

Abstract
Interest in bacterial proteasomes was sparked by the discovery that proteasomal degradation is required for the pathogenesis of Mycobacterium tuberculosis, one of the world's deadliest pathogens. Although bacterial proteasomes are structurally similar to their eukaryotic and archaeal homologs, there are key differences in their mechanisms of assembly, activation, and substrate targeting for degradation. In this article, we compare and contrast bacterial proteasomes with their archaeal and eukaryotic counterparts, and we discuss recent advances in our understanding of how bacterial proteasomes function to influence microbial physiology.
Keywords: Mycobacterium, proteasome, Pup, pupylation, regulated proteolysis
Introduction
Regulated protein degradation affects virtually every biological pathway across all domains of life. In eukaryotes, the majority of regulated proteolysis is carried out by proteasomes, which are compartmentalized proteases that primarily degrade proteins posttranslationally modified with the protein ubiquitin (reviewed in 22, 77). The proteolytic component of all proteasomes is the 20S core particle (CP) (Figure 1a), a complex composed of four heptameric rings that are stacked on one another to form a cylinder (27, 32, 46, 76, 78) that can cleave multiple types of substrates (27, 30, 52). The active site of the 20S CP is sequestered, which prevents folded proteins from being inappropriately degraded. Thus, protein degradation is highly dependent on proteasomal cofactors, which recruit specific substrates and unfold them so that they may enter the 20S CP.
Figure 1.
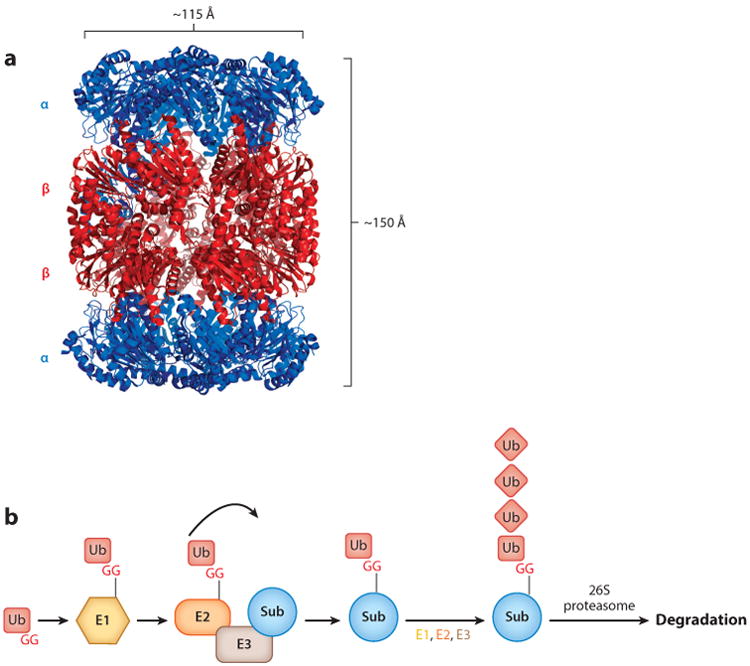
Structure of a proteasome 20S CP and schematic of the Ub-proteasome system. (a) Crystal structure of the 20S CP from Mycobacterium tuberculosis [Protein Data Bank (PDB) ID: 3MI0] (32). (b) Eukaryotic Ub-proteasome system. An E1 enzyme activates Ub by adenylating the C-terminal G of Ub and then forming a thioester bond. Ub is then transferred to an E2 Ub-conjugating enzyme, which then transfers Ub to an E3 ligase that conjugates Ub to a lysine on a target substrate. Additional rounds of ubiquitylation on Ub are generally required for receptors on the 26S proteasome to recognize the doomed protein for degradation. Abbreviations: CP, core particle; G, glycine; Sub, substrate; Ub, ubiquitin.
A cofactor complex responsible for degrading proteins in eukaryotes is the 19S regulatory particle (RP), which binds to either or both ends of the 20S CP to form the 26S proteasome (18). The 19S RP can be separated into two major components, the base and the lid, each of which is composed of several unique polypeptide subunits. The lid contains numerous components that play important functional roles, including removal of ubiquitin and recognition of proteins that are targeted for degradation (25, 79). The 19S RP base contains a hexameric ring of AAA ATPases (ATPases associated with diverse cellular activities) that unfolds proteins (4, 45) and pushes them into the active site of the 20S CP, and the base also has at least two receptors that help recruit substrates to the proteasome (33, 64).
In eukaryotes, proteins are marked for destruction by ubiquitin, a 76–amino acid protein that can be covalently linked by its carboxyl (C)-terminal glycine to the lysine of a doomed protein. The addition of ubiquitin to a target protein is carried out by an elegant coordinated pathway involving ubiquitin-activating and -conjugating enzymes (Figure 1b). Using ATP, an E1 ubiquitin-activating enzyme adenylates the C terminus of ubiquitin, allowing it to form a thioester bond with a cysteine in the E1 enzyme. Ubiquitin is then transferred to an E2 conjugating enzyme, which forms a more transient thioester bond with ubiquitin and, with the help of an E3 ligase, positions it for transfer to a lysine on a substrate protein (reviewed in 71). Because ubiquitin contains lysines, it can be ubiquitylated to form polyubiquitin chains that help target a substrate for proteasomal degradation (10). Eukaryotes encode numerous E3 ligases that can each target a specific protein or group of proteins for ubiquitylation. Thus, it is not surprising that the ubiquitin-proteasome system regulates many biological processes, and it is essential for eukaryotic life (reviewed in 60).
Proteasomes are also found in all archaea and in bacteria of the orders Actinomycetales and Nitrospirales. The mechanisms by which archaea target proteins for proteasomal degradation are still unclear; however, it is postulated that a posttranslational modification related to ubiquitylation called SAMPylation is involved in this process (reviewed in 49). In contrast, proteasome-containing bacteria utilize a system termed pupylation that is functionally analogous to but chemically distinct from ubiquitylation. Unlike eukaryotic proteasomes and ubiquitylation, bacterial proteasomes and pupylation are not absolutely required for viability; however, recent studies have begun to shed light on the important pathways that they regulate.
Bacterial 20S Core Particles
Core Particle Genes and Structure
The genes encoding 20S CP subunits and proteasomal cofactors are generally found in operons on bacterial chromosomes. The bacterial 20S CP is composed of just two subunits, α and β, which are encoded by proteasome core A (prcA) and proteasome core B (prcB) and are in an operon with pup (prokaryotic ubiquitin-like protein), which is described in detail in the eponymous section below (Figure 2a) (38, 76). Several other genes are also found near this locus, including dop (deamidase of Pup), pafA (proteasome accessory factor A), and mpa (mycobacterial proteasome ATPase), which are associated with proteasome function and are described in further detail below. pafB and pafC are cotranscribed with pafA but do not appear to contribute to proteasome function (21). Intriguingly, Rhodococcus species have two 20S CP operons, each encoding a distinct set of prcBA genes; the biological significance of this remains unknown.
Figure 2.
