

ABSTRACT
It is well established that influenza A virus (IAV) attachment to and infection of epithelial cells is dependent on sialic acid (SIA) at the cell surface, although the specific receptors that mediate IAV entry have not been defined and multiple receptors may exist. Lec2 Chinese hamster ovary (CHO) cells are SIA deficient and resistant to IAV infection. Here we demonstrate that the expression of the C-type lectin receptor langerin in Lec2 cells (Lec2-Lg) rendered them permissive to IAV infection, as measured by replication of the viral genome, transcription of viral mRNA, and synthesis of viral proteins. Unlike SIA-dependent infection of parental CHO cells, IAV attachment and infection of Lec2-Lg cells was mediated via lectin-mediated recognition of mannose-rich glycans expressed by the viral hemagglutinin glycoprotein. Lec2 cells expressing endocytosis-defective langerin bound IAV efficiently but remained resistant to IAV infection, confirming that internalization via langerin was essential for infectious entry. Langerin-mediated infection of Lec2-Lg cells was pH and dynamin dependent, occurred via clathrin- and caveolin-mediated endocytic pathways, and utilized early (Rab5+) but not late (Rab7+) endosomes. This study is the first to demonstrate that langerin represents an authentic receptor that binds and internalizes IAV to facilitate infection. Moreover, it describes a unique experimental system to probe specific pathways and compartments involved in infectious entry following recognition of IAV by a single cell surface receptor.
IMPORTANCE On the surface of host cells, sialic acid (SIA) functions as the major attachment factor for influenza A viruses (IAV). However, few studies have identified specific transmembrane receptors that bind and internalize IAV to facilitate infection. Here we identify human langerin as a transmembrane glycoprotein that can act as an attachment factor and a bone fide endocytic receptor for IAV infection. Expression of langerin by an SIA-deficient cell line resistant to IAV rendered cells permissive to infection. As langerin represented the sole receptor for IAV infection in this system, we have defined the pathways and compartments involved in infectious entry of IAV into cells following recognition by langerin.
INTRODUCTION
Influenza A viruses (IAV) enter and infect cells in a pH-dependent manner. In humans, epithelial cells lining the respiratory tract are the primary targets of IAV infection and support productive replication, resulting in virus amplification and spread. Seasonal IAV also infect airway macrophages (Mϕ) and dendritic cells (DC), generally resulting in abortive replication, although virulent strains such as highly pathogenic avian influenza can replicate productively in these cells (reviewed in reference 1).
It is generally accepted that binding of the IAV hemagglutinin (HA) to sialic acid (SIA) residues expressed at the cell surface is the first step in initiating infectious entry; however, binding to SIA residues per se does not induce virus internalization. Rather, induction of host cell signaling is required to sort IAV into specific entry routes, and this is likely to be a property of transmembrane receptors that may or may not bear SIA residues. Eierhoff et al. reported that multivalent binding of IAV to cell surface SIA resulted in clustering and activation of receptor tyrosine kinases to form a lipid raft-based signaling platform that stimulated internalization of virions (2). Infectious entry of IAV into epithelial cells can occur via endocytic pathways that are clathrin dependent, caveolin dependent, or independent of both clathrin and caveolin or by macropinocytosis (reviewed in reference 3). The sorting of IAV into specific entry pathways occurs at the plasma membrane and is likely to be determined by a specific adaptor protein(s) that binds to the cytoplasmic tails of IAV receptors and coreceptors, resulting in activation of intracellular signaling proteins and subsequent internalization of virus. Epsin-1, but not eps15, has been identified as a cargo-specific adaptor protein for clathrin-mediated internalization of IAV by BS-C-1 cells (4); however, specific transmembrane receptors linking adaptor proteins such as epsin-1 to virus internalization have not been identified.
In contrast to epithelial cells, significant progress has been made toward identifying transmembrane proteins that can function as attachment and entry receptors for IAV on Mϕ and DC. The macrophage mannose receptor (MMR) and macrophage galactose-type lectin (MGL) have been implicated as receptors for infectious entry of IAV into murine Mϕ (5–7), and human DC-SIGN has been reported to bind to IAV, resulting in enhanced infection of host cells (8–10). MMR, MGL, and DC-SIGN are C-type lectin receptors (CLRs) that express a conserved carbohydrate recognition domain that binds to derivatives of mannose (for MMR and DC-SIGN) or galactose (for MGL), and these sugars are commonly expressed on the surface of a range of pathogens, including viruses (11). The diversity of CLR expression on specific Mϕ and DC subsets in various tissues suggests the potential for different outcomes after CLR-mediated recognition by pathogens (12).
Langerin (CD207) (Lg) is a type II transmembrane CLR comprising an extracellular domain, a transmembrane region, and a cytoplasmic tail that contains a putative proline-rich signaling domain (PRD). Unlike other CLRs, langerin expression in cells is associated with formation of Birbeck granules, rod-shaped pentalamellar structures of the endosomal compartment implicated in the distribution, retention, and recycling of langerin itself (13–15). Langerin recognizes mannose-rich sugars expressed by bacterial and fungal pathogens, such as Mycobacterium spp. (16) and Candida albicans (17), and some viruses. Human immunodeficiency virus type 1(HIV-1) is recognized by a variety of CLRs, including DC-SIGN, MMR, and langerin (18, 19). While initial studies suggested that recognition by langerin resulted in uptake and degradation of HIV-1 in Birbeck granules (20), recent studies show that langerin-mediated uptake can facilitate transfer of the virus from DC to T cells, in a manner analogous to that reported for DC-SIGN (21–25). This occurs via two routes, either via plasma membrane-linked (neutral pH) intracellular compartments or via receptors for enhanced de novo replication in DC, thus facilitating virus transmission. While apparent contradictions have been reported regarding the role of langerin in HIV-1 infection, these likely reflect differences in the cell models used, the isolation methods and maturation status of primary cells, and the titers of inoculum used (20, 24). In addition to interactions with HIV-1, langerin has been reported to bind to herpes simplex virus 2 (26) and langerin-mediated capture of measles virus has been implicated in contributing to the processing and presentation of antigen to virus-specific CD4+ T cells (27).
Human langerin is expressed primarily by Langerhans cells (28), which in the airways tend to be present at a higher frequency in the epidermis and epithelia of the upper respiratory tract than in the lung (27). Murine langerin is also expressed by some DC subsets, including mucosal CD11b− CD103+ DC in the lungs, a population that can migrate to draining lymph nodes following IAV infection (29, 30). Moreover, depletion of langerin-positive DC from mice prior to IAV infection resulted in severe weight loss and delayed viral clearance, consistent with a role for langerin in protective immunity against IAV (30). As langerin-positive cells are present in the airways and have been implicated in protection against IAV, it was of interest to determine if langerin recognized IAV glycoproteins and if it could function as a cell surface receptor for IAV infection.
Our laboratory has demonstrated that a mutant CHO cell line (Lec2 cells) deficient in cell surface expression of SIA (31, 32) is resistant to IAV infection (9). Therefore, putative receptors can be expressed in Lec2 cells to assess their ability to function as attachment and/or entry receptors for IAV. Here we demonstrate that Lec2 cells expressing langerin (Lec2-Lg cells) bind IAV and are permissive for IAV infection. Unlike SIA-dependent infection of parental CHO cells, infection of Lec2-Lg cells is dependent on lectin-mediated recognition of IAV, which can be inhibited by mannan and modulated by the degree of glycosylation of the viral HA. Importantly, Lec2 cells expressing endocytosis-defective langerin mutants bound IAV efficiently but remained resistant to infection, demonstrating that langerin represented the only route of infectious entry into Lec2-Lg cells. Langerin-mediated IAV infection was shown to be pH dependent and occurred via dynamin-dependent routes, including either clathrin- or caveolin-dependent pathways. Finally, we confirmed the importance of Rab5+ early endosomes, but not Rab7+ late endosomes, in IAV infection of Lec2-Lg cells.
MATERIALS AND METHODS
Cell lines.
CHO Pro-5 (CHO; ATCC) and Lec2 CHO (Lec2; ATCC) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in alpha minimal essential medium (Gibco BRL, New York, NY) supplemented with 10% fetal calf serum (JRH Biosciences, Lenexa, KS), 4 mM l-glutamine, 100 IU penicillin (Gibco), 10 μg/ml streptomycin (Gibco), nonessential amino acids (Gibco), and 50 μM beta-mercaptoethanol. HeLa, Hep-2, and CV-1 cells were maintained in Dulbecco's minimal essential medium (Gibco) supplemented as described above.
Viruses.
The IAV strains used in this study were A/PR/8/34 (PR8, H1N1) and BJx109 (H3N2), a high-yielding reassortant of PR8 with A/Beijing/353/89 (Beij/89, H3N2) and expressing the H3N2 surface glycoproteins. Other viruses used in these studies were (i) H3N2 strains A/Memphis/1/71 (Mem/71), A/Beijing/353/89 (Beij/89), and A/New York/55/2004 (NY/04), and (ii) H1N1 strain A/Brazil/11/1978 (Braz/78) and A(H1N1)pdm09 strain A/California/7/2009 (Cal/09). Viruses were grown in 10-day embryonated eggs by standard procedures, and titers of infectious virus were determined by plaque assay on Madin-Darby canine kidney (MDCK) cells (33) and expressed as PFU per ml. Viruses were purified from allantoic fluid by rate zonal sedimentation on 25 to 75% (wt/vol) sucrose gradients as described previously (33). Reassortant IAV were generated by 8-plasmid reverse genetics (RG) as described previously (34) and included viruses which consisted of (i) 6 genes derived from PR8 with the HA and neuraminidase (NA) genes from Beij/89 (RG-PR8-Beij/89 HA NA), (ii) 7 genes derived from PR8 with the HA or NA gene from Beij/89 (RG-PR8-Beij/89 HA and RG-PR8-Beij/89 NA, respectively), (iii) 7 genes derived from PR8 with the HA gene from PR8 containing two mutations introduced by site-directed mutagenesis resulting in the addition of two potential sites of N-linked glycosylation (RG-PR8 + 94/131) (35), or (iv) 8 genes from PR8 (RG-PR8).
Respiratory syncytial virus (RSV) strain A2 (from Rosa Gualano, Department of Pharmacology, The University of Melbourne, Parkville, Australia) was propagated in HEp-2 cells by standard procedures (36). Titers of infectious RSV were determined by titration on CV-1 cells, followed by immunofluorescence staining with monoclonal antibody (MAb) against RSV fusion (F) protein (clone 133-1H; Millipore, MA, USA), and titers are expressed in fluorescent-forming units (FFU) per ml.
Generation of Lec2 cells expressing langerin.
Transfected Lec2 cells were generated using neomycin-resistant plasmids encoding human langerin (Lg). The pcDNA3-langerin plasmid was a kind gift from Stuart Turville (Westmead Millennium Institute for Medical Research, NSW, Australia). An Lg mutant containing a point mutation in its cytoplasmic domain (PREP to IREP) changing proline at position 23 to isoleucine (Lg P23I) and an Lg mutant lacking 30 amino acids from its cytoplasmic domain (ΔLg mutant) were generated by PCR using specific primers (for P23I, forward primer, 5′-AAA CAG AAC ATC TCC CTC TGG ATC CGA GAG CCT-3′, and reverse primer, 5′-AGG CTC TCG GAT CCA GAG GGA GAT GTT CTG TTT-3′; for ΔLg, forward primer, 5′-GGG GGA TCC AAG ATG GGT CCA TCT CTG GTC CCG-3′, and reverse primer, 5′-CCC CTC GAG CTG TCA CGG TTC TGA TGG GAC-3′). PCR-amplified products were purified following agarose gel electrophoresis and ligated into pcDNA3.1/V5-Hi-TOPO. Competent Escherichia coli (DH5α strain) cells were transformed, plasmid DNA was extracted using a miniprep kit (Qiagen), and langerin inserts were confirmed by sequencing.
