

ABSTRACT
Anti-HIV CD8 T cells included in therapeutic treatments will need to target epitopes that do not accumulate escape mutations. Identifying the epitopes that do not accumulate variants but retain immunogenicity depends on both host major histocompatibility complex (MHC) genetics and the likelihood for an epitope to tolerate variation. We previously found that immune escape during acute SIV infection is conditional; the accumulation of mutations in T cell epitopes is limited, and the rate of accumulation depends on the number of epitopes being targeted. We have now tested the hypothesis that conditional immune escape extends into chronic SIV infection and that epitopes with a preserved wild-type sequence have the potential to elicit epitope-specific CD8 T cells. We deep sequenced simian immunodeficiency virus (SIV) from Mauritian cynomolgus macaques (MCMs) that were homozygous and heterozygous for the M3 MHC haplotype and had been infected with SIV for about 1 year. When interrogating variation within individual epitopes restricted by M3 MHC alleles, we found three categories of epitopes, which we called categories A, B, and C. Category B epitopes readily accumulated variants in M3-homozygous MCMs, but this was less common in M3-heterozygous MCMs. We then determined that chronic CD8 T cells specific for these epitopes were more likely preserved in the M3-heterozygous MCMs than M3-homozygous MCMs. We provide evidence that epitopes known to escape from chronic CD8 T cell responses in animals that are homozygous for a set of MHC alleles are preserved and retain immunogenicity in a host that is heterozygous for the same MHC alleles.
IMPORTANCE Anti-HIV CD8 T cells that are part of therapeutic treatments will need to target epitopes that do not accumulate escape mutations. Defining these epitope sequences is a necessary precursor to designing approaches that enhance the functionality of CD8 T cells with the potential to control virus replication during chronic infection or after reactivation of latent virus. Using MHC-homozygous and -heterozygous Mauritian cynomolgus macaques, we have now obtained evidence that epitopes known to escape from chronic CD8 T cell responses in animals that are MHC homozygous are preserved and retain immunogenicity in a host that is heterozygous for the same MHC alleles. Importantly, our findings support the conditional immune escape hypothesis, such that the potential to present a greater number of CD8 T cell epitopes within a single animal can delay immune escape in targeted epitopes. As a result, certain epitope sequences can retain immunogenicity into chronic infection.
INTRODUCTION
Conventional cytotoxic CD8 T cells play an essential role in the control of human immunodeficiency virus (HIV)/simian immunodeficiency virus (SIV) replication. These potent immune cells recognize major histocompatibility complex (MHC) class I molecules presenting peptides derived from HIV or SIV on infected cells. Once detected, epitope-specific CD8 T cells secrete cytokines and enzymes, such as granzyme B, to destroy the infected target. Detection of infected targets depends on the existence of both the presentation of an immunogenic viral peptide and an effector epitope-specific CD8 T cell. During the course of infection, HIV/SIV accumulates mutations in many, but not all, targeted epitopes (1, 2). These mutations can prevent peptide presentation or disrupt recognition by the TCR of epitope-specific CD8 T cells. In both cases, these mutations help HIV/SIV evade CD8 T cells and, consequently, allow viruses to replicate more efficiently.
Long-term preservation of immunogenic T cell epitopes is highly relevant to ongoing HIV cure and vaccine research. Some have suggested that a functional cure for HIV should activate CD8 T cells targeting epitopes that remain immunogenic in the host (3). Others have proposed that a successful HIV vaccine will need to elicit CD8 T cells targeting regions that do not accumulate escape mutations (4–7). Ultimately, long-term maintenance of CD8 T cell responses is important for virus control. Generally, CD8 T cells will not target epitopes that have accumulated escape mutations. As a result, retaining original, immunogenic epitope sequences in HIV/SIV offers the potential to maintain long-term, effective CD8 T cells.
From a genome-wide association study of HIV elite controllers, there is support for the idea that host MHC genetics can play a key role in the development of effective antiviral T cell immunity (8). The presence of a single MHC allele, however, does not guarantee virus control, suggesting that other factors are required. Knowing that a single MHC allele is present is sufficient to predict whether specific epitopes will be targeted by CD8 T cells, but this information is not sufficient to predict when each of the targeted epitopes will accumulate escape mutations in a given host.
The differential rate of accumulation of escape mutations among animals that share a single MHC allele has been shown during acute SIV infection of MHC-homozygous and -heterozygous Mauritian cynomolgus macaques (MCMs). We found that the accumulation of variants in a single epitope targeted by a CD8 T cell response shared by multiple animals was delayed when there existed additional acute-phase epitope-specific CD8 T cell responses in an animal (9). Subsequent sequence changes that accumulate in the second wave of targeted epitopes need to be compatible with these initial adaptive mutations. In the previous study, we proposed that the SIV genome could tolerate a limited amount of simultaneous variation during acute infection, but we did not address whether increasing the number of targeted epitopes would affect the likelihood of accumulating point mutations in epitopes targeted during chronic infection when the virus population has further diversified. In the current study, we tested the hypothesis that there is similar conditional immune escape during chronic HIV/SIV infection, a time when the CD8 T cell response is thought to be less functional and newly emerging mutations need to be compatible within an SIV genome that has undergone further sequence evolution (10).
To test this hypothesis, circulating virus populations were deep sequenced from MCMs that were homozygous and heterozygous for the M3 MHC haplotype and infected with clonal SIV. We determined whether increasing the number of potential epitopes presented to CD8 T cells would blunt the accumulation of variants in epitope sequences restricted by M3 MHC molecules. If so, then there remain potentially immunogenic epitopes in MHC-heterozygous individuals that could be targeted by CD8 T cells during chronic infection and could be important in long term virus control.