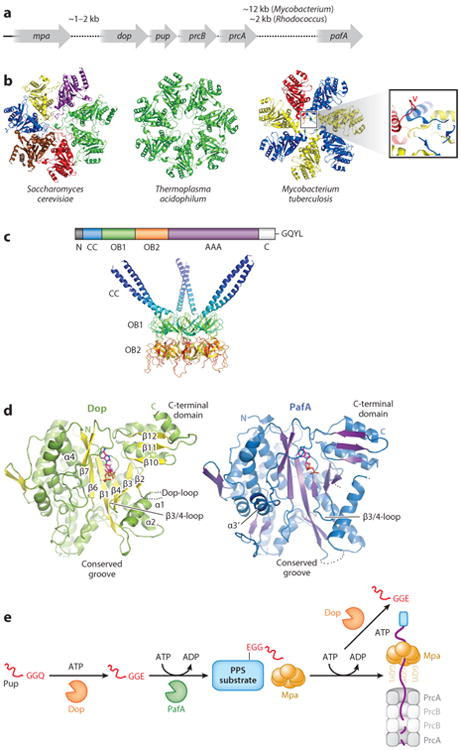
The Pup-proteasome system (PPS). (a) Genomic organization of bacterial proteasome operons in Mycobacterium tuberculosis and Rhodococcus erythropolis. (b) Proteasome gate structures differ among domains of life. Top-view comparison of the 20S CP α ring from Saccharomyces cerevisiae [Protein Data Bank (PDB) ID: 2F16] (27), Thermoplasma acidophilum (PDB: 1PMA) (65), and Mycobacterium tuberculosis (inset view is rotated and magnified) (32). Subunits that assume equivalent structures are colored similarly. (c) Mpa/ARC is a bacterial proteasomal ATPase. (Bottom) X-ray crystal structure of the coiled coil (CC)- and oligosaccharide/oligonucleotide-binding (OB) domains of Mycobacterium tuberculosis Mpa (PDB: 3M9B) (adapted from 80). (d) PafA and Dop have highly similar structures. X-ray crystal structure of PafA from Corynebacterium glutamicum (left) and Dop from Acidophilus cellulolyticus (right) (adapted from 53). (e) Schematic representation of the PPS.
The architecture of a bacterial 20S CP is similar to that of the eukaryotic and archaeal complexes. The core protease is composed of four heptameric rings, which stack on one another to form a barrel of ∼150 Å length and ∼115 Å diameter (32, 41, 44, 76). Each outer α ring is composed of seven identical PrcA subunits, and each inner β ring is composed of seven identical PrcB subunits to give an overall organization of α7β7β7α7 (Figure 1a). An opening of 23 Å in the α ring, which is wider than the 13 Å opening in archaea (46), leads to a pore that runs through the center of the entire complex and connects three large cavities, one at each α-ring/β-ring interface and one between the two β rings that contain the proteolytic active sites.
Assembly
To form a mature protease, the 20S CP must be assembled in a precisely ordered manner. In eukaryotes, this is accomplished with the aid of proteasomal chaperones that bind to 20S CP intermediates and facilitate their assembly (reviewed in 40). Bacterial CPs, however, are capable of self-assembly; coproduction of Rhodococcus PrcA and PrcB in Escherichia coli results in the formation of active 20S CPs without the need for any other Rhodococcus-specific factors (88). Interestingly, any combination of PrcA and PrcB from either Rhodococcus proteasome operon is capable of forming a mature 20S CP in vitro (87, 88).
Two intermediates of 20S CP assembly have been identified. The first is a half-proteasome, which consists of an α ring and a β ring, that forms spontaneously on coproduction of PrcA and PrcB. Two half-proteasomes then come together to form a preholoproteasome in which the PrcB subunits must undergo autoprocessing to become proteolytically active. This processing event involves removal of the PrcB amino (N)-terminal propeptides, leaving N-terminal threonines (Thr1) that act as the catalytic nucleophiles of the mature holoproteasome (87).
The PrcB propeptide appears to have several functions that are not always consistent among bacterial species. In Rhodococcus, the propeptide promotes 20S CP maturation; production of PrcB subunits lacking the propeptide results in the slow formation of preholoproteasomes from half-proteasomes. In contrast, production of a PrcB subunit that cannot remove its propeptide results in a more rapid formation of preholoproteasomes (87). The propeptide is normally buried within the central pore of a half-proteasome and makes inter-β-ring contacts that promote the apposition of two half-proteasomes (41, 84). These contacts can be provided even in trans, as addition of exogenously purified propeptide is sufficient to restore a wild-type maturation rate to a system using PrcB lacking the propeptide (87). Thus, as is observed for eukaryotic 20S CPs, the Rhodococcus PrcB propeptide promotes 20S CP maturation.
Interestingly, the exact opposite is true for the Mycobacterium tuberculosis 20S CP. This was first suggested by experiments that demonstrate the temperature dependence of 20S CP maturation: When PrcA and PrcB are produced in Escherichia coli at 37°C they form mature 20S CPs, but at 30°C they arrest at the half-proteasome state. This temperature dependence is overcome by deletion of the PrcB propeptide, which allows mature 20S CPs to form at 30°C (44). Thus, in Mycobacterium tuberculosis, the propeptide is a barrier to core particle maturation. Structural studies provided a mechanistic basis for this finding: The Mycobacterium tuberculosis propeptides extend from half-proteasomes (32), which is in contrast to the internal propeptides seen in Rhodococcus (41). Thus, it is proposed that these protruding propeptides prevent the apposition of two β rings to inhibit the progression from half-proteasome to preholoproteasome. Interestingly, on forming a preholoproteasome, the Mycobacterium tuberculosis propeptide retracts into the 20S core, taking a similar position to that which is seen in Rhodococcus (42). Thus, despite the contrasting effects of propeptides on 20S CP assembly, a similar structure is assumed for the final steps of maturation.
Catalytic Activities
The active sites of bacterial 20S CPs are similar to those described for archaea and eukaryotes (27, 46, 65). The hydroxyl group of PrcB Thr1 is the nucleophile that is responsible for the proteolytic activity of the proteasome (32, 44, 50). The amino group of PrcB Thr1 (Thr1N) acts as a proton acceptor that allows the side chain oxygen (Thr1γO) to attack an electrophilic center on a substrate. Aspartate 17 (Asp17) forms a salt bridge to a lysine (Lys33), and the Lys33 side chain amino group is thought to be protonated and form a hydrogen bond to Thr1γO that would further promote removal of its proton by Thr1N (27, 32, 46). Thus, Thr1γO, Thr1N, Lys33, and Asp17 form a catalytic tetrad to promote nucleophilic attack by Thr1γO.