Lec2 cells were transfected with pcDNA3.1/V5-His-TOPO expression vectors containing either full-length Lg, the P23I mutant, or the ΔLg mutant by using FuGene 6 transfection reagent (Roche Diagnostics, Switzerland) according to the manufacturer's instructions. As controls, CHO and Lec2 cells were transfected with pcDNA3.1/V5-His-TOPO expressing cytoplasmic hen egg ovalbumin (OVA) lacking the sequence for cell surface trafficking, as previously described (9). Stable transfectants expressing full-length Lg (Lec2-Lg), P23I mutant (Lec2-Lg P23I), Lg deletion mutant (Lec2-ΔLg), or cytoplasmic OVA (CHO-ctrl, Lec2-ctrl) were selected in the presence of 1 mg/ml Geneticin (G418; Invitrogen). Transfected cells were screened for cell surface expression of langerin using a phycoerythrin (PE)-labeled MAb specific for human langerin (clone DCGM4; Beckman Coulter). Single cells with high cell surface expression of langerin were isolated using a FACSAria cell sorter (BD Biosciences) and expanded in culture for use in experiments.
Western blotting.
Whole-cell lysates were prepared by adding 1 ml lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% Triton X-100, 1 mM CaCl2, 1 mM MgCl2, and broad-spectrum protease inhibitor cocktail; Roche, Mannheim, Germany) to a confluent TC75 flask for 1 h on ice. Cells were collected and clarified by centrifugation (10,000 × g, 3 min), and the protein concentration was determined by the Bradford assay (Bio-Rad protein dye; Bio-Rad, CA). Lysates (∼10 μg protein) were boiled for 5 min and analyzed by SDS-PAGE under nonreducing conditions using a 12.5% gels, followed by transfer to a polyvinylidene difluoride membrane (Millipore, MA) in Tris-glycine transfer buffer (25 mM Tris containing 192 mM glycine and 10% methanol; pH 8.3).
To detect langerin expression in lysates, membranes were blocked in 5% skim milk and 0.05% (vol/vol) Tween 20, and all subsequent wash and antibody binding steps were performed in Tris-buffered saline (TBS; 0.05 M Tris-HCl, 0.15 M NaCl [ph7.2]) with 5% (wt/vol) bovine serum albumin (BSA) and 0.05% Tween 20. Membranes were incubated for 1 h at room temperature with antilangerin antibody conjugated to biotin (clone eBioL31; eBioscience, CA), and bound antibody was detected using streptavidin-horseradish peroxidase (HRP) (Dako, Denmark) in conjunction with enhanced chemiluminescence (Western Lightning plus ECL; Perkin-Elmer, CT). Blots were developed using a Kodak Image Station 4000 MM digital imaging system, and images were managed using Adobe Photoshop software.
Binding of IAV to cells.
Adherent cell lines were detached from TC75 flasks using 0.75 mM EDTA in phosphate-buffered saline (PBS; 0.05 M Na2HPO4-12H2O, 0.14 M NaCl, pH 7.4) and washed in binding buffer (TBS containing 0.1%, [wt/vol] BSA and 20 mM CaCl2). Cells were incubated with 10 μg/ml of purified BJx109 on ice, followed by a biotinylated MAb that binds the HA glycoprotein of BJx109 (MAb C1/1; from L. E. Brown, Department of Microbiology and Immunology, The University of Melbourne, Parkville, Australia), and then washed and stained with streptavidin-allophycocyanin (BD Biosciences) before analysis using a FACSCalibur (BD Biosciences). To determine if binding was calcium dependent, CaCl2 was omitted from the binding buffer and replaced with 5 mM EDTA.
Virus infection assays.
CHO-ctrl, Lec2-ctrl, Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg cells (5 × 104 cells/250 μl) were seeded into eight-well chamber slides (Lab-Tek, Nunc, Denmark), cultured overnight, and infected with 107 PFU of IAV (corresponding to a multiplicity of infection [MOI] of 80), and the percentage of IAV-infected cells was determined as described previously (5, 6, 9). Briefly, after overnight culture, cell monolayers were washed and incubated with IAV or RSV in serum-free medium for 1 h at 37°C. Cells were then washed and incubated for a further 7 h (IAV) or 18 h (RSV) at 37°C in serum-free medium. Slides were washed with PBS, fixed with 80% (vol/vol) acetone, and stained using MAb MP3.10g2.1C7 (WHO Collaborating Centre for Reference and Research on Influenza, Melbourne, Australia), which is specific for the nucleoprotein (NP) of IAV, or with MAb 133-1H, which is specific for RSV F protein, followed by fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse Ig (Millipore, MA). The percentage of virus-infected cells was determined after costaining with propidium iodide (PI) and counting the total number of cells versus FITC-positive cells under ×100 magnification. A minimum of four random fields were selected for counting, with >200 cells assessed for each sample.
In some experiments, cell monolayers were pretreated with (i) 10 mg/ml mannan in serum-free medium for 30 min at 37°C to block C-type lectin receptors or (ii) 50 mU/ml bacterial sialidase from Vibrio cholerae (Sigma-Aldrich) for 60 min at 37°C to remove cell surface SIA prior to the addition of virus inoculum. In other experiments, ammonium chloride (NH4Cl), bafilomycin A1 (BafA1; Sigma-Aldrich), or dynasore (Dy; Sigma-Aldrich) was added to cell monolayers either with the virus inoculum or at various times after infection, as indicated.
In experiments designed to investigate endocytic uptake pathways, cells were pretreated at 37°C for 30 min with Pitstop2-100 (high-purity clathrin inhibitor; Abcam, United Kingdom) or genistein (Sigma-Aldrich). After 60 min at 37°C, virus inoculum and inhibitors were removed, cell monolayers were washed, and serum-free medium containing 10 mM NH4Cl was added to prevent further virus entry. The ability of chemical inhibitors to block uptake pathways was assessed using confocal microscopy and fluorescence-labeled ligands known to enter cells by clathrin-mediated endocytosis (transferrin [Tf]) (37) or caveolin-mediated endocytosis (cholera toxin subunit B [CTB]) (38), as described previously (39). Cell monolayers were pretreated with Pitstop2-100 or genistein for 30 min at 37°C prior to the addition of 15 μg/ml Tf-Alexa Fluor 594 (Life Technologies, CA) or 50 μg/ml CTB-Alexa Fluor 488 (Life Technologies, CA). After 15 min at 37°C, cells were washed and fixed with 2% paraformaldehyde (PFA), followed by staining with 4′,6-diamidino-2-phenylindole (DAPI) for double-stranded nucleic acid. Images were acquired with a Zeiss LSM710 microscope and processed using Fiji ImageJ software.
qRT-PCR for detection of vRNA and mRNA in IAV-infected cells.
Levels of viral RNA (vRNA) and mRNA in IAV-infected cells were determined using real-time quantitative reverse transcription-PCR (qRT-PCR). Briefly, 5 × 104 cells/250 μl seeded into eight-well chamber slides were cultured overnight and infected with 107 PFU BJx109 for 1 h at 37°C, washed twice, and incubated in serum-free medium. At 2 and 8 h postinfection, RNA was extracted from cells using an RNeasy minikit (Qiagen) and stored at −70°C. Reverse transcription was performed using an Omniscript reverse transcription kit (Qiagen) according to the manufacturer's instructions along with an oligo(dT) primer for mRNA and a primer complementary to the conserved 12 nt of the 3′ end of the vRNA (primer Uni12) for vRNA. Levels of matrix (M) gene vRNA and mRNA were determined via qRT-PCR using TaqMan chemistry. The primers and probes for the IAV M gene were as follows: forward primer, 5′-GAC CRA TCC TGT CAC CTC TGA C-3′; reverse primer, 5′-GGG CAT TYT GGA CAA AKC GTC TAC G-3′; and probe, 5′-FAM-TGC AGT CCT CGC TCA CTG GGC ACG-BHQ1. GreenStar (2×) master mix for real-time PCR was used (Bioneer, Australia) on an MX3005 real-time PCR instrument (Agilent Technologies, CA) using a standard program. vRNA and mRNA copy numbers were calculated by generating a standard curve using serial dilutions of plasmid containing DNA for the IAV M gene.
Antibody-mediated internalization of cell surface langerin.
To assess MAb internalization by confocal microscopy, cells cultured overnight in eight-well chamber slides were preincubated at 4°C for 20 min prior to the addition of a FITC-conjugated mouse antilangerin MAb (clone caa8-28H10; Miltenyi Biotec, Germany). Cells were then incubated at 4°C or 37°C for 30 min, washed and fixed with 2% PFA for 10 min, and then permeabilized with 1% Triton X-100 for 10 min at room temperature. After washing, cells were incubated with rabbit anti-mouse antibody–Alexa Fluor 488 for 30 min at room temperature and double-stranded DNA was stained using DAPI. Images were acquired using a Zeiss LSM710 microscope and processed using Fiji ImageJ software.
MAb internalization was also assessed using flow cytometry. First, purified antilangerin MAb was conjugated to the specific hybridization internalization probe (SHIP) as described previously (40). The SHIP sensor consists of a 5′ fluorescent probe (FIPCy5; Integrated DNA Technologies, IA) and a complementary sequence with a 3′ black hole quencher 2 (BHQ2; Integrated DNA Technologies, IA), which binds to exposed FIP, quenching its fluorescence. Cells were incubated with FIPCy5-conjugated antilangerin MAb at 4°C for 30 min, washed, and then incubated at either 4°C or 37°C for 30 min. After incubation, 1 μM BHQ2 was added for 5 min and then cells were washed and Cy5 expression was detected by flow cytometry using a FACSCanto analyzer (BD Biosciences).
IAV infection of cell lines transfected to express the wild type or dominant negative mutants of Rab5 or Rab7.
Plasmids of monomeric red fluorescent protein (mRFP)-Rab5 (14437), mCherry-Rab5DN (S34N) (35139), DsRed-Rab7 (12661), and DsRed-Rab7DN (T22N) (12662) were obtained from Addgene (Cambridge, MA, USA). CHO-ctrl, Lec2-ctrl, Lec2-Lg, and HeLa cells in 12-well plates were transfected with each plasmid for 24 h using FuGene6 transfection reagent (Roche Diagnostics, Switzerland) according to the manufacturer's instructions. Cells were then washed and infected with IAV in serum-free medium as described above. At 8 h postinfection, cells were detached following incubation with 0.75 mM EDTA in PBS, fixed with 2% PFA for 10 min at room temperature, and permeabilized in 1% Triton X-100 for 10 min at room temperature. Fixed, permeabilized cells were stained for IAV infection using anti-NP (MAb MP3.10g2.1C7), followed by FITC-conjugated goat anti-mouse IgG, as described above. Cells were analyzed for IAV infection (FITC+) or Rab5 expression (mRFP/mCherry+) by flow cytometry.
