10 | ✓ | ✓ | |
| qIL-3.1 | SLG3 | 41.10 | SLG3_bin53-SLG3_bin54 | 153.7 | 7.57 | 0.15 | 0.07 | ✓ | ✓ | |
| qIL-8.1 | SLG8 | 71.50 | SLG8_bin111-SLG8_bin112 | 360.2 | 10.52 | 0.17 | 0.09 | ✓ | ✓ | ✓ |
| qIL-9.1 | SLG9 | 83.70 | SLG9_bin105-SLG9_bin106 | 1255.3 | 9.76 | 0.17 | 0.10 | ✓ | ✓ | ✓ |
| qIL-12.1 | SLG12 | 53.50 | SLG12_bin69-SLG12_bin70 | 63.2 | 3.86 | 1.61 | 0.04 | ✓ | ✓ | |
| qNN-3.1 | SLG3 | 91.80 | SLG3_bin111-SLG3_bin112 | 193.0 | 4.62 | −0.91 | 0.05 | ✓ | ||
| qNN-3.2 | SLG3 | 100.10 | SLG3_bin126-SLG3_bin127 | 928.1 | 10.15 | −1.31 | 0.10 | ✓ | ||
| qNN-4.1 | SLG4 | 54.00 | SLG4_bin72-SLG4_bin73 | 466.0 | 3.96 | 0.94 | 0.04 | ✓ | ✓ | |
| qNN-8.1 | SLG8 | 66.50 | SLG8_bin110-SLG8_bin111 | 1154.1 | 7.99 | 1.13 | 0.07 | ✓ | ✓ | |
| qNN-13.1 | SLG13 | 24.40 | SLG13_bin32-SLG13_bin33 | 110.9 | 8.75 | 1.42 | 0.09 | ✓ | ✓ | |
| qTL-2.1 | SLG2 | 66.10 | SLG2_bin93-SLG2_bin94 | 686.3 | 3.04 | 4.68 | 0.03 | ✓ | ||
| qTL-3.1 | SLG3 | 94.40 | SLG3_bin124-SLG3_bin125 | 75.7 | 7.90 | −7.68 | 0.08 | ✓ | ||
| qTL-3.2 | SLG3 | 103.60 | SLG3_bin131-SLG3_bin132 | 83.2 | 7.38 | −6.58 | 0.08 | ✓ | ||
| qTL-4.1 | SLG4 | 74.10 | SLG4_bin93-SLG4_bin94 | 169.7 | 3.04 | 0.36 | 0.03 | ✓ | ||
| qTL-5.1 | SLG5 | 35.60 | SLG5_bin58-SLG5_bin59 | 71.1 | 3.19 | −4.24 | 0.03 | ✓ | ||
| qTL-13.1 | SLG13 | 8.80 | SLG13_bin22-SLG13_bin23 | 1362.3 | 2.67 | −0.35 | 0.03 | ✓ | ✓ | |
| qSCa-4.1 | SLG4 | 50.90 | SLG4_bin63-SLG4_bin64 | 199.9 | 15.41 | 0.84 | 0.13 | ✓ | ✓ | |
| qSCa-8.1 | SLG8 | 73.40 | SLG8_bin114-SLG8_bin115 | 2518.0 | 26.73 | 1.18 | 0.25 | ✓ | ✓ | |
| qSCa-8.2 | SLG8 | 62.60 | SLG8_bin105-SLG8_bin106 | 575.8 | 9.02 | 0.69 | 0.09 | ✓ | ||
| qSCb-4.1 | SLG4 | 50.90 | SLG4_bin63-SLG4_bin64 | 199.9 | 54.75 | 4.81 | 0.39 | ✓ | ✓ | |
| qSCb-8.1 | SLG8 | 72.40 | SLG8_bin114-SLG8_bin115 | 2518.0 | 27.08 | 3.11 | 0.21 | ✓ | ✓ | |
| qSCb-11.1 | SLG11 | 0.00 | SLG11_bin1-SLG11_bin2 | 367.8 | 5.56 | 1.38 | 0.03 | ✓ | ✓ | |
| qSCl-4.1 | SLG4 | 50.90 | SLG4_bin63-SLG4_bin64 | 199.9 | 30.34 | 5.62 | 0.21 | ✓ | ✓ | |
| qSCl-8.1 | SLG8 | 73.40 | SLG8_bin114-SLG8_bin115 | 2518.0 | 44.06 | 7.14 | 0.46 | ✓ | ✓ | |
| qSCl-11.1 | SLG11 | 0.00 | SLG11_bin1-SLG11_bin2 | 367.8 | 20.76 | 4.64 | 0.14 | ✓ | ✓ |
* Traits related to plant height including capsule zone length (CZL), height of the first capsule-bearing node (HFC), internode length (IL), node number (NN), and tip length without the capsule (TL) are also listed. “Flanking bin_marker” indicates the bin_markers flanking the related QTL; “Chr_space” indicates the distance between the flanking bin_markers on the sesame chromosome; “Additive effect” indicates the estimated value for the genotype transmitted stably to offspring; “R2” indicates the contribution rate of the locus to the phenotype; Check marks in the last three columns indicate that the QTL was detected at a specific trial site; WC, Wuchang field trial location; PY, Pingyu field trial location; YL, Yangluo field trial location