To hydrolyze a protein at a specific residue, a protease must accommodate a particular set of amino acids in its active site. In 20S CPs, a binding pocket formed by the β rings provides substrate specificity. Eukaryotes encode multiple types of β subunits with different substrate specificities and can therefore hydrolyze multiple distinct sites including sites after residues that are hydrophobic (chymotrypsin-like activity), basic (tryptic activity), and acidic (caspase-like activity) (27, 30). Because bacteria lack β-subunit diversity, they must rely on a single set of binding pocket residues to accommodate substrates. In Rhodococcus and Thermoplasma, these residues are hydrophobic and therefore have only chymotrypsin-like activity (12, 76). However, despite encoding only a single β subunit, Mycobacterium tuberculosis 20S CPs have all three catalytic activities of the eukaryotic protease (44). This was explained by the crystal structure, which revealed an active site lined by hydrophobic residues on one side and hydrophilic residues on the other (32). Thus, the Mycobacterium tuberculosis substrate-binding pocket resembles a hybrid of the three eukaryotic binding pockets.
Gating
In the absence of a proteasome activator, 20S CPs are normally in a closed-gate conformation with the N-terminal residues of each α subunit interacting to occlude the opening to the 20S CP active site. The gating residues of the Thermoplasma 20S CP are disordered, which may explain the observation that archaeal proteasomes have relatively high protease activity even in the absence of gate-opening cofactors (Figure 2b, center) (46). In contrast, each of the seven α-ring subunits occupies a specific position in the closed gate of the eukaryotic 20S CP (Figure 2b, left) (27), rendering it incapable of degrading even small peptides in the absence of a proteasome activator.
The formation of bacterial proteasome gates appears to be distinct from the formation of proteasome gates in other species. In contrast to the Thermoplasma complex, the Mycobacterium tuberculosis20S CP has a fully closed gate, which is achieved because each PrcA subunit assumes one of three different conformations. Three form an L shape (“L”), three form an extended linear shape (“E”), and one projects away from the 20S CP to avoid a steric clash (“V”) (Figure 2b, right) (42).
ATP-Dependent Proteasome Activators
In eukaryotes and archaea, the binding of proteasomal cofactors to 20S CPs leads to the activation of peptidase activity by repositioning α-ring gating residues into an open conformation. Structural studies of these activators have provided a wealth of mechanistic information that is described in detail elsewhere (58, 68, 69, 83). Like the eukaryotic and archaeal proteasome systems, bacteria possess AAA ATPases that target doomed proteins to 20S CPs for degradation. However, unlike eukaryotic 19S RPs that contain six unique ATPases, bacteria use homohexameric ATPase rings (16, 72). The first bacterial proteasomal ATPase described was a Rhodococcus ARC (ATPase forming a ring-shaped complex) (85), which was discovered on the basis of its sequence homology to the eukaryotic Rpt activators and the proximity of the arc gene to one of the Rhodococcus proteasome operons (Figure 2a). This was followed by the characterization of its Mycobacterium tuberculosis ortholog, Mpa (mycobacterial proteasome ATPase; the name arc was already in use for another gene in M. tuberculosis) (14).
Structurally, ARC/Mpa resembles other proteasomal ATPases. From its N terminus to its C terminus, it contains an α-helical domain followed by two oligonucleotide/oligosaccharide-binding (OB) domains and an AAA ATPase domain (Figure 2c). In an Mpa homohexamer, adjacent α-helical domains form three pairs of coiled coils. The OB domain alone is capable of self-assembly into a hexamer (81, 86), which suggests that this region determines the ringed structure of the mature protein. Like the related bacterial ATPase ClpX, Mpa likely utilizes internal pore loops to unfold and translocate a target protein toward its C-terminal end, as mutagenesis of pore loop residues in Mpa abrogates the degradation of substrates in Mycobacterium tuberculosis (48, 81).
Evidence that Mpa plays a direct role in proteasomal degradation came from the discovery of the first proteasome substrates. Two proteins, malonyl CoA-acyl carrier protein transacylase (FabD) and 3-methyl-2-oxobutanoate hydroxymethyltransferase (PanB), specifically accumulate in mutants lacking mpa or in wild-type Mycobacterium tuberculosis treated with a proteasome inhibitor. Mpa also accumulates upon chemical inhibition of the proteasome, which indicates that it is a proteasome substrate (54). Notably, Mpa has a highly conserved penultimate tyrosine (Tyr, Y), which is also found in the C-terminal HbYX (hydrophobic-tyrosine-any amino acid) motif that is required for several eukaryotic proteasomal cofactors to bind to 20S CPs. This penultimate tyrosine is essential for Mpa to facilitate proteasomal protein degradation, as substrate degradation of Mycobacterium tuberculosis mpa mutants could not be complemented by mpa alleles where the tyrosine was mutated (54). Notwithstanding a lack of data that show robust binding of Mpa to wild-type 20S CPs, it nonetheless appears that the C terminus of Mpa is an essential part of an interaction required for proteolysis.
Pupylation
Prokaryotic Ubiquitin-Like Protein
Although 20S CPs are conserved across domains of life, ubiquitin is found exclusively in eukaryotes. As a result, it was initially uncertain how substrates were targeted for degradation in bacteria. Pearce et al. (55) performed a bacterial two-hybrid screen to search for proteins that bound to Mpa; the goal was to identify cofactors required for proteolysis. This screen identified Pup, a 64–amino acid protein encoded upstream of prcBA that robustly interacts with Mpa. This study also determined that Pup covalently links to a specific lysine of the proteasome substrate FabD, and it showed that mutagenesis of the preferred lysine in FabD stabilizes the protein in vivo (55). Later in vitro degradation assays showed Mpa-, 20S CP–, and Pup-dependent degradation of both FabD and PanB, albeit very slowly (5, 72). Collectively, these data showed that pupylation directly targets proteins for proteasomal degradation in bacteria in a manner functionally analogous to eukaryotic ubiquitylation.
Studies assessing the structural characteristics of Pup elucidated further differences between Pup and ubiquitin. Ubiquitin folds into a characteristic compact structure, whereas Pup is not a compactly folded protein and for the most part lacks secondary structure (11, 43). Although Pup has been categorized as an intrinsically disordered protein, it has a propensity to form helices that are critical for protein-protein interactions necessary for protein degradation. Importantly, unlike ubiquitin, it does not appear that Pup forms poly-Pup chains.
Deamidation and Ligation of Pup to Substrates
Like ubiquitin, the C terminus of Pup forms a covalent bond with a lysine of a doomed protein. Pearce et al. (55) noted that Pup, although encoded with a C-terminal glutamine (Gln), forms an isopeptide bond with its target via a C-terminal glutamate (Glu), which suggests that the C terminus of Pup is deamidated prior to substrate ligation. Pearce et al. also noticed that a mutation in pafA prevents pupylation. PafA was identified at the same time as Mpa in a genetic screen for M. tuberculosis mutants that were hypersensitive to nitric oxide (NO) (13), but its enzymatic activity had not yet been elucidated. Not long after the discovery of Pup, the Aravind group (36) hypothesized that another protein, Rv2112c, encoded upstream of pup-prcBA, might also have a function in pupylation because PafA and Rv2112c share 32% amino acid sequence identity in addition to a predicted similar overall structure (Figure 2d). Both proteins were predicted to resemble carboxylate-amine ligases such as glutamine synthetase, but they have unique C-terminal domains found only in PafA/Rv2112c homologs.