MATERIALS AND METHODS
Animals.
All animals were used in previous studies (9, 11, 12). They were purchased from Charles River Laboratories or Bioculture Group, and cared for by the Wisconsin National Primate Research Center (WNPRC) according to protocols approved by the University of Wisconsin Graduate School Animal Care and Use Committee. We used samples from nine animals that were homozygous for the M3 MHC genotype, three animals that were heterozygous for the M1 and M3 MHC genotypes, and two animals that were heterozygous for the M5 and M3 MHC genotypes (Table 1). The time points used for deep sequencing are also indicated in Table 1.
TABLE 1.
Metrics of genome-wide sequences collected for virus populations isolated after infection with SIVmac239a
| Animal | Wk postinfection | No. of paired reads that mapped | Avg coverage | Viral load (copies/ml) | MHC genotype |
|---|---|---|---|---|---|
| CY0163 | 95 | 1,986,662 | 27,830 | 2,375,000 | M3/M3 |
| CY0164 | 51 | 1,772,494 | 25,487 | 3,190,000 | M3/M3 |
| CY0166 | 156 | 1,131,991 | 13,803 | 15,449,965 | M3/M3 |
| CY0332 | 61 | 277,492 | 2,789 | 183,000 | M3/M3 |
| CY0333 | 52 | 239,513 | 2,422 | 9,183,333 | M3/M3 |
| CY0334 | 59 | 294,169 | 2,887 | 261,333 | M3/M3 |
| CY0335 | 86 | 230,096 | 2,322 | 639,100 | M3/M3 |
| CY0336 | 70 | 348,279 | 3,063 | 1,834,800 | M3/M3 |
| CY0337 | 73 | 381,424 | 3,293 | 3,806,000 | M3/M3 |
| CY0328 | 57 | 291,338 | 2,894 | 1,043,555 | M1/M3 |
| CY0329 | 57 | 237,608 | 2,324 | 3,952,222 | M1/M3 |
| CY0331 | 82 | 2,005,862 | 26,124 | 963,600 | M1/M3 |
| CY0353 | 64 | 363,532 | 3,190 | 24,420 | M3/M5 |
| CY0354 | 72 | 441,630 | 3,815 | 2,508 | M3/M5 |
The total number of paired-end sequencing reads for each virus population is listed. The average coverage of total A, C, T, and G nucleotides at each position from 1,300 to 10,200 was quantified.
Deep sequencing SIV.
Genome-wide deep sequencing of replicating virus populations was performed as previously described (13). Briefly, viral RNA was isolated from plasma using the MinElute virus spin kit (Qiagen). Viral cDNA was generated and amplified using the Superscript III one-step reverse transcription-PCR (RT-PCR) system with high-fidelity Platinum Taq (Invitrogen) in four overlapping amplicons spanning the entire SIV coding sequence. The MinElute gel extraction kit (Qiagen) was used to purify PCR products, which were then quantified using the Quant-IT double-stranded DNA (dsDNA) HS assay kit (Invitrogen). Libraries were generated from 1 ng of pooled amplicons and then tagged with barcodes using the Nextera XT kit (Illumina). The tagged libraries were quantified with the Quant-IT dsDNA HS assay kit, and the quality of the library preparation was assessed with an Agilent Bioanalyzer. Libraries were pooled and sequenced on an Illumina MiSeq using either 2-by-250 or 2-by-300 sequencing kits.
Generation of sequence alignments.
FASTQ reads were imported into Geneious version 7.1 (Biomatters, Ltd.). Reads were quality trimmed, paired, and aligned with the SIVmac239 reference (GenBank no. M33262). The frequency of variants present at greater than 1% at each position with at least 20× coverage was determined using the SNP caller in Geneious. The variation at each position and the impact on the amino acid sequence was then exported as a CSV file and used to calculate mean numbers of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN).
Calculation of mean dS and dN at each site.
For each within-host virus population, SNPGenie was used to analyze SNP call data to determine dS and dN in comparison with the inoculum sequence (14). These were estimated by an adaptation of Nei and Gojobori's (15) method to pooled next-generation sequence data (16, 17). We estimated dS and dN separately for 14 CD8 T cell epitopes and for the remainders (excluding the epitopes) of each of the seven coding regions including one or more of the epitopes analyzed. All reported P values are two tailed.
Calculation of variant amino acid epitope sequences.
To determine the frequency of each amino acid sequence, we first annotated the reference genome in Geneious with the 14 CD8 T cell epitopes. The nucleotide sequences spanning each epitope were then extracted from the sequence alignments. Sequences spanning the entire length of epitope were selected. These reads were translated, and the number of times each amino acid variant was present was calculated using the “Find duplicates” feature in Geneious. The list of amino acid sequences and the number of times each was detected were exported from Geneious, and a custom Python script was used to convert it to a table of sequences. Microsoft Excel was used to quantify the frequency of each amino acid sequence within the replicating virus population.
IFN-γ ELISPOT analysis.
Cryopreserved peripheral blood mononuclear cells (PBMC) that had been collected during chronic infection were thawed and then subjected immediately to a gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assay, as previously described (18) and according to the manufacturer's protocol. For dates when the PBMC were originally cryopreserved, see Table 9. Briefly, a precoated monkey IFN-γ ELISpotPLUS plate (Mabtech, Mariemont, OH) was blocked, and peptides corresponding to each of the wild-type epitope sequences were added to each well at a final concentration of 1 μM. Peptides were tested in duplicate. Concanavalin A was used as a positive control at 10 μM. Wells were then imaged with an AID ELISPOT reader. Samples were considered positive if the number of spot-forming cells (SFCs) per 106 PBMC was greater than the average of four unstimulated samples plus two times the standard deviation of the unstimulated samples, or 100 SFCs, whichever was greater.