Statistical analyses.
Graphing and statistical analysis of data were performed using GraphPad Prism (GraphPad Software, San Diego, CA). An unpaired Student t test was used to compare two sets of data. When three or more sets of values were compared, the data were analyzed by one-way analysis of variance (ANOVA; nonparametric), followed by post hoc analysis using Tukey's multiple-comparison test. A P of ≤0.05 was considered significant.
RESULTS
SIA-deficient Lec2 cells expressing langerin are permissive to IAV infection.
SIA-deficient Lec2 cells were transfected with a construct expressing human langerin, and clones showing stable cell surface expression of langerin were selected. Flow cytometry confirmed cell surface expression of langerin on Lec2-Lg cells, compared to CHO-ctrl and Lec2-ctrl cells, respectively (Fig. 1Ai). Similarly, Western blot analysis confirmed the expression of an ∼38-kDa protein in lysates from Lec2-Lg but not CHO-ctrl or Lec2-ctrl cells, consistent with expression of langerin by Lec2-Lg cells (Fig. 1Aii).
FIG 1.

Expression of langerin by SIA-deficient Lec2 cells results in permissiveness to IAV infection. (A) (i) Langerin expression histograms showing cell surface expression of human langerin on Lec2-Lg, Lec2-ctrl, and CHO-ctrl cells. (ii) Western blot confirming expression of langerin in cell lysates from Lec2-Lg cells but not CHO-ctrl and Lec2-ctrl cells. (B and C) Monolayers of CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells were infected with 107 PFU of BJx109 at 37°C for 1 h, washed, and incubated as described in Materials and Methods. (B) At 2 h and 8 h, supernatants were removed and total RNA was extracted from cell monolayers and used for qRT-PCR measurements of vRNA (panel i) and mRNA (panel ii) levels of the IAV M gene. The amount of RNA at 2 h and 8 h postinfection is shown on the left panel, while the fold increase in copy number between 2 h and 8 h postinfection is shown on the right. Data represent the mean (±1 SD) from triplicate samples and are representative of two independent experiments. (C) (i) At 2 h and 8 h postinfection, cells were fixed and stained for expression of viral NP. Representative images show NP (FITC, green) and nucleic acid (PI, red). (ii) Data show the mean percent infection (±1 SD) and are representative of five independent experiments. The detection limit is indicated by a horizontal dotted line. (D) FACS histograms showing HA and NP expression 15 min and 8 h postinfection of Lec2-Lg (black filled histogram), CHO-ctrl (gray filled histogram), Lec2-ctrl (gray unfilled histogram), and uninfected (dotted lines) cells. For data in panels B and C, statistical significance was assessed using Student's t test to compare two sets of data and a one-way ANOVA to compare multiple data sets. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To examine IAV entry and replication in Lec2-Lg cells, we first assessed vRNA and mRNA expression. Cell monolayers were incubated for 1 h at 37°C with IAV strain BJx109, washed, and cultured for an additional 1 h or 7 h before cells were lysed, and expression of vRNA/mRNA for the IAV M gene was determined using qRT-PCR. As shown in Fig. 1Bi, vRNA levels increased between 2 h and 8 h postinfection in CHO-ctrl and Lec2-Lg cells. In contrast, vRNA levels were reduced in Lec2-ctrl cells at 8 h postinfection, possibly indicating degradation of cell-associated virus from the initial inoculum. Levels of mRNA increased significantly between 2 h and 8 h in CHO-ctrl and Lec2-Lg cells, with a modest but significant increase also observed in Lec2-ctrl cells (Fig. 1Bii). Ratios of vRNA at 8 h to that at 2 h in Lec2-Lg cells were significantly higher than in Lec2-ctrl cells but lower than in CHO-ctrl cells (Fig. 1Bi). Similar results were obtained when examining ratios of mRNA at 8 to 2 h (Fig. 1Bii). Together, these data demonstrate that expression of langerin provides a route of entry for IAV into Lec2 cells that results in initiation of replication and transcription of viral mRNA.
Next, we investigated the susceptibility of different cells to infection by IAV, as measured by detection of newly synthesized viral proteins. Cell monolayers were incubated for 1 h at 37°C with BJx109, washed, and cultured for an additional 1 h or 7 h before the cells were fixed and stained for expression of viral NP as described previously (7, 9). Representative images show negligible NP staining in any cell type at 2 h postinfection, compared to strong nuclear and cytoplasmic expression in CHO-ctrl and Lec2-Lg cells at 8 h (Fig. 1Ci). Counting NP+ (IAV-infected) and PI+ (total) cells to determine the percentage of NP+ cells demonstrated that CHO-ctrl cells were highly susceptible whereas SIA-deficient Lec2-ctrl cells were resistant to infection (Fig. 1Ci and ii). As shown in Fig. 1Cii, Lec2-Lg cells were also highly susceptible to infection, with a similar percentage of IAV-infected cells as that in CHO-ctrl cells. At 8 h postinfection, the percentage of Lec2-Lg cells infected with IAV was significantly greater than that of Lec2-ctrl cells (P < 0.001).
Flow cytometry confirmed negligible NP expression in all cell lines at 15 min postinfection compared to high levels at 8 h postinfection (Fig. 1Di). Consistent with immunofluorescence data, a high proportion of NP+ CHO-ctrl (∼70%) and Lec2-Lg cells (∼80%) were detected, compared to a low proportion of NP+ Lec2-ctrl cells (<10%), at 8 h (Fig. 1Di). Similar results were obtained when staining for expression of IAV HA, although we did note a higher expression of HA associated with CHO-ctrl cells at 15 min (Fig. 1Dii); however, cells expressed much higher levels of HA by 8 h. Thus, expression of langerin by SIA-deficient Lec2 cells results in permissiveness for IAV infection as measured by replication of the viral genome, transcription of viral mRNA, and expression of newly synthesized IAV proteins.
We and others have previously reported that CHO cells and CHO cell mutants do not support productive replication of IAV (7, 9, 41). Consistent with this, exposure of MDCK cells, but not CHO-ctrl or Lec2-Lg cells, to a low dose of BJx109 (104 PFU, corresponding to an MOI of 0.1) in the presence of trypsin resulted in increased titers of infectious virus at 72 h compared to 2 h postinfection (data not shown). Similarly, titers of infectious virus in supernatants from CHO-ctrl or Lec2-Lg cells exposed to 107 PFU (MOI = 80) of BJx109 did not increase between 2 and 24 h postinfection (data not shown).
The lectin activity of langerin facilitates SIA-independent infection of Lec2-Lg cells.
Given that langerin is a CLR, we next examined the mechanisms by which langerin expression facilitated IAV infection of Lec2-Lg cells. We used flow cytometry to examine the attachment of BJx109 to the surface of CHO-ctrl, Lec2-ctrl, or Lec2-Lg cells in the presence of 20 mM Ca2+ or 5 mM EDTA. Representative histograms (Fig. 2Ai), as well as the geometrical means from triplicate samples (Fig. 2Aii), are shown. BJx109 bound to CHO-ctrl cells, but not to Lec2-ctrl cells, in the presence of Ca2+ or EDTA, consistent with Ca2+-independent binding of IAV HA to cell surface SIA on CHO-ctrl cells. In contrast, EDTA abrogated the binding of BJx109 to Lec2-Lg cells, consistent with CLR-mediated recognition of IAV by cell surface langerin. While langerin is predicted to express two sites of N-linked glycosylation that may or may not be sialylated (GenBank accession number NM_015717), our findings confirm that langerin expressed on the surface of Lec2-Lg cells retains C-type lectin activity.
FIG 2.
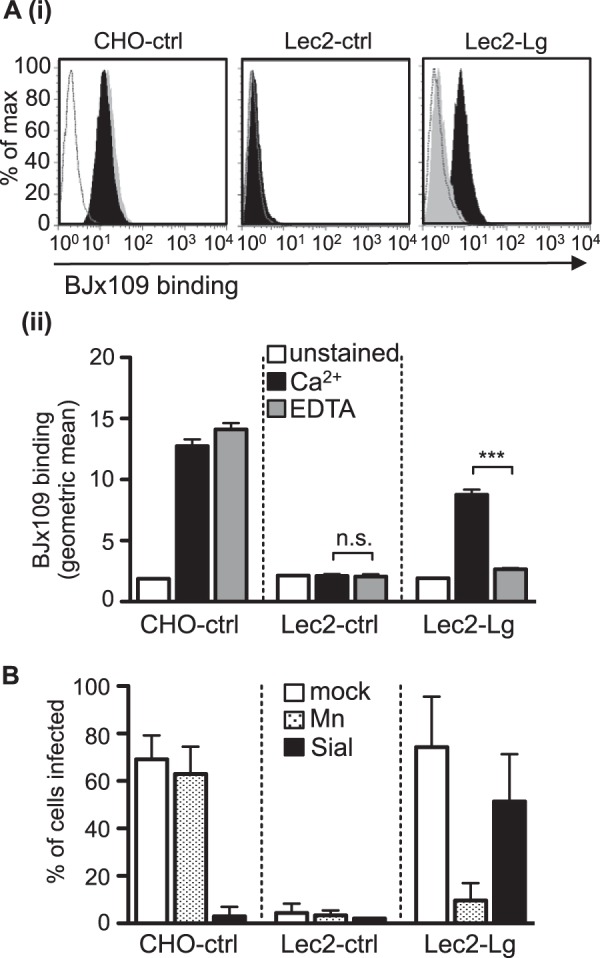
The lectin activity of langerin facilitates IAV infection of Lec2-Lg cells. (A) Binding of 10 μg/ml of BJx109 was determined by flow cytometry. (i) Representative histograms of BJx109 binding to CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells in the presence of 20 mM Ca2+ (black histograms) or 5 mM EDTA (gray histograms). Unstained cells were included as a negative control (white histograms). (ii) The geometrical mean from triplicate samples of BJx109 binding to cells in the presence of 20 mM Ca2+ (black bars) or 5 mM EDTA (gray bars). Unstained cells (white bars) are shown for comparison. (B) Infection of Lec2-Lg cells is blocked by mannan but not by pretreatment of cells with bacterial sialidase. Monolayers of CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells were treated with either 10 mg/ml mannan at 37°C for 30 min (Mn; stippled bars) or with 50 mU/ml of bacterial sialidase from Vibrio cholerae (Sial; black bars) at 37°C for 60 min prior to infection or incubated with serum-free medium alone (mock; white bars) before infection with 107 PFU of BJx109. The percentage of infected cells was determined by using immunofluorescence at 6 to 8 h postinfection. ***, P < 0.001; n.s., not significant.
Next, we investigated the role of cell surface SIA in langerin-mediated enhancement of infection in Lec2-Lg cells. Pretreatment of CHO-ctrl cells with bacterial sialidase completely abrogated susceptibility to BJx109 infection but had only a modest effect on IAV infection of Lec2-Lg cells (Fig. 2B). Moreover, pretreatment of cells with mannan, a complex polymer of mannose residues, prior to virus addition blocked IAV infection of Lec2-Lg cells but did not alter the susceptibility of CHO-ctrl cells to BJx109 (Fig. 2B). Together these data indicate that lectin-mediated IAV recognition of langerin can promote infection of Lec2 cells independently of SIA.