With these observations in place, the Weber-Ban group (73) made the key prediction that either Rv2112c or PafA was required for PupGln deamidation or PupGlu ligation to substrates. Striebel et al. (73) showed that deamidation by Rv2112c, now called Dop, activates Pup prior to substrate ligation by PafA (Figure 2d, e). Interestingly, Dop requires ATP as a cofactor but does not hydrolyze ATP during the deamidation reaction (73). Although Dop lacked any obvious catalytic sites, the use of a chemical trap showed that Dop utilizes an aspartate (Asp95 in Mycobacterium tuberculosis H37Rv Dop) as a nucleophile for deamidation, a chemically unusual mechanism for this reaction (6). The development of a Pup reporter with a fluorescent moiety on the terminal amino acid side chain confirmed that Dop hydrolyzes this bond. Importantly, this reporter could not be hydrolyzed using mycobacterial lysates or other cellular lysates (including macrophages) lacking Dop (51; K. Burns & K.H. Darwin, unpublished observations), which strongly suggests that the deamidation of Pup is a highly specific reaction that is performed only by Dop (51).
After Pup is deamidated, the γ-carboxylate of PupGlu is phosphorylated (and not adenylated like ubiquitin) by PafA using ATP, which produces an ideal leaving group for nucleophilic attack by the amino group of a substrate lysine side chain. The result is a complex of the two proteins covalently linked by an isopeptide bond (Figure 2e) (28). Ligation of Pup to a target substrate requires only the C-terminal 26 residues of Pup (7, 67), a finding that was supported by the crystal structure of the PafA-Pup complex. The normally disordered Pup C terminus folds into a helix upon binding to PafA, positioning the terminal Glu of Pup near ATP for phosphorylation (2).
PafA appears to be the only Pup ligase in bacteria; a pafA mutant of Mycobacterium tuberculosis is completely devoid of pupylated proteins (55). Three independent studies revealed that dozens if not hundreds of proteins are targeted for pupylation in Mycobacterium tuberculosis and Mycobacterium smegmatis (20, 56, 82), and the same appears to be true for Corynebacterium glutamicum (39). Despite this wealth of proteomic information, we still do not understand how proteins are selected for pupylation.
Depupylation
Two observations led to the hypothesis that Pup can be removed from pupylated proteins: Approximately 50% of bacterial species that have both dop and pup encode PupGlu and not PupGln, and partial purification of pupylated proteins (the pupylome) from Mycobacterium tuberculosis results in the rapid depupylation of these proteins in the presence of ATP (5). This led to the discovery that Dop has a second function as a depupylase. Similar to eukaryotic deubiquitylating enzymes (DUBs) that counteract the action of ubiquitin ligases, Dop can remove Pup from a pupylated protein, yielding PupGlu (Figure 2e) (5, 34). Unlike several DUBs, Dop cannot hydrolyze Pup from a linear sequence and is thus specific for removing Pup that is part of an isopeptide linkage with its substrate (5, 35).
An important function for Dop appears to be the recycling of Pup. In a Mycobacterium tuberculosis dop mutant producing PupGlu (to circumvent the need for PupGln deamidation), free or substrate-conjugated Pup is undetectable (9). This phenotype could be reversed by the addition of the proteasome inhibitor epoxomicin, which suggests that Pup-protein conjugates can be degraded by the proteasome without depupylation. Thus, the failure to remove Pup from substrates prior to proteasomal degradation results in the reduction of the overall Pup pool.
Mpa/Pup-Dependent Proteolysis
Regulated proteolysis requires that protease substrates be distinguished from nonsubstrates. In mycobacteria, this is accomplished through an interaction between Pup and Mpa. The N-terminal coiled coils of an Mpa hexamer serve as a template for the C-terminal half of Pup to fold into a helix, thus providing a mechanistic basis for substrate specificity (80). Although there are three coiled coils in an Mpa hexamer, only one Pup binds per hexamer (11, 75, 80). Pupylation of PanB with an N-terminally truncated Pup variant ('Pup) produces 'Pup-PanB that is capable of binding to Mpa but unable to be degraded by the proteasome in vitro (72). Similarly, production of an N-terminally truncated Pup in Mycobacterium smegmatis results in the modification of proteins with a truncated Pup that cannot facilitate protein degradation (7). We presume that, like in eukaryotes (57), the bacterial proteasome requires an unstructured region to initiate degradation and that this region is supplied by the N terminus of Pup. Finally, as in eukaryotes, where the removal of ubiquitin chains is coupled to degradation (79), it appears that Mpa and Dop function synergistically to unfold and depupylate a target protein (5, 72). There is currently no evidence that depupylation is required for substrate degradation. However, the observation that depupylation occurs prior to or concurrent with substrate degradation in Mycobacterium tuberculosis leaves us with an unsolved mystery: How does Pup avoid degradation after translocation through Mpa? Ubiquitin is not usually translocated through the proteasomal ATPase ring in eukaryotes, whereas several lines of evidence suggest Pup must be threaded through Mpa to target substrates for degradation. We speculate that Pup escapes through a gap between the Mpa hexamer and the 20S CP while a doomed substrate proceeds into the proteasome chamber.
ATP-Independent Proteasome Activators
Several in vivo phenotypes suggested that the 20S CP had Mpa/Pup-independent functions in mycobacteria. For one, disruption of mpa in Mycobacterium tuberculosis produces only a mild growth defect, whereas mutagenesis (23, 24) or chemical inhibition (13) of the 20S CP dramatically affects bacterial growth. In eukaryotes, the 20S CP interfaces with several activators in addition to the 19S RP, which allows it to degrade peptides and proteins in a ubiquitin- and ATP-independent manner (reviewed in 70); we thus hypothesized that the Mycobacterium tuberculosis 20S CP might work similarly with a pupylation-independent proteasome activator.
In an effort to identify such an activator, we looked for proteins that interacted with 20S CPs in Mycobacterium tuberculosis and identified PafE, a functional homolog of the eukaryotic 11S ATP-independent proteasome activators (37). Although PafE has no sequence homology to eukaryotic activators, it forms multimeric rings, caps the ends of 20S CPs, and enhances proteasomal degradation of peptides and a model unfolded protein β-casein in an ATP-independent manner. Interestingly, pafE is not found near the Pup-proteasome system (PPS) genes. Furthermore, PafE has C-terminal residues identical to those of Mpa (GQYL). Gly, Tyr, and Leu, but not Gln, are required for PafE and Mpa function (37) and thus constitute an essential motif for proteasomal activation; consistent with this observation, the three required residues are absolutely conserved in both proteins among proteasome-bearing bacteria. Furthermore, this motif serves as a functional homolog of the HbYX motif found in many eukaryotic cofactors.
In another study, Delley et al. (15) also reported the identification of PafE, which they named bacterial proteasome activator (Bpa). This study also showed the requirement of its GQYL motif for the degradation of β-casein and suggested that PafE/Bpa forms hexamers based on size-exclusion chromatography analysis. However, using size-exclusion chromatography coupled with multiangle light scattering, we found that PafE forms dodecamers (37). Because dodecameric proteasomal activators are unprecedented, it will be interesting to see the mechanistic basis of 20S CP activation by PafE.