Ten QTLs accounting for 3.03–23.32 % of the PH variation were detected. Four of these QTLs (qPH-3.2, qPH-3.3, qPH-8.2, and qPH-9.2) individually explained more than 10 % of the phenotypic variation (Table 2). We also detected eight HFC, eight CZL, and six TL QTLs. Of them, three of the HFC QTLs (qHFC-3.2, qHFC-9.1, and qHFC-9.3) and one CZL QTL (qCZL-8.1) were found to contribute more than 10 % of the phenotype variation at a minimum of two sites. TL might be easily affected by the environment because all of the TL QTLs were detected in only one or two of the three trial sites. Although five QTLs were detected for NN, none of them had a phenotype explanation rate of more than 10 %. Nevertheless, the qNN-13.1 locus accounted for approximately 9 % of NN and had a pleiotropic effect on CZL (qCZL-13.1) with a contribution rate of 6.89 %.
We also identified loci with pleiotropic effects in other QTLs based on their close or identical locations, particularly in QTLs with higher contribution rates. The qPH-8.2 and qPH-3.3 QTLs were the two major loci that accounted for 23.03 and 18.07 % of the sesame PH variation, respectively. The qPH-8.2 locus was derived from the tall parent with a positive effect of 10.18 % and was located between bin markers SLG8_bin111 and SLG8_bin112 in a 350-kb region. This region also contained three other QTLs (qHFC-8.2, qCZL-8.1, and qIL-8.1) that accounted for 8.91, 23.84, and 9.45 % of the phenotypic variation in HFC, CZL, and IL, respectively. The qPH-3.3 locus was located in a 928-kb region between bin126 and bin127 on SLG3. This region also contained three other QTLs (qHFC-3.2, qCZL-3.1, and qNN-3.2) that accounted for 22.00, 9.33, and 10.26 % of the phenotypic variation in HFC, CZL, and NN, respectively. The negative additive effects of these QTLs indicated that the ZZM2748 semi-dwarf parent contributes to a strong decrease in the sesame PH and related traits including HFC, CZL, and NN.
Plant height usually decides the plant architecture and contributes to the crop yield. Studies based on mutants with dwarf phenotypes had showed that plant hormones such as gibberellin (GA), auxin, cytokinin (CK), and brassinosteroid (BR) played important roles in determining stem elongation [28, 29]. In sesame, few studies have reported the locus or gene that encodes plant height; the exception is Wu et al.[5], who he detected two QTL with contribution rate no more than 6. However, the relationships of these QTLs were not clear for the inconsistent genetic maps and no sesame reference genomes available for the previous study. We predicted 53 and 102 candidate genes for the flanking bin-marker regions of qPH-8.2 and qPH-3.3, respectively, on the new assembled pseudomolecule chromosome according to the true confidence intervals (Additional file 1: Table S6). Annotation of these genes showed some of them may function in regulating of plant height. For example, SIN_1015931 in the site of qPH-3.3 site was predicted to encode an auxin-induced protein and SIN_1015910 encoded a brassinosteroid-insensitive protein. Further study in the future may be expected to verify their functions.