TABLE 9.
Time points of cryopreservation of cells used in IFN-γ ELISPOT assays (Table 8)
| Animal | Wk postinfectiona |
|---|---|
| CY0163 | 95 |
| CY0164 | 51 |
| CY0166 | 156 |
| CY0332 | 61 |
| CY0333 | 52 |
| CY0334 | 41 |
| CY0335 | 86 |
| CY0336 | 36 |
| CY0337 | 37 |
| CY0328 | 57 |
| CY0329 | 57 |
| CY0331 | 32 |
| CY0353 | 50 |
| CY0354 | 72 |
Time postinfection when PBMC were cryopreserved for the indicated animals that were used as part of IFN-γ ELISPOT assays, whose results are shown in Table 8.
RESULTS
Deep sequencing of SIV detects variants within CD8 T cell epitopes previously found to be restricted by M3 MHC molecules in MCMs.
The catalog of potential epitopes presented by MHC class I alleles in MCMs that are MHC identical and infected with clonal SIV can be defined (9, 12, 19). Fourteen SIV epitopes that are restricted by MHC class I molecules expressed by the M3 MHC class I haplotype and the CD8 T cell responses targeting these epitopes have been identified in M3+ MCMs (9, 12, 18, 20–22). Some of these 14 epitopes accumulate variants during acute SIV infection, some accumulate variants during chronic infection, and some rarely accumulate variants.
To characterize the spectrum and frequency of variants that accumulate in these epitopes by about 1 year postinfection, we deep sequenced virus populations isolated from plasma present in nine M3-homozygous MCMs during chronic SIV infection. By sequencing viruses replicating in this MHC-identical population of animals, viral sequence variation attributed to T cell escape was limited to epitopes restricted by M3 MHC alleles.
Viral RNA was isolated from animals whose viral loads ranged from 103 to 107 copies/ml (Table 1) (9, 11). Samples were sequenced using the Illumina MiSeq platform as described in Materials and Methods. Ranges of 230,096 to 2,005,862 paired-end reads were mapped to SIVmac239 (GenBank no. M33262) for each virus population. The average coverage at each nucleotide position for each genome ranged from 2,322 to 27,830 (Table 1). Variants greater than 1% were called throughout the coding sequences using the SNP caller in Geneious 7.1.
Categorizing variant accumulation in 14 SIV epitopes based on the value of dN.
Evidence for positive selection within CD8 T cell epitopes can be obtained by calculating dN (nonsynonymous substitutions per nonsynonymous site) and dS (synonymous substitutions per synonymous site) within and outside the targeted epitopes. Using the nucleotide frequency at each position, we calculated the mean dN and mean dS values (Tables 2 and 3) relative to an SIVmac239 inoculum that was sequenced in a similar manner. We chose a dN value of 0.0168 to distinguish epitopes that do accumulate mutations from those that do not accumulate mutations because it is the maximum dN value for the nonepitope regions of the seven SIV open reading frames evaluated in this study (Table 3). This suggests that SIV epitopes with a mean dN value less than 0.0168 are unlikely to accumulate variants under positive selection in M3-homozygous animals. Using this cutoff, we found that 10 epitopes (Gag146–154HL9, Gag386–394GW9, Pol592–599QP8, Tat42–49QA8, ARF130–40QL11, Env338–346RF9, Env620–628TL9, Rev59–68SP10, Nef103–111RM9, and Nef196–203HW8) had a mean dN value greater than 0.0168, whereas four epitopes (Gag28–37KA10, Gag221–229PR9, Gag459–467TV9, and Tat42–49CF9) had a mean dN value less than 0.0168.
TABLE 2.
Numbers of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN) in comparisons of epitopes with the inoculum sequence
| Category | Epitope | Mean ± SE for host typea |
|||||
|---|---|---|---|---|---|---|---|
| Homozygous (n = 9) |
Heterozygous (n = 5) |
All (n = 14) |
|||||
| dS | dN | dS | dN | dS | dN | ||
| A | Gag386–394GW9 | 0.0053 ± 0.0030 | 0.0495 ± 0.0021*** | 0.0015 ± 0.0010 | 0.0464 ± 0.0084*** | 0.0040 ± 0.0020 | 0.0484 ± 0.0014*** |
| Pol592–599QP8 | 0.0045 ± 0.0032 | 0.0493 ± 0.0023*** | 0.0015 ± 0.0015 | 0.0516 ± 0.0008*** | 0.0034 ± 0.0021 | 0.0501 ± 0.0015*** | |
| ARF130–40Q11 | 0.0000 ± 0.0000 | 0.0502 ± 0.0037*** | 0.0000 ± 0.0000 | 0.0457 ± 0.0031*** | 0.0000 ± 0.0000 | 0.0486 ± 0.0026*** | |