The degree of glycosylation on the head of the IAV HA glycoprotein is a critical determinant of susceptibility to langerin-mediated infection.
IAV strains differ in glycosylation of the HA and NA glycoproteins, in particular in the number sites expressed on the head of the viral HA (reviewed in reference 42). BJx109 is known to express four potential sites of N-linked glycosylation on the head of its HA glycoprotein (43), whereas the PR8 strain is notable for a complete lack of glycosylation on the head of its HA (44). Both BJx109 and PR8 infected CHO-ctrl but not Lec2-ctrl cells to high levels. Although Lec2-Lg cells were more susceptible to PR8 infection than Lec2-ctrl cells, the overall levels of PR8 infection of Lec2-Lg cells were significantly less than BJx109 infection levels of Lec2-Lg cells (Fig. 3A, gray columns, P < 0.001, one-way ANOVA). To refine these experiments and directly address the role of HA and NA as ligands for langerin-mediated infection, we used genetically defined viruses engineered by reverse genetics (RG) to express seven genes from PR8 in conjunction with the HA or the NA gene from Beij/89 (known as RG-PR8-Beij/89 HA and RG-PR8-Beij/89 NA, respectively). In addition, we utilized PR8 derived by RG (RG-PR8) as well as a virus expressing six genes from PR8 with both the HA and NA genes from Beij/89 (RG-PR8-Beij/89 HA/NA). All viruses infected CHO-ctrl but not Lec2-ctrl cells efficiently, and viruses expressing Beij/89 HA (i.e., RG-PR8-Beij/89 HA/NA and RG-PR8-Beij/89 HA) infected Lec2-Lg cells to high levels whereas viruses expressing PR8 HA (i.e., RG-PR8 and RG-PR8-Beij/89 NA) did not (Fig. 3Bi). Thus, these data indicate that expression of the HA protein of Beij/89 is associated with efficient infection of Lec2-Lg cells.
FIG 3.

Glycosylation on the head of HA modulates the efficiency of langerin-mediated infection of Lec2-Lg cells. (A) BJx109 infects Lec2-Lg cells efficiently, but the poorly glycosylated PR8 strain does not. Cells were infected with 107 PFU of BJx109 or PR8, and the percentages of infected cells were determined using immunofluorescence at 6 to 8 h postinfection. (B) Glycosylation of the IAV HA modulates the ability of IAV to infect Lec2-Lg cells. Cells were infected with 107 PFU of RG-PR8-Beij/89 HA/NA, RG-PR8, RG-PR8-Beij/89 HA, and RG-PR8-Beij/89 NA (panel i), or RG-PR8-Beij/89 HA, RG-PR8, and RG-PR8-Beij/89 HA + 94/131 (panel ii), and the percentages of infected cells were determined 6 to 8 h postinfection. (C) Lec2-ctrl or Lec2-Lg cell monolayers were infected with 5 × 106 PFU of H3N2 strains Mem/71, Beij/89, and NY/04 or with H1N1 strains PR8, Braz/78, and Cal/09, and the percentages of infected cells were determined 6 to 8 h postinfection. Numbers in parentheses at the bottom of the panel indicate the number of potential N-linked glycosylation sites present on the head of the viral HA. For panels B and C, data represent the mean percent infection (±1 SD) and are representative of at least two independent experiments. Statistical significance was assessed using one-way ANOVA with Tukey's post hoc analysis (*, P < 0.05; **, P < 0.01; ***, P < 0.001.; n.s., not significant).
Given that viruses expressing PR8 HA were particularly poor in their ability to infect Lec2-Lg cells, we next utilized an RG-engineered virus expressing seven genes from PR8 in conjunction with a modified form of the PR8 HA in which two N-linked glycosylation sites had been introduced using site-directed mutagenesis (RG-PR8 + 94/131) as described previously (45). RG-PR8-Beij/89 HA, RG-PR8, and RG-PR8 + 94/131 all infected CHO-ctrl cells, but not Lec2-ctrl cells, to high levels; however, only RG-PR8-Beij/89 HA infected Lec2-Lg cells efficiently (Fig. 3Bii). Of interest, RG-PR8 + 94/131 (two glycosylation sites on HA head) infected Lec2-Lg cells to significantly higher levels than RG-PR8 (no sites on HA head), and these were significantly lower than the infection levels of RG-PR8-Beij/89 HA (four sites on HA head). Together, these data confirm that the degree of HA glycosylation modulates the efficiency of langerin-mediated infection.
It is well established that “drift” strains of H1 and H3 subtype IAV differ in degree of glycosylation on the head of the viral HA, and this can be an important factor modulating the sensitivity of strains to soluble and cell surface CLR (reviewed in reference 42). H3N2 strains Mem/71, Beij/89, and NY/04 express two, four, and six potential sites, respectively, of N-linked glycosylation on the head of their HA molecules, and while Lec2-ctrl cells were largely resistant to infection by all viruses, infection of Lec2-Lg cells correlated with the degree of glycosylation of the viral HA (i.e., NY/04 > Beij/89 > Mem71 [Fig. 3C]). H1 strains PR8, Cal/09, and Braz/78 express 0, 1, and 4 potential glycosylation sites, respectively, on the head of HA and also infected Lec2-Lg cells in a manner that correlated with the degree of HA glycosylation (Braz/78 > Cal/09 > PR8). Together, these studies highlight the importance of HA glycosylation in determining the efficiency of langerin-mediated infection using virus strains relevant to human health.
The intracellular PRD of langerin is essential for langerin-mediated infection of Lec2 cells by IAV.
The cytoplasmic region of langerin contains a proline-rich domain (PRD) that has been suggested to function as a putative internalization and/or signaling domain (46), although its specific function is currently unknown. Therefore, we generated a deletion mutant of langerin (ΔLg mutant) lacking 30 amino acids from the cytoplasmic tail, including the PRD, as well as a mutated form where proline at position 23 was replaced with an isoleucine, the P23I mutant (Fig. 4A). Stable cell lines were selected, although Lec2-ΔLg cells consistently displayed higher levels of cell surface langerin (Fig. 4B). Next, we examined binding of BJx109 to Lec2 cells expressing wild-type (Lec2-Lg), mutated (Lec2-Lg P23I), or deleted (Lec2-ΔLg) forms of langerin. Representative histograms show that Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg bound BJx109 in a Ca2+-dependent manner (Fig. 4Ci). While Lec2-ΔLg and Lec2-Lg P23I bound significantly higher and lower amounts, respectively, of BJx109 than Lec2-Lg, these data confirm no major defects in IAV binding by mutated or deleted forms of langerin (Fig. 4Cii). Despite this, both Lec2-ΔLg and Lec2-Lg P23I cells were markedly less susceptible to infection by BJx109 (Fig. 4Di). Note that cells expressing mutant forms of langerin were equally susceptible to infection by RSV, a paramyxovirus that infects cells via fusion with the plasma membrane (47, 48), confirming that Lec2-ΔLg and Lec2-Lg P23I were not inherently resistant to viral infection.
FIG 4.

Mutations in the intracellular domain of langerin do not alter binding to IAV but do prevent IAV infection of Lec2-lg cells. (A) Nucleotide and deduced amino acid sequences of the cytoplasmic domain of langerin showing sequences for wild-type langerin (Lg WT), as well as mutated forms of langerin containing either a single mutation at position 23 which changes proline to isoleucine (Lg P23I) or a deletion corresponding to the first 30 amino acids of the intracellular domain (ΔLg). (B) Flow cytometry was used to determine cell surface expression of langerin on Lec2 cells expressing WT (Lec2-Lg, black histogram), P23I mutant (Lec2-Lg P23I, gray histogram), and ΔLg mutant (Lec2-ΔLg, dark gray histogram) forms of langerin. Lec2-ctrl cells were included to confirm the specificity of binding. (C) Binding of 5 μg/ml of purified BJx109 to Lec2 cells expressing mutated forms of langerin was determined by flow cytometry. (i) Representative histograms of BJx109 binding to Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg in the presence of 20 mM Ca2+ (black histograms) or 5 mM EDTA (gray histograms) are shown. Unstained cells were included as a negative control (white histograms). (ii) Geometric means (±1 SD) from triplicate samples of BJx109 incubated with cells in buffer containing 20 mM Ca2+ (black bars) or 5 mM EDTA (hatched bars) are shown. Unstained cells (white bars) were included for comparison. **, P < 0.01; ***, P < 0.001. (D) Cells were infected either with 107 PFU of BJx109 for 8 h and then fixed and stained for expression of IAV NP (panel i) or with 105 FFU of RSV for 18 h and then fixed and stained for expression of RSV F protein (panel ii). Data show the mean percent infection (±1 SD). Data were analyzed by one-way ANOVA with Tukey's post hoc analysis, ***, significantly reduced compared to results for Lec2-Lg (P < 0.001). No significant differences were observed between cells infected with RSV. n.s., not significant.
Lec2-ΔLg cells express endocytosis-defective langerin, whereas Lec2-Lg P23I cells show only a modest impairment in langerin-mediated endocytosis.
We used a langerin-specific MAb to compare the endocytic activities of langerin expressed by Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg cells. When examined by confocal microscopy, the anti-langerin MAb was shown to stain Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg cells but not Lec2-ctrl cells (Fig. 5A). At 4°C, langerin-MAb complexes were detected only at the cell surface. After incubation at 37°C, there was a marked increase in the intensity of intracellular staining in Lec2-Lg and Lec2-Lg P23I cells, consistent with receptor-mediated internalization of langerin-MAb complexes from the cell surface. However, the staining pattern of Lec2-ΔLg cells at 37°C remained very similar to that observed at 4°C, suggesting defective receptor-mediated endocytosis of langerin-anti-langerin MAb complexes.
FIG 5.

Deletion of the intracellular domain of langerin results in an endocytosis-defective mutant, whereas the P23I mutation results in less significant impairment in endocytic activity. Lec2-Lg, Lec2-Lg P23I, Lec2-ΔLg, and Lec2-ctrl cells were incubated with a mouse MAb specific for human langerin or with Alexa Fluor 488 donkey anti-mouse IgG alone (A488 only) on Lec2-Lg cells at 4°C or 37°C for 30 min. (A) After this time, cells were fixed, permeabilized, and stained with Alexa Fluor 488 donkey anti-mouse IgG to amplify the fluorescent signal associated with antilangerin MAb bound to ligand. Representative images show surface and cytoplasmic staining of langerin in a single plane. (B) Reduced endocytic capacity of langerin expressed by Lec2-ΔLg cells was confirmed using a flow cytometry-based assay. Cells were incubated with FIPCy5-labeled antilangerin MAb (LgCy5) for 30 min on ice, washed, and then placed on ice or at 37°C for 30 min. After incubation, cells were washed and surface-bound MAb was quenched using a DNA quencher probe as described in Materials and Methods. (i) Representative histograms show binding of labeled MAb to Lec2-Lg cells after incubation at either 4°C or 37°C, in the presence or absence of quenching. (ii) Data were used to calculate the percentage of antilangerin MAb internalized by each cell line at 37°C. Data show the mean (±1 SD) from triplicate samples and were analyzed by one-way ANOVA with Tukey's post hoc analysis. **, P < 0.01; ***, P < 0.001.