QTL for sesame seed coat color
The two parents Zhongzhi No. 13 and ZZM2748 were also distinguishable by seed coat color; the seeds of the former were white and the seeds of the latter were black (Fig. 4b). Genetic analysis showed that black was dominant to white in sesame seed coat color. The black seed coat color was characterized by delayed inheritance or predetermination because the F1 seeds (F2 organs in theory) produced from a cross between white and black seed sesame plants were always black. The RILs exhibited wide variations in seed coat color. The L*, a*, and b* values for the Zhongzhi No. 13 seed coat color were 64.35, 3.84, and 20.21, respectively, whereas those for ZZM2748 were 25.34, 0.73, and 2.87, respectively. The L* values for the 430 RILs ranged from 18.16 to 65.69 and the a* and b* values ranged from −0.12 to 12.62 and from 0.74 to 55.77, respectively (Additional file 2: Figure S7). Because “L*” represents brightness ranging from black to white, “a*” represents red and “b*” represents yellow for positive values [30], the measured values and distributions suggest that black, white, and yellow are predominant in the sesame seed coat color space, which is consistent with the observation that the seed coat color segregates in the RILs.
Across the Pingyu (PY) and Yangluo (YL) field trial locations, three QTLs (qSCl-8.1, qSCl-4.1, and qSCl-11.1) were detected repeatedly for the L* color space and made a cumulative contribution of 80 % to the phenotype. Among them, qSCl-8.1 explained approximately 46.0 % of the L* variation and was located at 73.40 cM on the SLG8 linkage group. This locus was flanked by the adjacent SLG8_bin114 (72.4 cM) and SLG8_bin115 (80.4 cM) bin markers that covered a 2.5-Mb region on pseudochromosome Chr8. The qSCl-4.1 locus was located in a 199.9-kb region on pseudochromosome Chr4 between the bin markers SLG4_bin63 (50.4 cM) and SLG4_bin64 (50.9 cM) and contributed 20.6 % of the L* variation. The qSCl-11.1 locus was located at the top of pseudochromosome Chr11 in a 367.8-kb region between bin markers SLG11_bin1 (0 cM) and SLG11_bin2 (0.15 cM) and explained 13.7 % of the L* variation. There two QTLs (qSCa-8.1 and qSCa-4.1) contributed 25.3 and 13.2 % of the color space a* value detected across the PY and YL field trial locations, respectively. Locus qSCa-8.2 was detected only at PY with a phenotype explanation rate of 9.2 %. Three QTLs (qSCb-4.1, qSCb-8.1, and qSCb-11.1) were detected for the color space b* across both locations with a total contribution rate of 64 %. The high contribution rate of the seed coat color locus to phenotype was also reported by Zhang et al., and he detected four QTL with contribution rates ranging from 9.6 to 39.9 % [24]. However, because the loci in the study of Zhang et al. were mainly located with AFLP markers in an independent genetic map, it is difficult to determine the relationship of the present loci to them.
Of the QTLs described above, qSCl-8.1, qSCa-8.1, and qSCb-8.1 were located in the same region on pseudochromosome Chr8, the qSCl-4.1, qSCb-4.1 and qSCa-4.1 QTLs were located in the same region on pseudochromosome Chr4, and qSCl-11.1 and qSCb-11.1 were located in the same region on pseudochromosome Chr11; thus, the three sites were pleiotropic for the sesame seed coat color. In other words, only 4 loci were detected here. In contrast, the intricate manifestation of seed coat color has been acknowledged to be due to the involvement of various pigments including flavonols, proanthocyanidin (carotenoid content condensed tannin), and possibly other phenolic relatives such as lignin and melanin [31]. Polyphenol oxidase (PPO) was reported to participate in the oxidation step in proanthocyanidin, lignin, and melanin biosynthesis, resulting in a dark seed coat color [32, 33]. Some genes encoding the enzymes participating in flavonoid biosynthesis have been cloned from Arabidopsis [34], grape [35], and soybean [36]. We checked the two sites of qSCl-8.1 and qSCl-4.1 with higher explanation rates (Additional file 1: Table S7 and S8) and found that the gene that encoded PPO (SIN_1016759), which was reported to account for the black color of the sesame seed coat, was also located at qSCl-4.1 [37]. For site qSCl-8.1, a gene (SIN_1022830) belonging to the cytochrome family that contained a ferredoxin reductase-type FAD-binding domain and flavoprotein transmembrane component was identified; this gene may contribute to the seed coat color [38]. These results also highlight the availability of the genome assembly.