| Rev59–68SP10 | 0.0098 ± 0.0047 | 0.0856 ± 0.0050*** | 0.0174 ± 0.0149 | 0.0826 ± 0.0068* | 0.0125 ± 0.0059 | 0.0845 ± 0.0039*** | |
| Nef103–111RM9 | 0.0016 ± 0.0012 | 0.0992 ± 0131*** | 0.0037 ± 0.0025 | 0.0987 ± 0171** | 0.0023 ± 0.0011 | 0.0990 ± 0100*** | |
| Nef196–203HW8 | 0.0041 ± 0.0029 | 0.0725 ± 0.0055*** | 0.0030 ± 0.0030 | 0.0604 ± 0.0033*** | 0.0037 ± 0.0021 | 0.0682 ± 0.0040*** | |
| B | Gag146–154HL9 | 0.0261 ± 0.0114 | 0.0230 ± 0.0067 | 0.0048 ± 0.0021 | 0.0098 ± 0.0074 | 0.0185 ± 0.0078 | 0.0183 ± 0.0052 |
| Tat42–49QA8 | 0.0132 ± 0.0088 | 0.0365 ± 0.0070 | 0.0006 ± 0.0006 | 0.0120 ± 0.0076b | 0.0087 ± 0.0058 | 0.0278 ± 0.0060 | |
| Env338–346RF9 | 0.0130 ± 0.0047 | 0.0342 ± 0.0064* | 0.0014 ± 0.0014 | 0.0059 ± 0.0037c | 0.0088 ± 0.0033 | 0.0241 ± 0.0056* | |
| Env620–628TL9 | 0.0020 ± 0.0017 | 0.0186 ± 0.0079 | 0.0040 ± 0.0027 | 0.0190 ± 0.0114 | 0.0026 ± 0.0014 | 0.0187 ± 0.0063* | |
| C | Gag28–37KA10 | 0.0095 ± 0.0031 | 0.0044 ± 0.0030 | 0.0015 ± 0.0011 | 0.0035 ± 0.0023 | 0.0067 ± 0.0031 | 0.0040 ± 0.0021 |
| Gag221–229PR9 | 0.0027 ± 0.0012 | 0.0050 ± 0.0028 | 0.0020 ± 0.0015 | 0.0000 ± 0.0000 | 0.0025 ± 0.0009 | 0.0032 ± 0.0019 | |
| Gag459–467TV9 | 0.0124 ± 0.0087 | 0.0018 ± 0.0013 | 0.0043 ± 0.0016 | 0.0000 ± 0.0000* | 0.0095 ± 0.0056 | 0.0012 ± 0.0008 | |
| Tat59–67CF9 | 0.0000 ± 0.0000 | 0.0012 ± 0.0012 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0008 ± 0.0007 | |
Paired t tests (2 tailed) of the hypothesis that dS is equal to dN were done: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Values for epitopes where no nonsynonymous differences were observed in heterozygous hosts are in bold.
A two-sample t test of the hypothesis that dN for heterozygous hosts equals that for homozygous hosts was done: P < 0.05.
A two-sample t test of the hypothesis that dN for heterozygous hosts equals that for homozygous hosts was done: P < 0.01.
TABLE 3.
Numbers of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN) in comparisons of the remainders of coding regions (excluding epitopes) with the inoculum sequence
| Coding region | Mean ± SE for host typea |
|||||
|---|---|---|---|---|---|---|
| Homozygous (n = 9) |
Heterozygous (n = 5) |
All (n = 14) |
||||
| dS | dN | dS | dN | dS | dN | |
| Gag | 0.0104 ± 0.0027 | 0.0055 ± 0.0005 | 0.0053 ± 0.0011 | 0.0047 ± 0.0001 | 0.0086 ± 0.0018 | 0.0052 ± 0.0004 |
| Pol | 0.0080 ± 0.0012 | 0.0018 ± 0.0002*** | 0.0090 ± 0.0010 | 0.0018 ± 0.0001** | 0.0084 ± 0.0009 | 0.0018 ± 0.0001*** |
| Tat | 0.0215 ± 0.0021 | 0.0038 ± 0.0006*** | 0.0188 ± 0.0013 | 0.0048 ± 0.0008** | 0.0206 ± 0.0014 | 0.0042 ± 0.0005*** |
| ARF1 | 0.0144 ± 0.0040 | 0.0168 ± 0.0020 | 0.0255 ± 0.0006 | 0.0117 ± 0.0048 | 0.0184 ± 0.0029 | 0.0150 ± 0.0021 |
| Env | 0.0178 ± 0.0019 | 0.0107 ± 0.0010*** | 0.0184 ± 0.0022 | 0.0093 ± 0.0006* | 0.0180 ± 0.0014 | 0.0102 ± 0.0007*** |
| Rev | 0.0057 ± 0.0014 | 0.0159 ± 0.0014** | 0.0049 ± 0.0011 | 0.0168 ± 0.0012** | 0.0055 ± 0.0009 | 0.0162 ± 0.0010*** |
| Nef | 0.0048 ± 0.0012 | 0.0074 ± 0.0011* | 0.0066 ± 0.0015 | 0.0071 ± 0.0014 | 0.0054 ± 0.0010 | 0.0073 ± 0.0008* |
Paired t tests (two tailed) of the hypothesis that dS is equal to dN were done: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further subdivide the 14 epitopes by the frequency of amino acid variants, we quantified amino acid variation within each epitope of each animal. The frequencies of amino acid sequences that were variant in each animal are shown in Tables 4, 5, and 6. We divided the 10 epitopes with a mean dN of >0.0168 into two groups. We found that the frequency of variant epitope sequences was greater than 95% for the epitopes Gag386–394GW9, ARF130–40QL11, Rev59–68SP10, Nef103–111RM9, and Nef196–203HW8 in all nine virus populations and for Pol592–599QP8 in eight of nine virus populations (Table 4). We classified these epitopes as category A. In contrast, we found that the frequency of variation in the other four epitopes (category B) (Table 5) ranged from 1 to 98% among all 9 virus populations. For the four epitopes with a mean dN of <0.0168 (Gag28–37KA10, Gag221–229PR9, Gag459–467TV9, and Tat42–49CF9), variant sequences were detected at a frequency of 10% or more in, at most, three of the nine virus populations (category C) (Table 6).
TABLE 4.