The defect in endocytic capacity caused by the deletion of the Lg cytoplasmic domain (ΔLg) on Lec2 cells was quantified by flow cytometry. In these assays, cells were incubated at 4°C with FIPCy5-conjugated antilangerin MAb, washed, and moved to 37°C to facilitate internalization of cell surface MAb. After 30 min, cells were washed in ice-cold buffer and 100 nM quencher probe was added to remove cell surface-bound MAb; cells were then analyzed by flow cytometry to determine the amount of MAb that had been internalized. Representative histograms in Fig. 5Bi show that FIPCy5-MAb was quenched at 4°C, consistent with cell surface expression of anti-langerin MAb. However, after incubation at 37°C, the geometric means of quenched and unquenched samples were quite similar, consistent with the internalization of FIPCy5-MAb rendering it resistant to effective quenching. Data from triplicate samples were analyzed to determine the percentage of MAb internalized, as described in Materials and Methods. After 30 min at 37°C, Lec2-Lg, Lec2-Lg P23I, and Lec2-ΔLg cells had internalized ∼90%, ∼60%, and ∼20% of MAb, respectively (Fig. 5Bii). Thus, the P23I point mutation conferred a modest, but significant, defect in the ability of langerin to endocytose ligand, whereas a major defect in langerin-mediated endocytosis occurred as a result of deletion of the cytoplasmic domain.
IAV infection of Lec2-Lg cells occurs via a pH-dependent, dynamin-dependent pathway.
IAV is known to enter cells via multiple pathways; however, the identity of specific entry receptors has remained elusive. Moreover, the existence of multiple entry receptors on sialylated cells may contribute to redundant and/or parallel entry pathways in different cell types. In contrast, IAV infection of Lec2-Lg cells is restricted to CLR-mediated entry via langerin, allowing us to investigate this specific pathway of infectious entry without the complications associated with the contribution of unknown (and possibly multiple) receptors.
Irrespective of the particular IAV cell surface receptor and entry pathway, it is well established that IAV infection is pH dependent. Endosomal acidification induces an irreversible conformational change in the viral HA (3), activating its membrane fusion activity and facilitating release of the viral capsid into the cytoplasm (49). Therefore, we incubated Lec2-Lg cells in increasing concentrations of NH4Cl, a weak base that inhibits endosomal acidification (50), and examined IAV infection 6 to 8 h later via immunofluorescence. CHO-ctrl cells were used as an epithelial cell control for SIA-dependent IAV infection. As shown in Fig. 6Ai, addition of ≥5 mM NH4Cl resulted in potent inhibition of IAV infection of Lec2-Lg (black bars) and CHO-ctrl cells (white bars). However, 10 mM NH4Cl had no effect on the ability of RSV to infect CHO-ctrl (white bars) or Lec2-Lg (black bars) cells (Fig. 6Aii), consistent with previous studies which reported pH-independent infection of epithelial cells by RSV (47, 48). Next, we examined the rate of virus entry (leading to IAV infection) by incubating cell monolayers with BJx109 at 4°C, washing them, and then culturing them at 37°C for various times before the addition of 10 mM NH4Cl to prevent further infection. All cells were then fixed at 8 h postinoculation and stained for expression of IAV NP (Fig. 6Aiii). Inoculation of CHO-ctrl (white bars) and Lec2-Lg (black bars) with no NH4Cl added (mock) resulted in 70% infection, whereas addition of NH4Cl immediately after incubation with virus (t = 0 h) reduced infection levels to <2%. Infectious entry of BJx109 into Lec2-Lg cells was significantly reduced at 0.5 and 1 h compared to that into CHO-ctrl cells; however, by 2 h, there were no significant differences between the two cell lines, and addition of NH4Cl after this time did not alter the overall percentage of infected cells (Fig. 6Aiii).
FIG 6.

IAV infection of Lec2-langerin cells occurs via a pH-dependent, dynamin-dependent pathway. (A) Monolayers of CHO-ctrl (white bars) and Lec2-Lg (black bars) cells either were infected with 107 PFU of BJx109 at 37°C for 1 h in the presence of increasing concentrations (mM) of NH4Cl, and after washing, incubated a further 6 to 8 h in the presence of NH4Cl and then fixed and stained for expression of viral NP (panel i), were infected with 105 FFU RSV at 37°C for 1 h in the presence of 10 mM NH4Cl, and after washing, were incubated a further 18 to 20 h in the presence of NH4Cl and then fixed and stained with a MAb specific for RSV F protein (panel ii), or were incubated with 107 PFU of BJx109 at 4°C for 30 min to allow virus binding and then moved to 37°C (panel iii). At various times, supernatants were removed and replaced with medium containing 10 mM NH4Cl to prevent further infection (panel iii). (B and C) Monolayers of CHO-ctrl (white bars) and Lec2-Lg (black bars) cells were infected with 107 PFU of BJx109 or 105 FFU RSV at 37°C for 1 h in the presence of increasing concentrations (nM) of bafilomycin A1 (BafA1) (B) or with increasing concentrations (μM) of dynasore (C). Cells were washed, incubated a further 6 to 8 h (IAV) or 18 to 20 h (RSV) in the presence 10 mM NH4Cl, and then fixed and stained as described in Materials and Methods. All data represent the mean percent infection (±1 SD) and are representative of two or more independent experiments. Data were analyzed by one-way ANOVA with Tukey's post hoc analysis. ***, P < 0.001; n.s., not significant.
Cells were also incubated in the presence of up to 80 nM bafilomycin A1 (BafA1), an inhibitor of the vacuolar-type H(+)-ATPase which also prevents endosomal acidification (51). Inclusion of BafA1 effectively blocked BJx109 infection of CHO-ctrl (Fig. 6Bi, white bars, upper panels) and Lec2-Lg (Fig. 6Bi, black bars, lower panels) cells but had no effect on RSV infection (Fig. 6Bii). Next, we examined the effects of dynasore on IAV infection of CHO-ctrl and Lec2-Lg cells. Dynasore is a small-molecule inhibitor of GTPase dynamin 2, which is essential for endocytic vesicle formation as it pinches the endosome from the plasma membrane to form early endosomes (52). Addition of 25 μM dynasore inhibited IAV infection of CHO-ctrl cells and was even more potent in its ability to block IAV infection of Lec2-Lg cells (Fig. 6Ci). IAV infection of both cell lines was completely blocked by 100 μM dynasore (data not shown), but this concentration of dynasore had no effect on RSV infection of CHO-ctrl and Lec2-Lg cells (Fig. 6Cii). Note that the highest concentrations of NH4Cl, BafA1, and dynasore used did not result in detachment of adherent cells (as determined by counting the total number of PI-positive cells after 8 h of incubation compared to mock-treated controls), confirming that these concentrations were not toxic (data not shown).
Langerin-mediated IAV infection can occur via clathrin- or caveolin-mediated endocytosis.
Multiple pathways of IAV entry have been reported, including both clathrin- and non clathrin-mediated endocytosis and macropinocytosis (reviewed in reference 53). Dissection of IAV entry pathways generally relies on the use of pharmacological inhibitors; however, the specificity of many of these inhibitors for particular entry pathways has been questioned (reviewed in reference 54). As IAV infection was strictly dynamin dependent, we focused on classical inhibitors of clathrin (Pitstop2-100)- and caveolin (genistein)-mediated entry to examine their ability to (i) block entry of known ligands of specific entry pathways such as transferrin (Tf; clathrin-mediated entry) and cholera toxin B (CTB; caveolin-mediated entry), (ii) inhibit IAV infection, and (iii) inhibit infection by RSV, which should occur independently of endocytic uptake.
Confocal microscopy was used to confirm the specificity of each chemical inhibitor. Tf and CTB were all taken up effectively by Lec2-Lg cells incubated in medium alone (Fig. 7A, mock), whereas 5 μM Pitstop2-100 blocked uptake of Tf but was less effective at blocking uptake of CTB (Fig. 7A, Pitstop2-100), consistent with potent inhibition of clathrin-mediated endocytosis and relatively modest effects on caveolin-mediated endocytosis. In contrast, 50 μg/ml genistein blocked uptake of CTB but had less pronounced effects on uptake of Tf (Fig. 7A, genistein), consistent with potent inhibition of caveolin-mediated endocytosis. To examine effects on IAV infection, cell monolayers were incubated with increasing concentrations of each inhibitor for 30 min prior to the addition of virus. After incubation at 37°C for 60 min, cells were washed to remove inhibitor and excess virus and then 10 mM NH4Cl was added to prevent further viral fusion and infection. Cells were then fixed and stained for expression of newly synthesized viral NP at 6 to 8 h postinfection. Incubation with either Pitstop2-100 or genistein inhibited IAV infection of CHO-ctrl cells (Fig. 7B, white bars) to significant levels. A similar pattern was observed using Lec2-Lg (Fig. 7B, black bars), although genistein appeared to be a particularly potent inhibitor of IAV infection. Pitstop2-100 and genistein did not affect the ability of RSV to infect CHO-ctrl or Lec2-Lg cells (Fig. 7B, right panel), nor were they toxic to cells at 8 h postinfection (data not shown).
FIG 7.
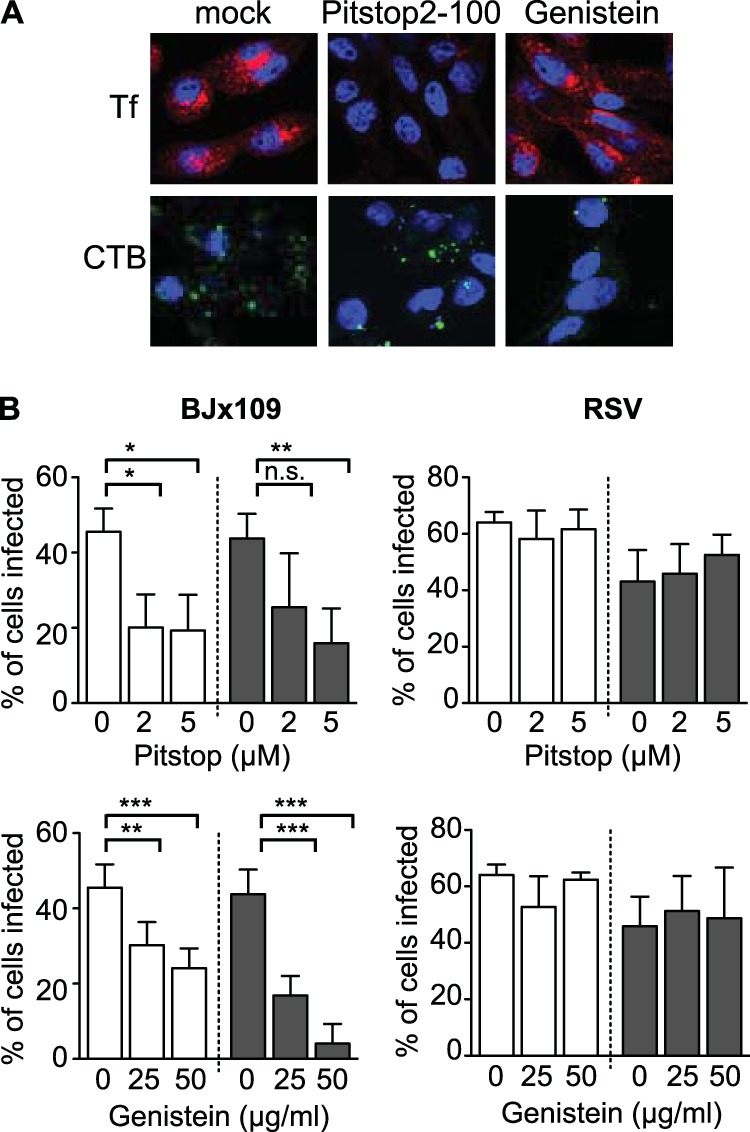
Langerin-mediated IAV infection can occur via multiple entry pathways. (A) Lec2-Lg cells were incubated at 37°C for 30 min in serum-free medium alone (mock) or serum-free medium containing either 5 μM Pistop2-100 or 50 μg/ml genistein. After the addition of 15 μg/ml of Alexa Fluor 594-conjugated transferrin (Tf; clathrin dependent) or 50 μg/ml of Alexa Fluor 488-conjugated cholera toxin B (CTB; caveolin dependent), cells were incubated at 37°C for a further 15 min and then fixed, stained for double-stranded nucleic acid (DAPI), and observed by confocal microscopy. Representative images are shown. (B) Effects of chemical inhibitors on IAV infection of CHO-ctrl and Lec2-Lg cells. Monolayers of CHO-ctrl (white bars) and Lec2-Lg (black bars) cells were incubated in serum-free medium alone (0) or in serum-free medium containing increasing concentrations of Pitstop2-100 or genistein at 37°C for 30 min. Next, 107 PFU BJx109 (left panels) or 105 FFU RSV (right panels) was added (while maintaining the concentration of chemical inhibitors) and incubated at 37°C for 1 h. Cells were then washed, 10 mM NH4Cl was added to prevent further infection, and the cells were cultured for a further 7 h (BJx109) or 18 h (RSV) and then fixed and stained for virus-infected cells. Data show the mean percent infection (±1 SD). Statistical significance was assessed using one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
Rab5, but not Rab7, is implicated in langerin-mediated infection of Lec2 cells by IAV.