Proteasomes and Bacterial Physiology
Nitric Oxide Resistance of Mycobacterium tuberculosis
Mycobacterium tuberculosis primarily resides in macrophages, a first line of defense against numerous infectious agents (62). In mice, macrophages produce NO, which can form a variety of reactive nitrogen intermediates leading to the damage of proteins, nucleic acids, and lipids (3). Several studies suggest that NO is an important component of the immune response to Mycobacterium tuberculosis, as mice lacking inducible nitric oxide synthase (iNOS), which catalyzes the production of NO in macrophages, are highly susceptible to infection (47). As discussed earlier, a screen identified pafA and mpa as essential for NO resistance in Mycobacterium tuberculosis (13). Moreover, although these strains are attenuated for growth in animals, the growth defect of an mpa mutant is partially ameliorated by genetic disruption of iNOS in mice (13, 14). This result suggested that part of the effect of Mpa on virulence is due to its contribution to NO resistance, but it must also affect bacterial pathogenesis in other ways.
The first link between the PPS and NO resistance was established through the use of a genetic suppressor screen. Operating with the hypothesis that NO sensitivity was due to the accumulation of a specific PPS substrate, Samanovic et al. (63) mutagenized a Mycobacterium tuberculosis mpa mutant and screened for secondary mutations that suppressed its NO-sensitive phenotype. Remarkably, genetic disruption of Rv1205 restores NO resistance to an mpa mutant. Rv1205 was previously uncharacterized, but a structural homology search indicated that it has high similarity to the plant enzyme LONELY GUY (LOG), which catalyzes the final step in the synthesis of cytokinins, a family of plant hormones. In vitro studies demonstrated that Rv1205 indeed has LOG activity, and it therefore was named Log in Mycobacterium tuberculosis. Supporting the original hypothesis of this study, Samanovic et al. (63) determined that Log is a pupylated proteasome substrate, the accumulation of which sensitizes Mycobacterium tuberculosis to NO. Importantly, Samanovic et al. (63) showed that Mycobacterium tuberculosis synthesizes and secretes several cytokinins. An mpa mutant accumulates Log and therefore accumulates cytokinins, whereas log mutants have dramatically reduced cytokinin production (63). Metabolomic studies of these strains demonstrated that it is likely the breakdown of accumulated cytokinins into aldehydes that kills M. tuberculosis in the presence of NO. Thus, the PPS is required to posttranslationally regulate Log levels to prevent sensitizing mycobacteria to an antimicrobial agent (Figure 3a).
Figure 3.
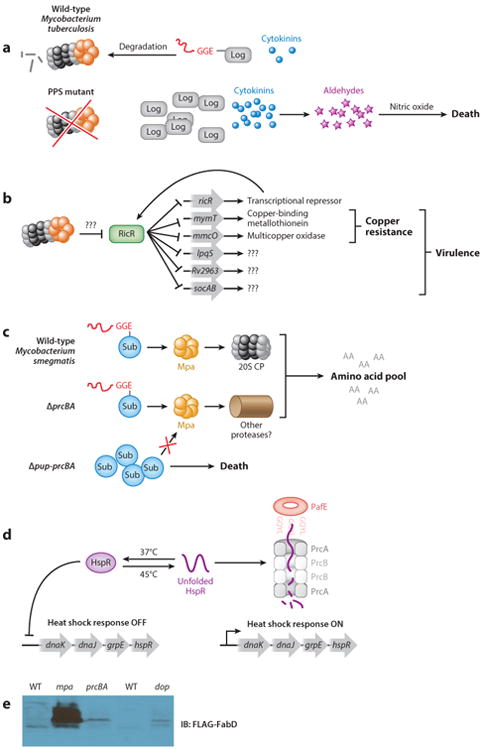
Biological roles of bacterial proteasomes. (a) Accumulation of Log sensitizes PPS-deficient Mycobacterium tuberculosis to nitric oxide. (b) The RicR regulon mediates copper resistance of Mycobacterium tuberculosis. (c) The PPS is required to survive nitrogen starvation in Mycobacterium smegmatis. (d) PafE-dependent proteasomal degradation of HspR contributes to the heat shock response of Mycobacterium tuberculosis. (e) Mpa, Dop, and the 20S CP contribute differently to the degradation of pupylated substrates in Mycobacterium smegmatis. Ectopically produced FLAG-FabD-His6 detected in Mycobacterium smegmatis strains mc2 155 and in isogenic mpa∷kan and ΔprcBA strains, as well as in SMR5 and isogenic Δdop∷kan strains. Panel e is reproduced with permission. Abbreviations: AA, amino acid; CP, core particle; HspR, heat shock protein repressor; IB, immunoblot; PPS, Pup-proteasome system; Sub, substrate; WT, wild-type.
Copper Resistance of Mycobacterium tuberculosis
Proteasomes in eukaryotes are intimately associated with transcriptional regulation (reviewed in 29). It was thus a logical step to determine if proteasomal degradation was important for gene expression in Mycobacterium tuberculosis. Microarray analysis identified a novel copper-responsive regulon that is repressed in both mpa and pafA mutants when compared with wild-type Mycobacterium tuberculosis. One of these genes, ricR (regulated in copper repressor), encodes a transcriptional repressor of copper homeostasis genes (19). Under low copper concentrations, RicR represses transcription by binding to a palindromic element in the promoters of five loci: its own gene, mymT (mycobacterial metallothionein), the putative membrane protein genes Rv2963 and lpqS, mmcO (mycobacterium multi-copper oxidase), and two small open reading frames, socA and socB (small ORF induced by copper A and B), which are encoded together in an operon (Figure 3b) (19). Metallothioneins are small, usually cysteine-rich proteins that bind metals to protect against toxicity (reviewed in 1). Thus, it is not surprising that disruption of mymT results in copper sensitivity in vitro (26). MmcO is a membrane-anchored multi-copper oxidase, and like a mymT mutant, an MmcO-deficient strain is sensitive to copper killing (61, 66). Although the functions of the other RicR-regulated genes are currently unknown, Shi et al. (66) determined that disruption of any single RicR-regulated gene was not sufficient to attenuate bacterial growth in mice. Only upon the construction of a Mycobacterium tuberculosis strain producing a mutant RicR protein that constitutively represses all genes in the RicR regulon can one observe an attenuated phenotype in a mouse infection model (66).
How the proteasome is linked to copper resistance is not known. RicR does not appear to be a substrate of the proteasome under any conditions tested so far. Interestingly, several genes that are associated with iron and zinc metabolism are expressed at increased levels in mpa and pafA-deficient Mycobacterium tuberculosis strains (19), which suggests that metal homeostasis in general may be affected by proteasomal degradation.