Percentage of category A epitopes with amino acid variation in M3-homozygous and -heterozygous MCMs
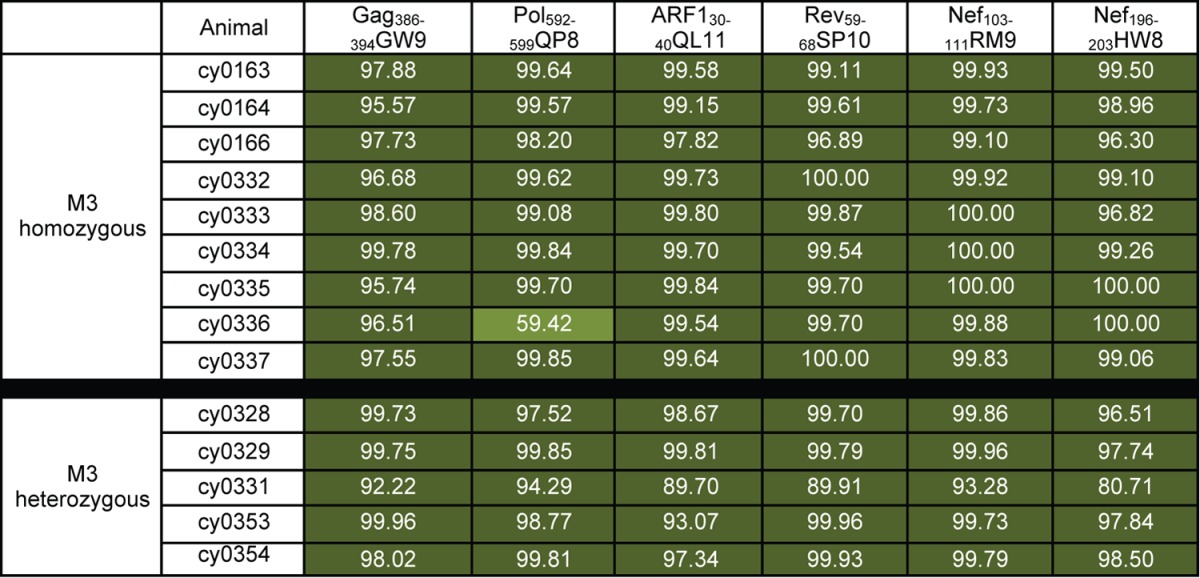
Amino acid variation was calculated as described in Materials and Methods. Values are percentages of variant sequences. Dark green, >75% variant; medium green, 50% to 75% variant.
TABLE 5.
Percentage of category B epitopes with amino acid variation in M3-homozygous and -heterozygous MCMs
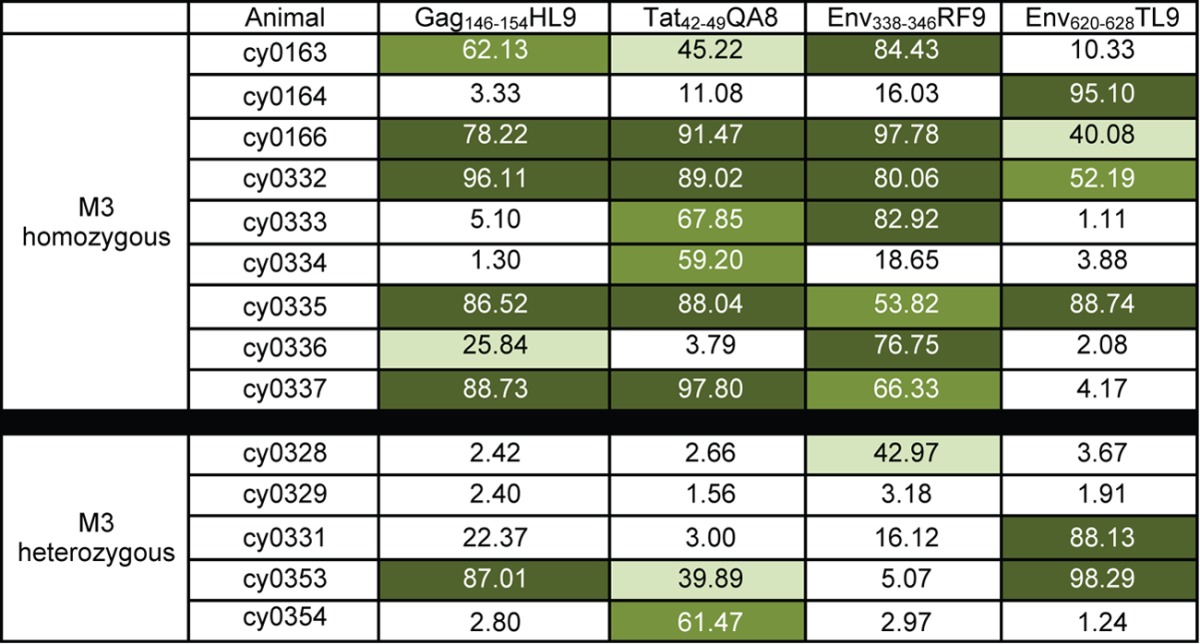
Amino acid variation was calculated as described in Materials and Methods. Values are percentages of variant sequences. Dark green, >75% variant; medium green, 50% to 75% variant; light green, 25% to 50%; white, <25% variant.
TABLE 6.