IAV exhibits a conventional endocytic uptake pattern and is internalized into Rab5+ early endosomes, which acidify during maturation to reach a suitable pH that facilitates viral fusion and release into the cytoplasm (3). Lec2-Lg cells were transfected with constructs encoding RFP-Rab5, cultured for 24 h, and then incubated with purified virus for 30 min at 4°C or 37°C. After this time, cells were fixed and stained for viral HA. Colocalization between Rab5 and BJx109 HA was observed after incubation at 37°C, but not 4°C, confirming that langerin can deliver IAV to early endosomes in Lec2-Lg cells (data not shown).
Lec2-Lg, CHO-ctrl, and Lec2-ctrl cells were transfected with constructs encoding RFP-Rab5 (to mark Rab5+ early endosomes and to retain functionality) or dominant negative (DN) RFP-Rab5 S34N (to mark Rab5+ early endosomes and to impair functionality), cultured for 24 h, and then infected with BJx109 before quantitation of viral NP at 8 h postinfection. Confocal microscopy confirmed expression of RFP-Rab5 and RFP-Rab5 S34N (Fig. 8A, Rab5) and that CHO-ctrl (Fig. 8Ai) and Lec2-Lg (Fig. 8Aiii) but not Lec2-ctrl (Fig. 8Aii) cells were highly susceptible to infection (Fig. 8A, NP). For CHO-ctrl and Lec2-Lg cells, many of the Rab5+ cells were also NP+; however, it appeared that fewer Rab5 S34N+ cells also stained NP+ (Fig. 8A, merged).
FIG 8.

Langerin-mediated IAV infection involves entry via Rab5+ early endosomes. Cells transfected with expression vectors encoding Rab5 or DN Rab5 S34N were incubated at 37°C for 24 h to allow for protein expression. After incubation, cells were infected with 107 PFU BJx109 at 37°C for 1 h, washed, and incubated a further 6 to 8 h before they were fixed and stained for viral NP. (A) Representative images from confocal microscopy show RFP-labeled Rab5 and Rab5 S34N expression in all cell lines (red, left panels) and newly synthesized viral NP (green, middle panels), as well as a merged image (right panels), including staining for double-stranded nuclei acid using DAPI (blue). White arrows indicate examples of cells showing colocalization of Rab5 and viral NP. Note that cells are present in the middle panel for Lec2-ctrl cells but did not stain for viral NP. (B) Following transfection and IAV infection, cells were detached using 0.75 mM EDTA in PBS, fixed, and stained for viral NP prior to analysis via flow cytometry. Representative dot plots showing RFP-Rab5/RFP-Rab5 S34N expression (vertical axis) and NP expression (horizontal axis) are shown. Gates have been included to identify Rab5/Rab5 S34N+ cells (Q1 and Q4) as well as NP+ cells (Q2 and Q3). Arrows indicate cells with high expression levels of Rab5 or Rab5 S34N. (C) Analysis of transfected and untransfected CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells shows the percentage of IAV-infected cells in the RFP− fraction of cells transfected with constructs encoding Rab5 (white bars) or DN Rab5 S34N+ (black bars) (panel i) and the percentage of IAV-infected cells in the RFP+ fraction of cells transfected with constructs encoding Rab5 (white bars) or DN Rab5 S34N+ (black bars) (panel ii). Data represent the mean (±1 SD) of triplicate samples and are representative of two independent experiments. Statistical significance was assessed using one-way ANOVA with Tukey's post hoc analysis. ***, P < 0.001; n.s., not significant.
Flow cytometry was used to quantitate NP+ cells as well Rab5+ or Rab5 S34N+ cells (Fig. 8B). CHO-ctrl and Lec2-Lg but not Lec2-ctrl cells were susceptible to IAV infection (see Fig. 8B, untransfected BJx109, for representative dot plots, Q2/Q3). Next, we gated on cells expressing Rab5 or Rab5 S34N to investigate the effects of the double-negative (DN) mutant on IAV infection (Fig. 8B, Q1/Q2). Expression of Rab5 (particularly very high levels) had a modest effect on sensitivity to IAV infection; however, high levels of Rab5 S34N had a much more profound inhibitory effect (Fig. 8B, compare sections marked by arrows between Rab5 and Rab5 S34N panels). To quantitate data, we determined the percentages of NP+ cells from Rab5 or Rab5 S34N untransfected and transfected cells. When cultures of cells transfected with Rab5 and Rab5 S34N were compared, no difference was observed in the percentages of NP+ cells in the untransfected cell gate (e.g., see Fig. 8Ci, Rab5− versus Rab5 S34N−). Of all Rab5+ transfected cells, ∼50% of CHO-ctrl and Lec2-Lg cells were also NP+, whereas for DN Rab5 S34N+ transfected cells, significantly less were NP+ (∼20 to 25%) (Fig. 8Cii). Together, these data indicate that IAV entry through Rab5+ early endosomes is important for IAV infection of both CHO-ctrl and Lec2-Lg cells.
Rab5 marks early endosomes, while Rab7 and Rab11 mark late and recycling endosomes, respectively (reviewed in reference 55). Previous studies highlighted the importance of Rab7+ late endosomes in facilitating effective infection of HeLa cells by IAV (56). Therefore, cells were transfected with constructs encoding RFP-labeled Rab7 or DN Rab7 (T22N), with the latter construct designed to impair the function of Rab7+ late endosomes. Confocal microscopy confirmed expression of RFP-Rab7 and RFP-Rab7 T22N (Fig. 9A, Rab7) and susceptibility to IAV infection (Fig. 9A, NP). However, we did not observe major differences in the frequencies of NP+ cells between Rab7+ and Rab7 T22N+ cells (Fig. 9A, merged). Furthermore, flow cytometry confirmed that untransfected cells (Rab7− versus Rab7 T22N−) showed no differences in susceptibility to IAV infection (Fig. 9Ci). Expression of DN Rab7 T22N had a modest but significant effect on the proportion of NP+ cells in CHO-ctrl cells; however, no differences were observed in the proportion of NP+ cells between Rab7+ and Rab7 T22N+ populations of Lec2-Lg cells (Fig. 9Cii). Confirming previous studies (56), we found that both DN Rab5 S34N+ or DN Rab7 T22N+ HeLa cells were significantly less susceptible to IAV infection (data not shown). Together, these data define the importance of Rab5+ early endosomes, but not Rab7+ late endosomes, in langerin-mediated infection of Lec2 cells by IAV.
FIG 9.

Rab7+ late endosomes are not required for langerin-mediated IAV infection. CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells transfected with expression vectors encoding Rab7 or DN Rab7 T22N were incubated at 37°C for 24 h to allow for protein expression. After incubation, cells were infected with 107 PFU BJx109 at 37°C for 1 h, washed, and incubated a further 6 to 8 h before they were fixed and stained for viral NP. (A) Representative images from confocal microscopy show RFP-labeled Rab7 and Rab7 T22N expression in all cell lines (red, left panels), newly synthesized viral NP (green, middle panels), and a merged image (right panels), including staining for double-stranded nuclei acid using DAPI (blue). White arrows indicate examples of cells showing colocalization of Rab7/Rab7 T22N and viral NP. Note that cells are present in the middle panel for Lec2-ctrl cells but did not stain for viral NP. (B) Following transfection and IAV infection, cells were detached using 0.75 mM EDTA in PBS, fixed, and stained for viral NP prior to analysis via flow cytometry. Representative dot plots showing RFP-Rab7/RFP-Rab7 T22N expression (vertical axis) and NP expression (horizontal axis) of CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells are shown. Gates have been included to identify all Rab7/Rab7 T22N+ cells (Q1 and Q4) as well as NP+ cells (Q2 and Q3). Arrows indicate cells with high expression levels of Rab7 or Rab7 T22N. (C) Analysis of transfected and untransfected of CHO-ctrl, Lec2-ctrl, and Lec2-Lg cells shows the percentage of IAV-infected cells in the RFP− fraction of cells transfected with constructs encoding Rab7 (white bars) or DN Rab7 T22N (black bars) (panel i) and the percentage of IAV-infected cells in the RFP+ fraction of cells transfected with constructs encoding Rab7 (white bars) or DN Rab7 T22N (black bars) (panel ii). Data represent the mean (± 1 SD) of triplicate samples and are representative of 2 independent experiments. Statistical significance was assessed using one-way ANOVA with Tukey's post hoc analysis. *, P < 0.05; n.s., not significant.
DISCUSSION
As Lec2 cells are deficient in cell surface SIA (31, 32) and largely resistant to IAV infection (9), they provide a convenient system to assess the ability of putative receptors to promote IAV attachment and/or infectious entry in the absence of multivalent interactions between IAV HA and cell surface SIA. Here we demonstrate that expression of human langerin enhanced the susceptibility of Lec2 cells to IAV entry and infection. Langerin was the sole receptor for infectious entry of IAV into Lec2-Lg cells, as cell surface expression of endocytosis-defective langerin mutants was associated with recognition of IAV, but cells remained resistant to infection (Fig. 3C and D). Our studies are the first to demonstrate that recognition by cell surface langerin can also lead to infectious IAV entry. Langerin functions not only as an attachment factor but also as a bone fide endocytic receptor for IAV delivery to early endosomes, resulting in viral fusion and infection.