Survival Under Nutrient-Limiting Conditions of Mycobacterium smegmatis
In eukaryotes, the proteasome is required for recycling amino acids (74). Thus, studies were undertaken to determine if the proteasome played a similar role in Mycobacterium smegmatis, a nonpathogenic, environmental saprophyte and distant relative of Mycobacterium tuberculosis. Gur and coworkers (17) determined that proteasome-associated genes are induced and that pupylated proteins accumulate during the transition from logarithmic to stationary phase growth; it was thus hypothesized that the Mycobacterium smegmatis proteasome was required for survival under starvation conditions. A Mycobacterium smegmatis Δpup prcBA mutant has a survival defect under nitrogen starvation conditions, which suggests that the proteasome is required for amino acid recycling. However, mitigation of this defect by supplementation with amino acids was not shown, and it is possible that the proteasome is instead required to regulate other pathways that are necessary to survive under these conditions. Intriguingly, complementation of the Δpup prcBA strain with pup, but not with prcBA, restores resistance to the starvation conditions, which suggests that another protease is capable of degrading pupylated proteins (Figure 3c).
Heat Shock Protein Repressor Regulation in Mycobacterium tuberculosis
A pafE strain has a growth defect on solid and in liquid media, and wild-type pafE, but not an allele with a mutated GQYL motif, can complement these phenotypes (37). Importantly, this mutant is attenuated for growth in mice, which indicates a role in tuberculosis pathogenesis. We performed several proteomic studies to identify putative PafE-dependent substrates and identified a set of proteins that are completely distinct from known Mpa-dependent substrates; the most highly accumulated protein was heat shock protein repressor (HspR). We found that PafE promotes the robust proteasomal degradation of HspR in an ATP-independent manner in vitro. Consistent with a role for PafE in promoting the degradation of unfolded proteins and HspR, a pafE mutant is sensitive to heat shock (Figure 3d). Thus, PafE contributes to a distinct proteasomal degradation pathway that has a significant role in Mycobacterium tuberculosis biology.
Unanswered Questions
What Additional Cofactors Are Needed for Robust Pup-Dependent Proteasomal Degradation?
In sharp contrast to the eukaryotic proteasome system, no group has yet recapitulated the robust in vitro degradation of an endogenous, pupylated bacterial proteasome substrate using wild-type proteasomes. Although the 20S CP and Mpa could in theory provide all of the activity necessary for degradation of a pupylated protein, the best attempts at reconstitution using wild-type 20S CPs achieve substrate degradation on a timescale of ∼12–24 h (5, 72). A possibility is that an additional protein is needed to facilitate the entry of a protein or the exit of peptides, as is proposed for the eukaryotic ATP-independent activator PA28α/β (59). Alternatively, the weak interaction between 20S CPs and Mpa may require an adaptor protein to enhance binding for efficient degradation.
How Is the Pup-Proteasome System Regulated?
Hundreds of proteins are potentially pupylated in Mycobacterium species. Remarkably, expression of Mycobacterium tuberculosis pafA and pupGlu in Escherichia coli, which does not have a PPS, results in the pupylation of an almost equal number of proteins in Escherichia coli as are modified in mycobacteria (8). However, although a crystal structure of PafA has now been solved both alone (53) and in complex with Pup (2), these structures provide only clues to the structural constraints of Pup binding and phosphorylation and no information about how PafA chooses one substrate over another.
Levels of Mpa, PafA, Dop, and 20S CP proteins in Mycobacterium tuberculosis do not significantly change between logarithmic and stationary phase growth (N. Bode & K.H. Darwin, unpublished observations), which suggests the regulation of Pup-dependent degradation is not necessarily dependent on protein levels. We currently have data that show Ino1 (inositol-1-phosphate synthase) is degraded only in deep stationary phase in both Mycobacterium tuberculosis and Mycobacterium smegmatis (K.E. Burns & K.H. Darwin, unpublished observations). It is possible that Ino1 is pupylated only during stationary phase or that Pup∼Ino1 is not delivered to the proteasome until stationary phase. Both scenarios could be due to an intrinsic change in Ino1 or due to the presence of one or more growth phase–specific adaptor proteins that promote pupylation or delivery to the proteasome. Determination of whether or not this occurs will be a key advance toward understanding how the PPS is regulated.
How Is PafE-Dependent Degradation Regulated?
Although several putative PafE-dependent substrates have been identified in Mycobacterium tuberculosis, we do not yet know how they are targeted for degradation. We speculate that PafE enhances the degradation of small, somewhat unstructured proteins. This is supported by the observation that β-casein and HspR are robust substrates of PafE-proteasome mediated proteolysis. PafE does not further enhance the degradation of HspR when using an open-gate 20S CP, which suggests that it is unlikely to provide substrate specificity by binding and recruiting individual substrates (37). The discovery of additional substrates may help us to understand how substrates are targeted for PafE-dependent degradation as well as reveal the pathways to which PafE contributes. This may also provide clues to conditions that may regulate its levels or activity.
Another question that remains to be answered: Do Mpa and PafE compete for access to 20S CPs? PafE was shown to block Mpa-mediated protein degradation in vitro (15), whereas Mpa was unable to inhibit PafE-dependent activation of peptide degradation (37), suggesting that PafE binds to the 20S CP with greater affinity than Mpa. However, neither activator binds to 20S CPs with high affinity, unlike their eukaryotic counterparts. This might suggest that the CPs only interact with the relevant activator when absolutely needed. To date, we do not know with any certainty the relative amounts of 20S CPs, PafE, or Mpa/ARC in any bacterial species.
Can Pupylation and the 20S Core Particle Serve Different Functions in Different Species?
Accumulating evidence suggests proteasomes play distinct roles in the biology of Mycobacterium tuberculosis and Mycobacterium smegmatis. This is most evident when comparing the phenotypes of 20S CP–deficient strains. Genetic disruption of prcBA in Mycobacterium smegmatis or Streptomyces does not affect normal axenic growth (31, 38), whereas deletion of the Mycobacterium tuberculosis 20S CP produces a severe growth defect under most conditions tested (23, 24). Interestingly, these phenotypic data correlate with an effect on the degradation of pupylated proteins: Known Pup-proteasome substrates accumulate only mildly in a Mycobacterium smegmatis strain deficient in the 20S CP, whereas deletion of mpa leads to substantial substrate accumulation, which suggests that the stability of pupylated proteins is only partially proteasome dependent in Mycobacterium smegmatis (Figure 3e). In contrast, pupylated proteins accumulate to a similar degree in PafA-, Dop-, Mpa-, and PrcBA-deficient strains of Mycobacterium tuberculosis. Together, these data indicate that Mycobacterium smegmatis may be able to use additional proteases to degrade pupylated proteins, whereas Mycobacterium tuberculosis cannot. Thus, it appears that pupylation and the 20S CP play distinct roles in Mycobacterium tuberculosis and Mycobacterium smegmatis physiology. Going forward, it will be critical to avoid the assumption that proteasome data acquired from one species can necessarily be extended to that of others.
Finally, not all bacteria that encode a pupylation system have proteasomes (39). It is therefore possible that Pup also acts as an inhibitor or facilitator of protein activity or localization. Alternatively, Pup may direct proteins to a different protease for degradation, as hinted at by Mycobacterium smegmatis data from our lab and the Gur group (17); the intrinsically disordered nature of Pup may make it a target for other compartmentalized proteases such as ClpP, HslUV, or Lon. It is curious that Corynebacterium also encodes ARC/Mpa homologs in the absence of proteasomes; this might be explained by the observation that Mpa facilitates depupylation (7, 72), or it might imply that Mpa is capable of interfacing with one or more other proteases in these organisms.