Conclusions
No previously reported sesame genetic map has contained the same number of linkage groups as the number of chromosomes. The high number of linkage groups (even in the sesame genome assembly version 1.0) has caused confusion in the numbering of linkage groups and the assembled pseudomolecules in various studies. The present study brought the total number of assembled pseudomolecules equals the chromosome number of Sesamum indicum, and that 94.3 % of the de novo assembly and 97.2 % of the predicted gene models were anchored successfully. Thus, the number of sesame pseudochromosomes is fixed, which is important and helpful for comparative genomics and genetics studies such as sesame genome resequencing, marker development, genetic map integration, gene mapping, allele analysis, and evolutionary analyses.
We provide a high-density sesame bin map containing 1,522 bins spanning 1090.99 cM. The map covers the entire assembled genome and is useful for mapping QTLs or genes. Aside from the investigation of traits important for breeding sesame cultivars, such as indehiscent capsule, definite growth habit, PH, and seed coat color [24, 39, 40], few agronomic traits have been studied in the sesame. The present study not only mapped 41 QTLs for sesame PH and related traits but also detected nine QTLs for sesame seed coat color. In particular, two major QTLs (qPH-8.2 and qPH-3.3) associated with PH were located in a 350-kb region on Chr8 and in a 928-kb region on Chr3, respectively. We identified 53 and 102 candidate genes, respectively, in the two regions for future study. Notably, the qPH-3.3 locus is predicted to be responsible for the semi-dwarf sesame plant phenotype. This is the first report of a sesame semi-dwarf locus that can be used in an architectural study of sesame. The major qSCl-8.1 pleiotropic seed coat color locus for the L*, a*, and b* color space values was located on Chr8 in a 2.5-Mb region. Another major locus qSCb-4.1 located on Chr4 in a 199.9-kb region contributed 39 % to the b* values and contained 32 candidate genes. Collectively, the combination of the high-density genetic map and the sesame genome assembly update will serve as an essential platform for future comparative genomics and genetic studies.
Methods
Plant material and field trials
The ZZM2748 semi-dwarf sesame strain with a black seed coat was discovered and identified from among the 6,000 sesame germplasms preserved at the National Medium-Term Sesame Genebank of China (Wuhan, China). The mapping population consisted of 430 recombinant inbred lines (RILs, F8) and was derived from a cross between the Zhongzhi No. 13 (female parent) and the semi-dwarf ZZM2748 (male parent) cultivars. Zhongzhi No. 13 was de novo sequenced in 2014 [2] and was characterized by a high plant height (PH) and white seed coat color.
Three field trials were conducted in 2013 (F8) and 2014 (F8:9) during the normal sesame planting season (June–September) in three environments (Wuchang (2013), Pingyu (2014), and Yangluo (2014)) in China to evaluate trait variations in the RIL population. The 430 lines were grown in a randomized complete block design with three replicate plots in each trial. Each trial plot comprised three 2-m rows spaced 40 cm apart with a plant spacing of 10 cm. Sesame PH and related traits including height of the first capsule-bearing node (HFC), capsule zone length (CZL), tip length without the capsule (TL), node number (NN), and internode length (IL) were measured in each plot at a stage near maturity.
The seeds harvested from the Pingyu (2014) and Yangluo (2014) trial location environments were used to evaluate the sesame seed coat color. The seeds from three replicate plots of each line at each site were mixed together and evaluated using a Chroma Meter CR-400 (Konica Minolta, Japan). The Chroma Meter measures color by decomposing it into the L*, a*, and b* color space values, where “L*” represents brightness (0 for black, 100 for white), “a*” represents the color red when positive and the color green when negative, and “b*” represents the color yellow when positive and the color blue when negative [30].
DNA extraction and restriction-site associated DNA (RAD) sequencing
Healthy young leaves were collected from the two parental lines and the 430 RILs (F8) in 2013 and used for DNA extraction. The cetyltrimethylammonium bromide (CTAB) method was used with modifications to prepare total genomic DNA [4, 41]. DNA concentrations and quality were estimated using an ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA) and 0.8 % agarose gel electrophoresis with a lambda DNA standard [4].