Percentage of category C epitopes with amino acid variation in M3-homozygous and -heterozygous MCMs
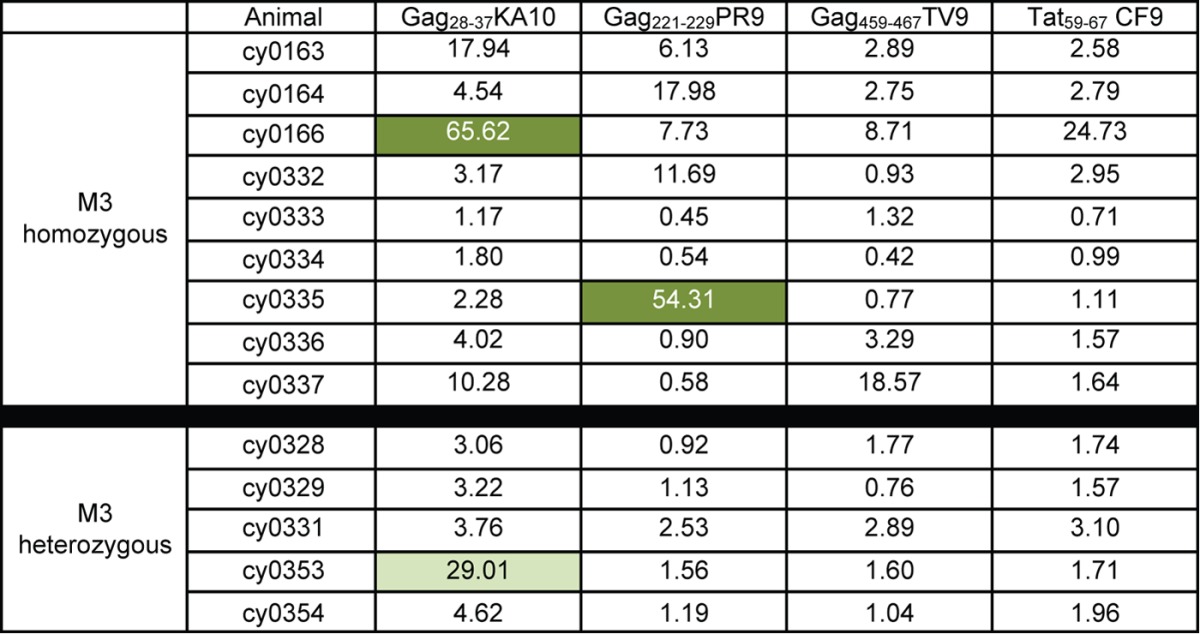
Amino acid variation was calculated as described in Materials and Methods. Values are percentages of variant sequences. Medium green, 50% to 75% variant; light green, 25% to 50%; white, <25% variant.
Preservation of category B T cell epitopes in MHC-heterozygous MCMs.
With the recognition that animals that are heterozygous for M3 MHC alleles have the capacity to present the same epitopes as their M3 MHC-homozygous counterparts, it is logical to hypothesize that CD8 T cells in M3-heterozygous animals select for variants in the same epitopes targeted in M3-homozygous animals. We tested this hypothesis by deep sequencing virus populations from M3-heterozygous animals at approximately 1 to 1.5 years postinfection (Table 1). We calculated the mean value of dN in all 14 epitopes and found that for the category A epitopes, it remained greater than 0.0168 in the M3-heterozygous animals. In contrast, the dN value for the category B epitopes was less than 0.0168 for three of the four epitopes, thus resembling the value for category C epitopes (Table 2). We further compared the frequency of category B epitopes with variant amino acids in the homozygous and heterozygous animals. We found that only 30% of these epitopes (6 of the 20 sequences examined) had variation in >25% of the amino acid sequences in the heterozygous animals, whereas 67% of the category B epitopes (24 of the 36 sequences examined) had variation in >25% of the amino acid sequences in the homozygotes. The samples of the M3 heterozygotes used for sequencing ranged from 57 to 82 weeks postinfection, whereas the samples of the M3 homozygotes ranged from 51 to 95 weeks postinfection, with one sample taken at 156 days postinfection (Table 1). There is thus no overt difference in the time of infection that would have led to the difference in variant accumulation. Together, these data suggest that the category B epitopes are more likely to retain the original wild-type sequence in MHC-heterozygous animals than in MHC-homozygous animals during chronic infection.
To determine whether these category classifications extend to epitopes restricted by other MHC alleles, we assessed the frequency of amino acid variant sequences in two epitopes restricted by MHC class I alleles present on the M1 MHC haplotype (23, 24): Gag191–199NA9 and Nef254–262LT9. We assessed the frequency of variant amino acid sequences in the three M1-heterozygous animals in this study and compared them to the frequency of variant amino acid sequences present in M1-homozygous animals from previous studies (9, 12). We found that the Nef254–262LT9 epitope tended to accumulate mutations in both M1 homozygotes and heterozygotes, whereas the Gag191–199NA9 epitope tended to accumulate variants more frequently in the M1 homozygotes than the heterozygotes (Table 7). Even though the sample size was small, we suggest that Nef254–262LT9 may be a category A epitope, whereas Gag191–199NA9 more closely resembles a category B epitope. Importantly, it suggests that epitopes restricted by MHC alleles on other haplotypes follow a pattern similar to the one we have observed among the epitopes restricted by M3 MHC alleles.
TABLE 7.