Our data provide evidence that in the absence of SIA, langerin represents a specific transmembrane receptor that activates cellular signaling pathways to induce virus uptake, although the particular signaling components involved are yet to be elucidated. The cytoplasmic domain of langerin does not contain the “classical” tyrosine-based internalization motifs such as those expressed by related CLRs, such as DC-SIGN and MGL (reviewed in reference 57). Rather, the PRD (WPREPPP) in the cytoplasmic tail of langerin has been proposed to interact with Src homology 3 (SH3) domains found on adaptor proteins (58), which have been implicated in a diverse array of cellular processes, including vesicular trafficking, cytoskeleton movement, and signaling through receptor tyrosine kinases (59). While little is currently known regarding interactions between langerin and specific adaptor proteins, the PRD of dynamin is known to interact with specific SH3 domain-containing accessory proteins to regulate the recruitment, assembly, and GTPase activity of dynamin during endocytosis (60). Removal of the intracellular domain (ΔLg) or mutation within the langerin PRD (P23I) abrogated langerin-mediated infection of Lec2 cells by IAV; however, the P23I mutation was associated with only a modest, but significant, defect in internalization of langerin-specific MAb. These findings argue that the major effect of this mutation might be to disrupt interactions between the PRD of langerin and SH3 domain-containing accessory proteins downstream of internalization, which could disrupt the GTPase function of Rab5 (61) or interfere with endosomal trafficking and maturation (62).
For epithelial cells, it is well established that clathrin-mediated endocytosis is the primary route of IAV entry, although uptake of IAV can also occur via clathrin-independent pathways (reviewed in reference 3). It has been difficult to dissect IAV entry mechanisms further, as specific transmembrane receptors are not known, multiple receptors may contribute to infection, and the particular receptors used are likely to differ markedly between cell types, as well as between cells derived from distinct species. The selection of a particular entry pathway is often linked to a specific receptor, and some receptors can promote entry via more than one pathway. Langerin-mediated internalization has been reported to occur via clathrin-mediated endocytosis (63) or by caveolin-mediated endocytosis (64), consistent with our findings that infection of Lec2-Lg cells was strictly dynamin dependent but that blocking of either clathrin- or caveolin-mediated endocytic pathways with chemical inhibitors reduced the susceptibility of Lec2-Lg cells to IAV infection.
Live cell imaging, in conjunction with small interfering RNA and/or a range of DN mutants, has been used to identify entry pathways and adaptor molecules involved in IAV uptake by epithelial cells (4, 65); however, many of the adaptor proteins expressed by CHO cells are quite distinct from those expressed by humans and mice (e.g., CHO and human epsin-1 share only ∼50% sequence identity), limiting the utility of such approaches in our SIA-deficient model. The transfection/expression approaches described here allowed for characterization of interactions between langerin and IAV in the absence of additional cell surface interactions with SIA or other IAV receptors. However, future studies examining IAV uptake into primary Langerhans cells from humans or mice would benefit from live-cell imaging analysis to define cellular processes associated with recognition, internalization, signaling, and intracellular trafficking of IAV in Langerhans cells. As experimental culture conditions can modulate entry pathways, including induction of macropinocytosis as a route of infectious entry for IAV into epithelial cells in the presence of fetal calf serum (66), it would also be of interest to examine the pathways utilized for IAV infection of primary Langerhans cells in the presence and absence of serum and/or bronchoalveolar lavage fluids.
The transfection/expression approaches described in this study overcome barriers that have limited identification and characterization of IAV receptors for many years. Namely, Lec2 CHO cells lacking cell surface SIA are largely resistant to IAV infection such that langerin represented the only route of infectious entry into Lec2-Lg cells, allowing us to define the mechanisms underlying enhanced IAV infection (i.e., via generation of endocytosis-defective mutants of langerin), as well as specific pathways and endosomal compartments. However, caution must be exercised when extrapolating data from our experiments to our understanding of virus-CLR interactions in general. For example, parental CHO cells are inherently resistant to IAV infection and we required a high infectious dose (MOI of 80) to achieve 60 to 80% infection, which is much higher than doses required to obtain similar levels of infection in highly susceptible epithelial cell lines such as MDCK cells. Furthermore, we and others have reported that parental CHO cells (and mutant CHO cell lines) do not support productive IAV replication, as infectious progeny are not released (7, 9, 41), although the mechanisms underlying this block in replication are not known. While replication of seasonal IAV in Mϕ and DC is also abortive, different mechanisms may well limit the infectious cycle in such different cell types. Therefore, the studies presented here have relied on the use of a high inoculum dose of IAV to examine a single cycle of virus attachment, entry, and infection.
Birbeck granules are cytoplasmic organelles present in Langerhans cells but can be induced by ectopic expression of langerin in heterologous cells (13, 27). The function of Birbeck granules is still not clear, although most studies suggest an active role in receptor-mediated endocytosis, intracellular trafficking, and/or processing and presentation of antigen (63, 67). In addition to Rab11+ recycling compartments (14), langerin colocalizes with early, but not late, endosomes (13, 14). Consistent with this, langerin-mediated IAV infection of Lec2 cells was inhibited by expression of DN Rab5+ but not DN Rab7+ (Fig. 7 and 8). We did, however, confirm previous findings (56) that functional Rab5+ and Rab7+ endosomes were required for efficient trafficking and subsequent IAV infection of HeLa cells (data not shown). Both the pathway selected for endocytic entry and the endosomal requirements for IAV infection will be modulated by the nature of receptor(s) used to infect distinct cell types. Differences in endosomal pH between cell types may also impact the efficiency of infectious entry of IAV (68). The optimal pH required to trigger IAV fusion (∼5.0 to 6.0) can vary considerably between different IAV strains (69). Thus, the efficiency of langerin-mediated infection could be restricted for some strains, as langerin does not traffic from early to late endosomes, where the pH is further reduced (70). Accordingly, strains that do not fuse effectively with early endosomes may be trafficked on to Rab11+ compartments, including Birbeck granules. The fate of such virions is unclear but may be analogous to HIV-1, where a proportion of internalized virions can be trafficked into Birbeck granules and degraded, likely via the caveolin-1 degradation pathway (20, 64).
An additional level of restriction may also occur at the level of virus recognition, as IAV strains expressing poorly glycosylated HA are not recognized efficiently by the langerin carbohydrate recognition domain (Fig. 2C and D). Of interest, when pandemic IAV strains enter the human population, they tend to express a poorly glycosylated HA but acquire additional glycans as they persist and circulate in the human population (reviewed in reference 42). Thus, the antiviral activities of soluble CLRs, such as mannose-binding lectin (MBL) and surfactant protein D (SP-D), and cell-associated CLRs, such as DC-SIGN, MGL, and langerin, are likely to be limited against emerging pandemic strains but are more likely to contribute to effective innate defense against disease severity following infection with seasonal IAV.
ACKNOWLEDGMENTS
This study was supported by project grant 1027545 from the National Health and Medical Research Council (NHMRC) of Australia. P.C.R., S.L.L., and A.G.B are all recipients of funding from the NHMRC. W.C.N. is a recipient of a NHMRC Dora Lush Biomedical Research Scholarship. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health.
We thank Robert Webster, St. Jude Children's Research Hospital, Memphis, Tennessee, USA, for provision of the plasmid vector used to create the reverse engineered viruses for this study.
Funding Statement
The work was supported by grant 1027545 from the NHMRC, Australia.
REFERENCES
- 1.Short KR, Brooks AG, Reading PC, Londrigan SL. 2012. The fate of influenza A virus after infection of human macrophages and dendritic cells. J Gen Virol 93:2315–2325. doi: 10.1099/vir.0.045021-0. [DOI] [PubMed] [Google Scholar]
- 2.Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. 2010. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog 6:e1001099. doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect 6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C, Zhuang X. 2008. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci U S A 105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reading PC, Miller JL, Anders EM. 2000. Involvement of the mannose receptor in infection of macrophages by influenza virus. J Virol 74:5190–5197. doi: 10.1128/JVI.74.11.5190-5197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Upham JP, Pickett D, Irimura T, Anders EM, Reading PC. 2010. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J Virol 84:3730–3737. doi: 10.1128/JVI.02148-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng WC, Liong S, Tate MD, Irimura T, Denda-Nagai K, Brooks AG, Londrigan SL, Reading PC. 2014. The macrophage galactose-type lectin can function as an attachment and entry receptor for influenza virus. J Virol 88:1659–1672. doi: 10.1128/JVI.02014-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S-F, Huang JC, Lee Y-M, Liu S-J, Chan Y-J, Chau Y-P, Chong P, Chen YMA. 2008. DC-SIGN mediates avian H5N1 influenza virus infection in cis and in trans. Biochem Biophys Res Commun 373:561–566. doi: 10.1016/j.bbrc.2008.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, Reading PC. 2011. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol 85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillaire MLB, Nieuwkoop NJ, Boon ACM, de Mutsert G, Vogelzang-van Trierum SE, Fouchier RAM, Osterhaus ADME, Rimmelzwaan GF. 2013. Binding of DC-SIGN to the hemagglutinin of influenza A viruses supports virus replication in DC-SIGN expressing cells. PLoS One 8:e56164. doi: 10.1371/journal.pone.0056164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drickamer K. 1992. Engineering galactose-binding activity into a C-type mannose-binding protein. Nature 360:183–186. doi: 10.1038/360183a0. [DOI] [PubMed] [Google Scholar]
- 12.Turville SG, Cameron PU, Handley A, Lin G, Pöhlmann S, Doms RW, Cunningham AL. 2002. Diversity of receptors binding HIV on dendritic cell subsets. Nat Immunol 3:975–983. doi: 10.1038/ni841. [DOI] [PubMed] [Google Scholar]
- 13.McDermott R, Bausinger H, Fricker D, Spehner D, Proamer F, Lipsker D, Cazenave J-P, Goud B, de la Salle H, Salamero J, Hanau D. 2004. Reproduction of Langerin/CD207 traffic and Birbeck granule formation in a human cell line model. J Investig Dermatol 123:72–77. doi: 10.1111/j.0022-202X.2004.22728.x. [DOI] [PubMed] [Google Scholar]
- 14.Uzan-Gafsou S, Bausinger H, Proamer F, Monier S, Lipsker D, Cazenave J-P, Goud B, de la Salle H, Hanau D, Salamero J. 2007. Rab11A controls the biogenesis of Birbeck granules by regulating Langerin recycling and stability. Mol Biol Cell 18:3169–3179. doi: 10.1091/mbc.E06-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thepaut M, Valladeau J, Nurisso A, Kahn R, Arnou B, Vivès C, Saeland S, Ebel C, Monnier C, Dezutter-Dambuyant C, Imberty A, Fieschi F. 2009. Structural studies of langerin and Birbeck granule: a macromolecular organization model. Biochemistry 48:2684–2698. doi: 10.1021/bi802151w. [DOI] [PubMed] [Google Scholar]