RAD sequencing was performed according to the method of Baird et al. with some modifications [20]. Briefly, the extracted DNA was digested with the Taq I restriction enzyme (Takara, Dalian, China), and adapters containing a multiplex identifier (MID) were added to the samples. The ligation products were combined in appropriate multiplex pools (either two parental samples or seven RIL samples per library pool). Each of the library samples was size-selected by gel electrophoresis for DNA fragments that were 350–600 bp in length. Finally, the libraries were enriched by PCR amplification, quantified on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and sequenced on an Illumina HiSeq2000 instrument (San Diego, CA, USA) using paired-end reads (115 bp).
Sequence analysis and SNP discovery
Raw reads from multiple Illumina sequence channels were separated using the appropriate MID and assigned to each sample. The reads without unique barcodes and those with low quality in which 10 % of the nucleotides had a quality value less than Q30 (equals to 0.1 % sequencing error) were discarded. The clean pair-end reads were further trimmed to the RAD tags with a uniform length of 94 nucleotides.
All high-quality reads were aligned against the Zhongzi No. 13 scaffold sequences using BWA software [21]. The “mpileup” function in SAMtools software [42] was used to detect the SNPs between the Zhongzhi No. 13 and ZZM2748 parental lines using reads with mapping quality values greater than or equal to 20. The detected SNPs were assessed in the RILs and used for genotyping. The RIL genotypes were assigned to the Zhongzhi No. 13 or ZZM2748 homozygous genotypes or scored as heterozygous if the genotype was the same as both parents.
SNP marker development and linkage map construction
Prior to the construction of the linkage map, low quality SNP markers that were not homozygous in both parents or with a proportion of missing data greater than 70 % in the RIL population were filtered out. For each scaffold, 1 to 30 high-quality SNPs were selected from the head, middle and end. The segregation ratios of the SNP markers were evaluated using the chi-square test, and significantly distorted (P < 0.01) markers were removed. The remaining SNPs were grouped and ordered according to pairwise recombination frequencies at a logarithm of minimum odds (LOD) of 4.0 using JoinMap 4 software [43]. The Kosambi mapping function was chosen to translate the recombination frequencies into map distances in centiMorgans (cM). The goodness-of-fit of the calculated regression map for each tested position was checked against the default parameter. When the SNPs from the same scaffold were dispersed by other SNP on the map, all of the SNPs on the interactive scaffolds were regrouped and reordered and the related scaffolds were split at the recombinant breakpoints. The scaffolds were anchored to the pseudomolecules by relying on the order of the SNPs in the primary genetic map.
The bin map was constructed after the scaffolds were anchored according the method of Huang et al.[23]. Briefly, the consecutive genotypic SNPs on each pseudomolecule were scanned with a sliding window size of 15 SNPs and a step size of 1. For each window, the SNP from either parent were calculated. The windows containing 13 or more SNPs from either parent were considered to be homozygous for an individual, whereas those with less were classified as heterozygous. Adjacent windows with the same genotype were combined into blocks based on the recombinant breakpoints, and the blocks were considered to be bins [22, 44]. The bins on each pseudomolecule were taken as genetic markers and analyzed for linkage groups using JoinMap 4 with the setup described above. Finally, a sesame bin map was constructed.
QTL analysis
The frequency distributions of the mean phenotypic data for all 430 lines in each trial were analyzed using the R package software (https://www.r-project.org/). The QTLs underlying the six traits were detected using the composite interval mapping method implemented in Windows QTL Cartographer 2.5 software (Microsoft, Inc. Redmond, WA, USA) [27]. A 1,000 permutation parameter was set to determine the statistical significance of the QTL effects and a minimum LOD score of 2.5 was used to judge the presence of a QTL. QTLs were named according to the trait and linkage group locations. QTL names were designated based on the rules of the wheat gene nomenclature (http://wheat.pw.usda.gov/ggpages/wgc/98/Intro.htm).
Availability of supporting data
The RAW data of RAD-seq had been submitted to NCBI under the BioProject PRJNA301193 with SRA accession number SRA308937. The data sets supporting the results of this article including the updated genome assembly and annotation are available in http://ocri-genomics.org/Sinbase_v2.0 (genome assembly) and http://ocri-genomics.org/Sin_SNP_430RIL.tar.gz (SNP information). The candidate gene sequences near the mapped QTL can be accessed from sesame genome database (http://ocri-genomics.org/Sinbase).