Percentage of epitopes restricted by M1 MHC class I B molecules with amino acid variation in M1-homozygous and -heterozygous MCMs
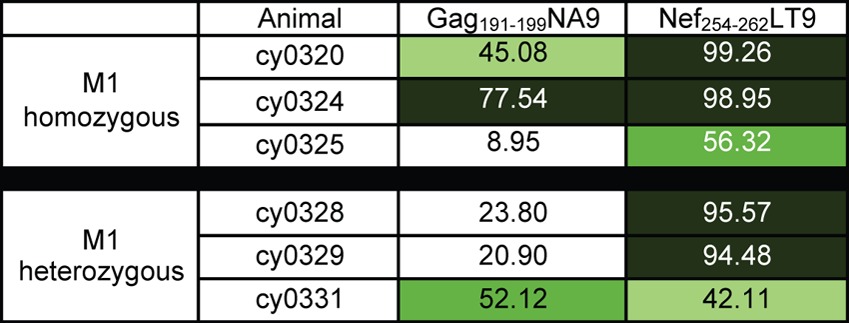
Amino acid variation was calculated as described in Materials and Methods. Values are percentages of variant sequences. Dark green, >75% variant; medium green, 50% to 75% variant; light green, 25% to 75%; white, <25% variant.
Preservation of category B T cell responses in MHC-heterozygous MCMs.
Given the preservation of wild-type sequence in the category B epitopes, we wanted to determine if the corresponding epitope-specific CD8 T cell responses were preserved in chronic infection in the M3 heterozygotes compared to the M3 homozygotes. We performed IFN-γ ELISPOT assays using cryopreserved PBMC that had been collected during chronic infection (Tables 8 and 9). Samples were tested in duplicate for all 14 animals. A sample was considered positive if its value was greater than the average for four unstimulated samples plus two times the standard deviation of the unstimulated samples, or 100 SFCs, whichever was greater. For the category B epitopes Gag146–154HL9, Tat42–49QA8, and Env338–346RF9, we found a greater percentage of samples that were positive in the M3-heterozygous animals than M3-homozygous animals (Table 8). We performed a similar comparison of positive responses specific for category A epitopes. We found very few responses to the category A epitopes during chronic infection for both M3 homozygotes and heterozygotes. These results suggest that T cell responses to the category A epitopes in nearly all M3+ animals at late stages of infection are absent but that T cell responses to category B epitopes are more likely to be preserved in the M3-heterozygous animals in the chronic phase.
TABLE 8.
Frequency of samples with positive IFN-γ ELISPOT responses from M3 homozygotes and heterozygotes
| Category | Epitope | No. of positive wells/total |
|
|---|---|---|---|
| Homozygotes | Heterozygotes | ||
| A | Gag386–394GW9 | 2/18 | 0/10 |
| Pol592–599QP8 | 2/18 | 0/10 | |
| ARF130–40QL11 | 2/18 | 0/10 | |
| Rev59–68SP10 | 0/18 | 1/10 | |
| Nef103–111RM9 | 1/18 | 0/10 | |
| Nef196–203HW8 | 2/18 | 4/10 | |
| B | Gag146–154HL9 | 8/18 | 9/10 |
| Tat42–49QA8 | 3/18 | 4/10 | |
| Env338–346RF9 | 3/18 | 4/10 | |
| Env620–628TL9 | 4/18 | 2/10 | |
| C | Gag28–37KA10 | 2/18 | 0/10 |
| Gag221–229PR9 | 2/18 | 5/10 | |
| Gag459–467TV9 | 1/18 | 0/10 | |
| Tat59–67CF9 | 2/18 | 0/10 | |
IFN-γ ELISPOT assays were performed in duplicate from cryopreserved PBMC, as described in Materials and Methods. Results are based on each well tested, rather than each animal tested. Overall, cryopreserved cells from the nine homozygous animals and the five heterozygous animals were tested. Each peptide was tested in duplicate.
Poor detection of T cell responses targeting category C epitopes.
During the chronic phase, we found that the circulating sequences of category C epitopes frequently matched the inoculum (Table 6). Notably, detection of CD8 T cells specific for these epitopes were rarely detected in the blood by IFN-γ ELISPOT (Table 8). Even though it has been shown that epitopes in category C are restricted by M3 MHC class I molecules (20), our results call into question their immunogenicity during a natural SIV infection. It is possible that category C epitopes are not presented well by MHC molecules, CD8 T cells specific for category C epitopes are located in inaccessible sites, or the immune system is too compromised at the later stage of infection, when category C epitopes could otherwise be successfully targeted. Even if CD8 T cells targeting these epitopes develop poorly during natural SIV infection of M3+ animals, the low mean dN value of these epitope sequences in the circulating virus of all M3+ animals suggests that they remain viable targets for CD8 T cells during chronic infection.
DISCUSSION
In a previous study (9), we found that immune escape in epitopes targeted during acute infection was conditional. We found that variant accumulation in an epitope presented by an MHC allele shared between two groups of animals was delayed in the animals that had the potential to present more distinct SIV epitopes during acute infection. This observation led us to hypothesize that similar conditional escape may occur during chronic SIV infection, when the comprehensive set of sequences present in the virus population is more complex and diverse than during acute infection. In this study, we deep sequenced SIV replicating in MCMs that were homozygous and heterozygous for the M3 MHC haplotype, and we evaluated variant accumulation in the 14 epitopes previously shown to be restricted by M3 MHC class I molecules (18, 20).
We found that the 14 epitopes restricted by M3 MHC class I alleles could be placed into three separate categories at about 1 year postinfection, based on their proclivity for accumulating variants. We found that epitopes in category A accumulate variants by this time among animals expressing the restricting MHC allele, and the corresponding CD8 T cell responses are absent. It is likely that the epitope variants are not detected by the host immune response, making them poor targets for CD8 T cell-dependent virus control during chronic infection. At the other end of the spectrum, we found that epitopes in category C rarely accumulate variants in the same animals. Unfortunately, T cells targeting category C epitopes were rarely detectable, calling their immunogenicity into question. In the middle, category B epitopes were less likely to accumulate variants in MHC-heterozygous hosts than in MHC-homozygous animals. Correspondingly, the MHC-heterozygous animals were more likely to maintain T cell responses targeting category B epitopes than MHC-homozygous animals. Overall, these results suggest that conditional escape extends into the category B epitopes during the chronic phase of SIV infection.