Future Directions
The characterization of Mpa/Pup- and PafE-dependent proteasomal degradation has finally begun to reveal how bacterial 20S CPs that are highly similar to eukaryotic 20S CPs carry out regulated proteolysis in the absence of ubiquitin. We still do not understand how the bacterial systems select particular substrates; elucidation of the basis of PafA substrate selection will undoubtedly be essential to fully appreciate proteasomal degradation in bacteria. In addition, structural studies of how ARC/Mpa and PafE interact with 20S CPs will be critical to reveal the mechanisms of gate opening, which may also contribute to a better understanding of gate activation in eukaryotes and archaea. Finally, much remains to be determined about the biological roles of pupylation, with or without proteasomes. Careful studies of various bacterial species will ultimately reveal what are likely to be numerous functions of pupylation and proteasomal degradation in bacterial physiology.
Acknowledgments
Proteasome research in the Darwin lab is supported by NIH grants HL92774 and AI088075 awarded to K.H.D. and T32 AI007180 and F30 AI110067 awarded to J.B.J. K.H.D. was supported by the Irma T. Hirschl Trust and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Glossary
- Pup
prokaryotic ubiquitin-like protein
- Dop
deamidase of Pup
- PafA,-B, -C, -E
proteasome accessory factor A, B, C, E
- Mpa
mycobacterial proteasome ATPase
- PPS
Pup-proteasome system
Footnotes
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this article.
Literature Cited
- 1.Babula P, Masarik M, Adam V, Eckschlager T, Stiborova M, et al. Mammalian metallothioneins: properties and functions. Metallomics. 2012;4:739–50. doi: 10.1039/c2mt20081c. [DOI] [PubMed] [Google Scholar]
- 2.Barandun J, Delley CL, Ban N, Weber-Ban E. Crystal structure of the complex between prokaryotic ubiquitin-like protein and its ligase PafA. J Am Chem Soc. 2013;135:6794–97. doi: 10.1021/ja4024012. [DOI] [PubMed] [Google Scholar]
- 3.Bowman LA, McLean S, Poole RK, Fukuto JM. The diversity of microbial responses to nitric oxide and agents of nitrosative stress close cousins but not identical twins. Adv Microb Physiol. 2011;59:135–219. doi: 10.1016/B978-0-12-387661-4.00006-9. [DOI] [PubMed] [Google Scholar]
- 4.Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, et al. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol. 1999;1:221–26. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- 5.Burns KE, Cerda-Maira FA, Wang T, Li H, Bishai WR, Darwin KH. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell. 2010;39:821–27. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns KE, McAllister FE, Schwerdtfeger C, Mintseris J, Cerda-Maira F, et al. Mycobacterium tuberculosis prokaryotic ubiquitin-like protein-deconjugating enzyme is an unusual aspartate amidase. J Biol Chem. 2012;287:37522–29. doi: 10.1074/jbc.M112.384784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns KE, Pearce MJ, Darwin KH. Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J Bacteriol. 2010;192:2933–35. doi: 10.1128/JB.01639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cerda-Maira FA, McAllister F, Bode NJ, Burns KE, Gygi SP, Darwin KH. Reconstitution of the Mycobacterium tuberculosis pupylation pathway in Escherichia coli. EMBO Rep. 2011;12:863–70. doi: 10.1038/embor.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:1123–35. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Solomon WC, Kang Y, Cerda-Maira F, Darwin KH, Walters KJ. Prokaryotic ubiquitin-like protein Pup is intrinsically disordered. J Mol Biol. 2009;392:208–17. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlmann B, Kopp F, Kuehn L, Hegerl R, Pfeifer G, Baumeister W. The multicatalytic proteinase (prosome, proteasome): comparison of the eukaryotic and archaebacterial enzyme. Biomed Biochim Acta. 1991;50:465–69. [PubMed] [Google Scholar]
- 13.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–66. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 14.Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55:561–71. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- 15.Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. Bacterial proteasome activator Bpa (Rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLOS ONE. 2014;9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, et al. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell. 2009;34:580–90. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 17.Elharar Y, Roth Z, Hermelin I, Moon A, Peretz G, et al. Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J. 2014;33:1802–14. doi: 10.15252/embj.201387076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eytan E, Ganoth D, Armon T, Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. PNAS. 1989;86:7751–55. doi: 10.1073/pnas.86.20.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, et al. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol Microbiol. 2011;79:133–48. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, et al. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLOS ONE. 2010;5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Festa RA, Pearce MJ, Darwin KH. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J Bacteriol. 2007;189:3044–50. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandotra S, Lebron MB, Ehrt S. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLOS Pathog. 2010;6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med. 2007;13:1515–20. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, et al. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–23. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 26.Gold B, Deng H, Bryk R, Vargas D, Eliezer D, et al. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat Chem Biol. 2008;4:609–16. doi: 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, et al. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–71. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 28.Guth E, Thommen M, Weber-Ban E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated Pup intermediate. J Biol Chem. 2011;286:4412–19. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammond-Martel I, Yu H, Affar el B. Roles of ubiquitin signaling in transcription regulation. Cell Signal. 2012;24:410–21. doi: 10.1016/j.cellsig.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997;272:25200–9. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 31.Hong B, Wang L, Lammertyn E, Geukens N, Van Mellaert L, et al. Inactivation of the 20S proteasome in Streptomyces lividans and its influence on the production of heterologous proteins. Microbiology. 2005;151:3137–45. doi: 10.1099/mic.0.28034-0. [DOI] [PubMed] [Google Scholar]
- 32.Hu G, Lin G, Wang M, Dick L, Xu RM, et al. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59:1417–28. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 33.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–88. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imkamp F, Rosenberger T, Striebel F, Keller PM, Amstutz B, et al. Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol Microbiol. 2010;75:744–54. doi: 10.1111/j.1365-2958.2009.07013.x. [DOI] [PubMed] [Google Scholar]
- 35.Imkamp F, Striebel F, Sutter M, Ozcelik D, Zimmermann N, et al. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 2010;11:791–97. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyer LM, Burroughs AM, Aravind L. Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biol Direct. 2008;3:45. doi: 10.1186/1745-6150-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, et al. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. PNAS. 2015;112:E1763–72. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knipfer N, Shrader TE. Inactivation of the 20S proteasome in Mycobacterium smegmatis. Mol Microbiol. 1997;25:375–83. doi: 10.1046/j.1365-2958.1997.4721837.x. [DOI] [PubMed] [Google Scholar]
- 39.Kuberl A, Franzel B, Eggeling L, Polen T, Wolters DA, Bott M. Pupylated proteins in Corynebac-terium glutamicum revealed by MudPIT analysis. Proteomics. 2014;14:1531–42. doi: 10.1002/pmic.201300531. [DOI] [PubMed] [Google Scholar]