Acknowledgements
The studies conducted by the Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, were supported by the National Basic Research Program of China (973 Program) (no. 2011CB109300), the National Natural Science Foundation of China (no. 31271766), China Agriculture Research System (no. CARS − 15), Chinese National Programs for High Technology Research and Development (863 program) (2014AA10A602-4), and Shenzhen Engineering Laboratory of Crop Molecular design breeding.
Abbreviations
- CZL
capsule zone length
- FISH
fluorescent in situ hybridization
- HFC
height of the first capsule-bearing node
- IL
internode length
- LOD
logarithm of odds
- MID
multiplex identifier
- NN
node number
- PH
plant height
- PY
Pingyu field trial location
- QTL
quantity trait loci
- RAD
restriction-site associated DNA
- SLAF
specific length amplified fragment
- SLG
sesame linkage groups
- TL
tip length without capsule
- WC
Wuchang field trial location
- YL
Yangluo field trial location
Additional files
Consists of Tables S1–S8. Table S1. Information and relationships of the sesame high-density genetic map to genome assembly; Table S2. Bin marker information for the high-density genetic map; Table S3. List of the scaffolds anchored onto chromosomes; Table S4. The 6 split scaffolds and the 4 relocated scaffolds; Table S5. Variation in plant height and related traits in the population across field trials. Table S6. List of genes in QTLs qPH-8.2 and qPH-3.3; Table S7. List of genes in the qSCl-8.1 QTL; Table S8. List of genes in the qSCb-4.1 QTL. (XLSX 152 kb)
Consists of Figures S1–S7. Figure S1. Distributions of the interval distances between adjacent markers on the genetic map; Figure S2. Length distributions of the anchored scaffolds; Figure S3. Genetic distance vs. physical distance. The genetic position of the markers was plotted against the corresponding physical position; Figure S4. Variations in sesame plant height and the height of the first capsule-bearing node in the two parents and the RILs; Figure S5. Boxplots of sesame plant height and related traits of the population across three trial sites; Figure S6. Histograms of the segregation of sesame plant height and related traits across three trial sites; Figure S7. Distributions of the L*, a*, and b* color space values across two trial sites. (DOCX 1406 kb)
Footnotes
Linhai Wang and Qiuju Xia contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XRZ, LHW and ZWQ contributed to the design of the research. QJX XMN, and HTX participated in sequencing and data analysis. YXZ, XFZ, YG and XDZ prepared plant samples. YXZ, XDZ XW and DHL performed the field trial and evaluated the phenotypes. QJX, XMN, LHW, and HTX participated in the genome assembly and QTL mapping. QJX, JYY and LHW participated in statistical analysis. LHW wrote the manuscript. XRZ and ZWQ participated in co-ordination and finalisation of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Linhai Wang, Email: linhai827@163.com.
Qiuju Xia, Email: xiaqiuju@genomics.org.cn.
Yanxin Zhang, Email: 13277065857@163.com.
Xiaodong Zhu, Email: 554656956@qq.com.
Xiaofeng Zhu, Email: xfsunny90@163.com.
Donghua Li, Email: ldh360681@163.com.
Xuemei Ni, Email: nixuemei@genomics.cn.
Yuan Gao, Email: gaoyuan0612@163.com.
Haitao Xiang, Email: xianghaitao@genomics.cn.
Xin Wei, Email: weixin8002@gmail.com.
Jingyin Yu, Email: yujyinfor@gmail.com.
Zhiwu Quan, Email: Quanzhiwu@genomics.org.cn.
Xiurong Zhang, Email: zhangxr@oilcrops.cn.