- 16.Kim HJ, Brennan PJ, Heaslip D, Udey MC, Modlin RL, Belisle JT. 2015. Carbohydrate-dependent binding of langerin to SodC, a cell wall glycoprotein of Mycobacterium leprae. J Bacteriol 197:615–625. doi: 10.1128/JB.02080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Jong MAWP, Vriend LEM, Theelen B, Taylor ME, Fluitsma D, Boekhout T, Geijtenbeek TBH. 2010. C-type lectin Langerin is a β-glucan receptor on human Langerhans cells that recognizes opportunistic and pathogenic fungi. Mol Immunol 47:1216–1225. doi: 10.1016/j.molimm.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turville SG, Arthos J, Donald KM, Lynch G, Naif H, Clark G, Hart D, Cunningham AL. 2001. HIV gp120 receptors on human dendritic cells. Blood 98:2482–2488. doi: 10.1182/blood.V98.8.2482. [DOI] [PubMed] [Google Scholar]
- 19.Harman AN, Bye CR, Nasr N, Sandgren KJ, Kim M, Mercier SK, Botting RA, Lewin SR, Cunningham AL, Cameron PU. 2013. Identification of lineage relationships and novel markers of blood and skin human dendritic cells. J Immunol 190:66–79. doi: 10.4049/jimmunol.1200779. [DOI] [PubMed] [Google Scholar]
- 20.de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong MAWP, de Gruijl T, Piguet V, van Kooyk Y, Geijtenbeek TBH. 2007. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med 13:367–371. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 21.Turville SG, Santos JJ, Frank I, Cameron PU, Wilkinson J, Miranda-Saksena M, Dable J, Stössel H, Romani N, Piatak M, Lifson JD, Pope M, Cunningham AL. 2004. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood 103:2170–2179. doi: 10.1182/blood-2003-09-3129. [DOI] [PubMed] [Google Scholar]
- 22.Smed-Sorensen A, Lore K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz AL, Koup RA. 2005. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol 79:8861–8869. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fahrbach KM, Barry SM, Ayehunie S, Lamore S, Klausner M, Hope TJ. 2007. Activated CD34-derived Langerhans cells mediate transinfection with human immunodeficiency virus. J Virol 81:6858–6868. doi: 10.1128/JVI.02472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawamura T, Koyanagi Y, Nakamura Y, Ogawa Y, Yamashita A, Iwamoto T, Ito M, Blauvelt A, Shimada S. 2008. Significant virus replication in Langerhans cells following application of HIV to abraded skin: relevance to occupational transmission of HIV. J Immunol 180:3297–3304. doi: 10.4049/jimmunol.180.5.3297. [DOI] [PubMed] [Google Scholar]
- 25.Nasr N, Lai J, Botting RA, Mercier SK, Harman AN, Kim M, Turville S, Center RJ, Domagala T, Gorry PR, Olbourne N, Cunningham AL. 2014. Inhibition of two temporal phases of HIV-1 transfer from primary Langerhans cells to T cells: the role of langerin. J Immunol 193:2554–2564. doi: 10.4049/jimmunol.1400630. [DOI] [PubMed] [Google Scholar]
- 26.de Jong MAWP, de Witte L, Taylor ME, Geijtenbeek TBH. 2010. Herpes simplex virus type 2 enhances HIV-1 susceptibility by affecting Langerhans cell function. J Immunol 185:1633–1641. doi: 10.4049/jimmunol.0904137. [DOI] [PubMed] [Google Scholar]
- 27.van der Vlist M, de Witte L, de Vries RD, Litjens M, de Jong MAWP, Fluitsma D, de Swart RL, Geijtenbeek TBH. 2011. Human Langerhans cells capture measles virus through Langerin and present viral antigens to CD4+ T cells but are incapable of cross-presentation. Eur J Immunol 41:2619–2631. doi: 10.1002/eji.201041305. [DOI] [PubMed] [Google Scholar]
- 28.Van Pottelberge GR, Bracke KR, Demedts IK, De Rijck K, Reinartz SM, van Drunen CM, Verleden GM, Vermassen FE, Joos GF, Brusselle GG. 2010. Selective accumulation of Langerhans-type dendritic cells in small airways of patients with COPD. Respir Res 11:35. doi: 10.1186/1465-9921-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung S-SJ, Fu SM, Rose CE, Gaskin F, Ju S-T, Beaty SR. 2006. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol 176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 30.GeurtsvanKessel CH, Willart MAM, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus ADME, Rimmelzwaan GF, Lambrecht BN. 2008. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J Exp Med 205:1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanley P, Sudo T, Carver JP. 1980. Differential involvement of cell surface sialic acid residues in wheat germ agglutinin binding to parental and wheat germ agglutinin-resistant Chinese hamster ovary cells. J Cell Biol 85:60–69. doi: 10.1083/jcb.85.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.North SJ, Huang H-H, Sundaram S, Jang-Lee J, Etienne AT, Trollope A, Chalabi S, Dell A, Stanley P, Haslam SM. 2010. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J Biol Chem 285:5759–5775. doi: 10.1074/jbc.M109.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders EM, Hartley CA, Jackson DC. 1990. Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc Natl Acad Sci U S A 87:4485–4489. doi: 10.1073/pnas.87.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tate MD, Brooks AG, Reading PC. 2011. Specific sites of N-linked glycosylation on the hemagglutinin of H1N1 subtype influenza A virus determine sensitivity to inhibitors of the innate immune system and virulence in mice. J Immunol 187:1884–1894. doi: 10.4049/jimmunol.1100295. [DOI] [PubMed] [Google Scholar]
- 36.Ghildyal R, Hartley C, Varrasso A, Meanger J, Voelker DR, Anders EM, Mills J. 1999. Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J Infect Dis 180:2009–2013. doi: 10.1086/315134. [DOI] [PubMed] [Google Scholar]
- 37.Rappoport JZ, Simon SM. 2003. Real-time analysis of clathrin-mediated endocytosis during cell migration. J Cell Sci 116:847–855. doi: 10.1242/jcs.00289. [DOI] [PubMed] [Google Scholar]
- 38.Pang H, Le PU, Nabi IR. 2004. Ganglioside GM1 levels are a determinant of the extent of caveolae/raft-dependent endocytosis of cholera toxin to the Golgi apparatus. J Cell Sci 117:1421–1430. doi: 10.1242/jcs.01009. [DOI] [PubMed] [Google Scholar]
- 39.Heikkila O, Susi P, Tevaluoto T, Harma H, Marjomaki V, Hyypia T, Kiljunen S. 2010. Internalization of coxsackievirus A9 is mediated by 2-microglobulin, dynamin, and Arf6 but not by caveolin-1 or clathrin. J Virol 84:3666–3681. doi: 10.1128/JVI.01340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, Johnston APR. 2013. A programmable sensor to probe the internalization of proteins and nanoparticles in live cells. Angew Chem Int Ed Engl 52:5744–5748. doi: 10.1002/anie.201301243. [DOI] [PubMed] [Google Scholar]
- 41.Kumari K, Gulati S, Smith DF, Gulati U, Cummings RD, Air GM. 2007. Receptor binding specificity of recent human H3N2 influenza viruses. Virology J 4:42. doi: 10.1186/1743-422X-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tate M, Job E, Deng Y-M, Gunalan V, Maurer-Stroh S, Reading P. 2014. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses 6:1294–1316. doi: 10.3390/v6031294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reading PC, Morey LS, Crouch EC, Anders EM. 1997. Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J Virol 71:8204–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 45.Tate MD, Brooks AG, Reading PC. 2011. Receptor specificity of the influenza virus hemagglutinin modulates sensitivity to soluble collectins of the innate immune system and virulence in mice. Virology 413:128–138. doi: 10.1016/j.virol.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 46.van der Vlist M, Geijtenbeek TBH. 2010. Langerin functions as an antiviral receptor on Langerhans cells. Immunol Cell Biol 88:410–415. doi: 10.1038/icb.2010.32. [DOI] [PubMed] [Google Scholar]
- 47.Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ, Karpilow JM, Davey RA. 2007. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J Virol 81:7786–7800. doi: 10.1128/JVI.02780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309. doi: 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo M. 2011. Influenza virus entry, p 201–221. In Advances in experimental medicine and biology. Springer US, Boston, MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohkuma S, Poole B. 1978. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A 75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pérez L, Carrasco L. 1994. Involvement of the vacuolar H(+)-ATPase in animal virus entry. J Gen Virol 75:2595–2606. doi: 10.1099/0022-1317-75-10-2595. [DOI] [PubMed] [Google Scholar]
- 52.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Sieczkarski SB, Whittaker GR. 2002. Dissecting virus entry via endocytosis. J Gen Virol 83:1535–1545. doi: 10.1099/0022-1317-83-7-1535. [DOI] [PubMed] [Google Scholar]
- 54.Ivanov AI. 2008. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol Biol 440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- 55.Rodman JS, Wandinger-Ness A. 2000. Rab GTPases coordinate endocytosis. J Cell Sci 113:183–192. [DOI] [PubMed] [Google Scholar]
- 56.Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 57.Sancho D, Reis e Sousa C. 2012. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol 30:491–529. doi: 10.1146/annurev-immunol-031210-101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12:71–81. doi: 10.1016/S1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 59.Cohen GB, Ren R, Baltimore D. 1995. Modular binding domains in signal transduction proteins. Cell 80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 60.Sundborger AC, Hinshaw JE. 2014. Regulating dynamin dynamics during endocytosis. F1000Prime Rep 6:85. doi: 10.12703/P6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinu L, Santiago-Walker A, Qi H, Chou MM. 2002. Endocytosis of epidermal growth factor receptor regulated by Grb2-mediated recruitment of the Rab5 GTPase-activating protein RN-tre. J Biol Chem 277:50996–51002. doi: 10.1074/jbc.M204869200. [DOI] [PubMed] [Google Scholar]
- 62.Raychaudhuri M, Roy K, Das S, Mukhopadhyay D. 2012. The N-terminal SH3 domain of Grb2 is required for endosomal localization of AβPP. J Alzheimers Dis 32:479–493. doi: 10.3233/JAD-2012-120388. [DOI] [PubMed] [Google Scholar]
- 63.Mc Dermott R, Ziylan U, Spehner D, Bausinger H, Lipsker D, Mommaas M, Cazenave J-P, Raposo G, Goud B, de la Salle H, Salamero J, Hanau D. 2002. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Mol Biol Cell 13:317–335. doi: 10.1091/mbc.01-06-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Berg LM, Ribeiro CM, Zijlstra-Willems EM, de Witte L, Fluitsma D, Tigchelaar W, Everts V, Geijtenbeek TBH. 2014. Caveolin-1 mediated uptake via langerin restricts HIV-1 infection in human Langerhans cells. Retrovirology 11:123. doi: 10.1186/s12977-014-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rust MJ, Lakadamyali M, Zhang F, Zhuang X. 2004. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11:567–573. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jiménez V, Scholte F, García-Sastre A, Rottier PJM, de Haan CAM. 2011. Dissection of the influenza A virus endocytic routes reveals Macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bartosik J. 1992. Cytomembrane-derived Birbeck granules transport horseradish peroxidase to the endosomal compartment in the human Langerhans cells. J Investig Dermatol 99:53–58. doi: 10.1111/1523-1747.ep12611845. [DOI] [PubMed] [Google Scholar]
- 68.Murakami S, Horimoto T, Ito M, Takano R, Katsura H, Shimojima M, Kawaoka Y. 2012. Enhanced growth of influenza vaccine seed viruses in Vero cells mediated by broadening the optimal pH range for virus membrane fusion. J Virol 86:1405–1410. doi: 10.1128/JVI.06009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 70.Piper RC, Luzio JP. 2001. Late endosomes: sorting and partitioning in multivesicular bodies. Traffic 2:612–621. doi: 10.1034/j.1600-0854.2001.20904.x. [DOI] [PubMed] [Google Scholar]