- 40.Kunjappu MJ, Hochstrasser M. Assembly of the 20S proteasome. Biochim Biophys Acta. 2014;1843:2–12. doi: 10.1016/j.bbamcr.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon YD, Nagy I, Adams PD, Baumeister W, Jap BK. Crystal structures of the Rhodococcus proteasome with and without its pro-peptides: implications for the role of the pro-peptide in proteasome assembly. J Mol Biol. 2004;335:233–45. doi: 10.1016/j.jmb.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 42.Li D, Li H, Wang T, Pan H, Lin G, Li H. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 2010;29:2037–47. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao S, Shang Q, Zhang X, Zhang J, Xu C, Tu X. Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem J. 2009;422:207–15. doi: 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- 44.Lin G, Hu G, Tsu C, Kunes YZ, Li H, et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol. 2006;59:1405–16. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 45.Liu CW, Millen L, Roman TB, Xiong H, Gilbert HF, et al. Conformational remodeling of proteasomal substrates by PA700, the 19 S regulatory complex of the 26 S proteasome. J Biol Chem. 2002;277:26815–20. doi: 10.1074/jbc.M201782200. [DOI] [PubMed] [Google Scholar]
- 46.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science. 1995;268:533–39. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 47.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. PNAS. 1997;94:5243–48. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin A, Baker TA, Sauer RT. Pore loops of the AAA + ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–51. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maupin-Furlow JA. Prokaryotic ubiquitin-like protein modification. Annu Rev Microbiol. 2014;68:155–75. doi: 10.1146/annurev-micro-091313-103447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mc Cormack T, Baumeister W, Grenier L, Moomaw C, Plamondon L, et al. Active site-directed inhibitors of Rhodococcus 20 S proteasome. Kinetics and mechanism. J Biol Chem. 1997;272:26103–9. doi: 10.1074/jbc.272.42.26103. [DOI] [PubMed] [Google Scholar]
- 51.Merkx R, Burns KE, Slobbe P, El Oualid F, El Atmioui D, et al. Synthesis and evaluation of a selective fluorogenic Pup derived assay reagent for Dop, a potential drug target in Mycobacterium tuberculosis. ChemBioChem. 2012;13:2056–60. doi: 10.1002/cbic.201200460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nussbaum AK, Dick TP, Keilholz W, Schirle M, Stevanovic S, et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. PNAS. 1998;95:12504–9. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozcelik D, Barandun J, Schmitz N, Sutter M, Guth E, et al. Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat Commun. 2012;3:1014. doi: 10.1038/ncomms2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearce MJ, Arora P, Festa RA, Butler-Wu SM, Gokhale RS, Darwin KH. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25:5423–32. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–7. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poulsen C, Akhter Y, Jeon AH, Schmitt-Ulms G, Meyer HE, et al. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol. 2010;6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–37. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 58.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008;30:360–68. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raule M, Cerruti F, Benaroudj N, Migotti R, Kikuchi J, et al. PA28αβ reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem Biol. 2014;21:470–80. doi: 10.1016/j.chembiol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 60.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–90. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rowland JL, Niederweis M. A multicopper oxidase is required for copper resistance in Mycobacterium tuberculosis. J Bacteriol. 2013;195:3724–33. doi: 10.1128/JB.00546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. 2011;240:252–68. doi: 10.1111/j.1600-065X.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samanovic MI, Tu S, Novak O, Lakshminarayan I, McAllister F, et al. Proteasomal control of cytokinin synthesis protects Mycobacterium tuberculosis against nitric oxide. Mol Cell. 2015;57:984–94. doi: 10.1016/j.molcel.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, et al. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–52. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Proteasome from Thermoplasma acidophilum: a threonine protease. Science. 1995;268:579–82. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 66.Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, et al. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio. 2014;5:e00876. doi: 10.1128/mBio.00876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smirnov D, Dhall A, Sivanesam K, Sharar RJ, Chatterjee C. Fluorescent probes reveal a minimal ligase recognition motif in the prokaryotic ubiquitin-like protein from Mycobacterium tuberculosis. J Am Chem Soc. 2013;135:2887–90. doi: 10.1021/ja311376h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–44. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell. 2005;20:687–98. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 70.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011;41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Streich FC, Jr, Lima CD. Structural and functional insights to ubiquitin-like protein conjugation. Annu Rev Biophys. 2014;43:357–79. doi: 10.1146/annurev-biophys-051013-022958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO J. 2010;29:1262–71. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16:647–51. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 74.Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell. 2012;48:242–53. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sutter M, Striebel F, Damberger FF, Allain FH, Weber-Ban E. A distinct structural region of the prokaryotic ubiquitin-like protein (Pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett. 2009;583:3151–57. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 76.Tamura T, Nagy I, Lupas A, Lottspeich F, Cejka Z, et al. The first characterization of a eubacterial proteasome: the 20S complex of Rhodococcus. Curr Biol. 1995;5:766–74. doi: 10.1016/s0960-9822(95)00153-9. [DOI] [PubMed] [Google Scholar]
- 77.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–45. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, et al. The structure of the mammalian 20S proteasome at 2.75 Å resolution. Structure. 2002;10:609–18. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 79.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–15. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 80.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17:1352–57. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang T, Li H, Lin G, Tang C, Li D, et al. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure. 2009;17:1377–85. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Watrous J, Burns K, Liu WT, Patel A, Hook V, et al. Expansion of the mycobacterial “PUPylome”. Mol Biosyst. 2010;6:376–85. doi: 10.1039/b916104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, et al. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature. 2000;408:115–20. doi: 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- 84.Witt S, Kwon YD, Sharon M, Felderer K, Beuttler M, et al. Proteasome assembly triggers a switch required for active-site maturation. Structure. 2006;14:1179–88. doi: 10.1016/j.str.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 85.Wolf S, Nagy I, Lupas A, Pfeifer G, Cejka Z, et al. Characterization of ARC, a divergent member of the AAA ATPase family from Rhodococcus erythropolis. J Mol Biol. 1998;277:13–25. doi: 10.1006/jmbi.1997.1589. [DOI] [PubMed] [Google Scholar]
- 86.Zhang X, Stoffels K, Wurzbacher S, Schoofs G, Pfeifer G, et al. The N-terminal coiled coil of the Rhodococcus erythropolis ARC AAA ATPase is neither necessary for oligomerization nor nucleotide hydrolysis. J Struct Biol. 2004;146:155–65. doi: 10.1016/j.jsb.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 87.Zuhl F, Seemuller E, Golbik R, Baumeister W. Dissecting the assembly pathway of the 20S proteasome. FEBS Lett. 1997;418:189–94. doi: 10.1016/s0014-5793(97)01370-7. [DOI] [PubMed] [Google Scholar]
- 88.Zuhl F, Tamura T, Dolenc I, Cejka Z, Nagy I, et al. Subunit topology of the Rhodococcus proteasome. FEBS Lett. 1997;400:83–90. doi: 10.1016/s0014-5793(96)01403-2. [DOI] [PubMed] [Google Scholar]