References
- 1.The Angiosperm Phylogeny G An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc. 2009;161(2):105–121. doi: 10.1111/j.1095-8339.2009.00996.x. [DOI] [Google Scholar]
- 2.Wang L, Yu S, Tong C, Zhao Y, Liu Y, Song C, et al. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 2014;15(2):R39. doi: 10.1186/gb-2014-15-2-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Yu J, Li D, Zhang X: Sinbase: An Integrated Database to Study Genomics, Genetics and Comparative Genomics in Sesamum indicum. Plant Cell Physiol. 2014. [DOI] [PubMed]
- 4.Zhang Y, Wang L, Xin H, Li D, Ma C, Ding X, et al. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 2013;13:141. doi: 10.1186/1471-2229-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu K, Liu H, Yang M, Tao Y, Ma H, Wu W, et al. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014;14(1):274. doi: 10.1186/s12870-014-0274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vales MI, Schon CC, Capettini F, Chen XM, Corey AE, Mather DE, et al. Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theor Appl Genet. 2005;111(7):1260–1270. doi: 10.1007/s00122-005-0043-y. [DOI] [PubMed] [Google Scholar]
- 7.Tanksley SD, Ganal MW, Prince JP, de Vicente MC, Bonierbale MW, Broun P, et al. High density molecular linkage maps of the tomato and potato genomes. Genetics. 1992;132(4):1141–1160. doi: 10.1093/genetics/132.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirata F, Fujita K, Ishikura Y, Hosoda K, Ishikawa T, Nakamura H. Hypocholesterolemic effect of sesame lignan in humans. Atherosclerosis. 1996;122(1):135–136. doi: 10.1016/0021-9150(95)05769-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen PR, Chien KL, Su TC, Chang CJ, Liu T-L, Cheng H, et al. Dietary sesame reduces serum cholesterol and enhances antioxidant capacity in hypercholesterolemia. Nutr Res. 2005;25(6):559–567. doi: 10.1016/j.nutres.2005.05.007. [DOI] [Google Scholar]
- 10.Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet. 2010;42(6):541–544. doi: 10.1038/ng.591. [DOI] [PubMed] [Google Scholar]
- 11.Jain N, Yadav R. Genetic diversity analysis among pre-green revolution, post-green revolution era cultivars, and wheat landraces as revealed by microsatellite markers. Crop Pasture Sci. 2009;60(4):373–381. doi: 10.1071/CP08310. [DOI] [Google Scholar]
- 12.Hedden P. The genes of the Green Revolution. Trends Genet. 2003;19(1):5–9. doi: 10.1016/S0168-9525(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 13.Saville RJ, Gosman N, Burt CJ, Makepeace J, Steed A, Corbitt M, et al. The ‘Green Revolution’ dwarfing genes play a role in disease resistance in Triticum aestivum and Hordeum vulgare. J Exp Bot. 2012;63(3):1271–1283. doi: 10.1093/jxb/err350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cagirgan MI, Uzun B. Comparison of determinate and indeterminate lines of sesame for agronomic traits. Field Crops Res. 2006;96(1):13–18. doi: 10.1016/j.fcr.2005.06.010. [DOI] [Google Scholar]
- 15.Uzun B, Cagirgan MI. Identification of molecular markers linked to determinate growth habit in sesame. Euphytica. 2009;166(3):379–384. doi: 10.1007/s10681-008-9818-y. [DOI] [Google Scholar]
- 16.Wang L, Zhang Y, Li P, Wang X, Zhang W, Wei W, et al. HPLC analysis of seed sesamin and sesamolin variation in a sesame germplasm collection in China. J Am Oil Chem Soc. 2012;89(6):1011–1020. doi: 10.1007/s11746-011-2005-7. [DOI] [Google Scholar]
- 17.Kanu PJ. Biochemical analysis of black and white sesame seeds from China. Am J Biochem Mol Biol. 2011;1(2):145–157. doi: 10.3923/ajbmb.2011.145.157. [DOI] [Google Scholar]
- 18.Suh Y, Vijg J. SNP discovery in associating genetic variation with human disease phenotypes. Mutat Res. 2005;573(1–2):41–53. doi: 10.1016/j.mrfmmm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, Cistue L, Corey A, Filichkina T, et al. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12:4. doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen ZL, Wang BB, Dong XM, Liu H, Ren LH, Chen J, Hauck A, Song WB, Lai JS. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F-2 maize population. BMC Genomics. 2014;15(1):433. [DOI] [PMC free article] [PubMed]
- 23.Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19(6):1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Miao H, Wei L, Li C, Zhao R, Wang C. Genetic analysis and QTL mapping of seed coat color in sesame (Sesamum indicum L.) PLoS One. 2013;8(5):e63898. doi: 10.1371/journal.pone.0063898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu X, Leung HC, Chin FY, Yiu SM, Quan G, Liu B, et al. PERGA: a paired-end read guided de novo assembler for extending contigs using SVM and look ahead approach. PLoS One. 2014;9(12):e114253. doi: 10.1371/journal.pone.0114253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Argyris JM, Ruiz-Herrera A, Madriz-Masis P, Sanseverino W, Morata J, Pujol M, et al. Use of targeted SNP selection for an improved anchoring of the melon (Cucumis melo L.) scaffold genome assembly. BMC Genomics. 2015;16(1):4. doi: 10.1186/s12864-014-1196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva Lda C, Wang S, Zeng ZB. Composite interval mapping and multiple interval mapping: procedures and guidelines for using Windows QTL Cartographer. Methods Mol Biol. 2012;871:75–119. doi: 10.1007/978-1-61779-785-9_6. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Li J. Molecular basis of plant architecture. Annu Rev Plant Biol. 2008;59:253–279. doi: 10.1146/annurev.arplant.59.032607.092902. [DOI] [PubMed] [Google Scholar]
- 29.Durbak A, Yao H, McSteen P. Hormone signaling in plant development. Curr Opin Plant Biol. 2012;15(1):92–96. doi: 10.1016/j.pbi.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 30.Imtiaz M, Ogbonnaya FC, Oman J, van Ginkel M. Characterization of quantitative trait loci controlling genetic variation for preharvest sprouting in synthetic backcross-derived wheat lines. Genetics. 2008;178(3):1725–1736. doi: 10.1534/genetics.107.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu CY. Molecular mechanism of manipulating seed coat coloration in oilseed Brassica species. J Appl Genet. 2013;54(2):135–145. doi: 10.1007/s13353-012-0132-y. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Tang T, Qian Q, Wang Y, Yan M, Zeng D, et al. Independent losses of function in a polyphenol oxidase in rice: differentiation in grain discoloration between subspecies and the role of positive selection under domestication. Plant Cell. 2008;20(11):2946–2959. doi: 10.1105/tpc.108.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer AM. Polyphenol oxidases in plants and fungi: going places? A review. Phytochemistry. 2006;67(21):2318–2331. doi: 10.1016/j.phytochem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Saito K, Yonekura-Sakakibara K, Nakabayashi R, Higashi Y, Yamazaki M, Tohge T, et al. The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol Biochem. 2013;72:21–34. doi: 10.1016/j.plaphy.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot. 2011;62(8):2465–2483. doi: 10.1093/jxb/erq442. [DOI] [PubMed] [Google Scholar]
- 36.Yang K, Jeong N, Moon JK, Lee YH, Lee SH, Kim HM, et al. Genetic analysis of genes controlling natural variation of seed coat and flower colors in soybean. J Hered. 2010;101(6):757–768. doi: 10.1093/jhered/esq078. [DOI] [PubMed] [Google Scholar]
- 37.Wei X, Liu K, Zhang Y, Feng Q, Wang L, Zhao Y, et al. Genetic discovery for oil production and quality in sesame. Nat Commun. 2015;6:8609. doi: 10.1038/ncomms9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatlestad GJ, Sunnadeniya RM, Akhavan NA, Gonzalez A, Goldman IL, McGrath JM, et al. The beet R locus encodes a new cytochrome P450 required for red betalain production. Nat Genet. 2012;44(7):816–820. doi: 10.1038/ng.2297. [DOI] [PubMed] [Google Scholar]
- 39.Uzun B, Lee D, Donini P, ÇAǧirgan M. Identification of a molecular marker linked to the closed capsule mutant trait in sesame using AFLP. Plant Breed. 2003;122(1):95–97. doi: 10.1046/j.1439-0523.2003.00787.x. [DOI] [Google Scholar]
- 40.Uzun B, Yol E. Inheritance of number of capsules per leaf axil and hairiness on stem, leaf and capsule of sesame (Sesamum indicum L.). Australian Journal of Crop Science 2011;5(1):78–81.
- 41.Doyle J, Doyle J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry Bulletin, 1987;19:11–15.
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.VAN Ooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res. 2011;93(5):343–349. doi: 10.1017/S0016672311000279. [DOI] [PubMed] [Google Scholar]
- 44.Ren R, Ray R, Li P, Xu J, Zhang M, Liu G, et al. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genomics. 2015;290(4):1457–1470. doi: 10.1007/s00438-015-0997-7. [DOI] [PubMed] [Google Scholar]