Typically, during acute HIV/SIV infection, host immune responses are directed at a small number of epitopes, such that variants can accumulate sequentially. As a result, if more epitopes are targeted at a given time, such as in MHC-heterozygous animals, then it is logical to propose that variant accumulation in certain epitopes may be delayed while other epitopes are accumulating mutations so that the total combination of mutations is compatible with successful virus replication. This concept is similar to the increase in waiting time experienced for multiple mutations to arise during evolution or carcinogenesis (25, 26). We do not mean to imply that two mutations need to occur in the same virus gene so that an individual protein has enhanced function, rather that the total compilation of mutations is replication competent. In our study, MHC-heterozygous animals have the potential to present a greater number of discrete peptides to T cells than MHC-homozygous animals. Consequently, the variant sequences that accumulate in viruses replicating in the MHC-heterozygous animals need to be compatible with a larger number of other variants so that the virus population can remain viable. The data we present here support the hypothesis that targeting more epitopes can delay the accumulation of variants, as the epitopes we classified as category B are less likely to accumulate variants in the MHC-heterozygous animals than the MHC-homozygous animals at 1 year postinfection. It is possible that these category B epitopes would accumulate variants at a later time, but many of these samples were obtained at necropsy, and we could not continue following virus evolution in these animals.
We also found that when the accumulation of variants in category B epitopes was blunted in MHC-heterozygous animals, CD8 T cell responses were preserved. Because preserving epitope-specific CD8 T cell responses did not necessarily portend better viral control in our cohort, it is possible that the preserved CD8 T cell responses we observed were not effectively engaging their cognate antigen. This opens up a potential opportunity to therapeutically enhance the magnitude, functionality, and efficacy of these CD8 T cells specifically for epitopes that retain immunogenicity. Importantly, though, our data suggest that by directing the immune response to target more epitopes, variant accumulation can be delayed and immune responses can be preserved.
Conclusions.
In our study, we found that conditional immune escape extends into chronic SIV infection. We also found that by increasing the number of discrete peptide epitopes that can be presented to the immune system, it is possible to delay the accumulation of variants in specific targeted epitopes. This may increase the likelihood that epitope-specific CD8 T cell responses can be preserved, suggesting that eliciting these CD8 T cell populations may be useful in therapeutic interventions. This study provides evidence that CD8 T cell epitopes differentially tolerate variant nucleotides and that the rate of variant accumulation during chronic infection can be affected by the number of potential epitopes that are targeted in an individual animal. This may explain, at least in part, why some epitopes do not universally accumulate variants in individuals who express the known restricting MHC class I allele. Additionally, we suggest that there is a unique category of epitopes that may retain immunogenicity during chronic infection, and therefore, therapeutic interventions aimed at eliciting anti-HIV/SIV-specific CD8 T cells should focus on these specific targets.
ACKNOWLEDGMENTS
We thank members of the WNPRC for their support with the animal care and the labs of S.L.O. and David O'Connor for critical discussions about the manuscript.
This work was supported by NIH R01 AI108415 and by University of Wisconsin—Madison startup funds to S.L.O. A.J.E.'s work was also supported by NIH grant R01 AI084787 to David O'Connor and grant T32 AI078985 to A.J.E. The research was conducted in part at a facility constructed with support from Research Facilities Improvement Program grant numbers RR15459-01 and RR020141-01. This project was also funded by the National Institutes of Health (grant R01AI084787). This was also supported by an NSF GRF (DGE-0929297), a University of South Carolina (USC) Presidential Fellowship, and a USC Department of Biological Sciences Kathryn Hinnant-Johnson, M.D. Memorial Fellowship to C.W.N. We also thank members of the Wisconsin National Primate Research Center, a facility supported by grants P51RR000167 and P51OD011106.
D.D.G. and M.S. performed the sequencing experiments. A.J.B. performed IFN-γ ELISPOT assays. C.W.N. and A.L.H. calculated dN and dS values. A.J.E. made key observations contributing to our hypothesis. S.L.O. conceived of the study, generated nucleotide alignments, and wrote the manuscript.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Funding Statement
The research was conducted in part at a facility constructed with support from Research Facilities Improvement Program grants RR15459-01 and RR020141-01.
REFERENCES
- 1.O'Connor DH, McDermott AB, Krebs KC, Dodds EJ, Miller JE, Gonzalez EJ, Jacoby TJ, Yant L, Piontkivska H, Pantophlet R, Burton DR, Rehrauer WM, Wilson N, Hughes AL, Watkins DI. 2004. A dominant role for CD8+-T-lymphocyte selection in simian immunodeficiency virus sequence variation. J Virol 78:14012–14022. doi: 10.1128/JVI.78.24.14012-14022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goulder PJ, Watkins DI. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol 4:630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 3.Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, Margolick JB, Gurer C, Murphy AJ, Valenzuela DM, Yancopoulos GD, Deeks SG, Strowig T, Kumar P, Siliciano JD, Salzberg SL, Flavell RA, Shan L, Siliciano RF. 2015. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rolland M, Manocheewa S, Swain JV, Lanxon-Cookson EC, Kim M, Westfall DH, Larsen BB, Gilbert PB, Mullins JI. 2013. HIV-1 conserved elements vaccines: relationship between sequence conservation and replicative capacity. J Virol 87:5461–5467. doi: 10.1128/JVI.03033-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulkarni V, Valentin A, Rosati M, Alicea C, Singh AK, Jalah R, Broderick KE, Sardesai NY, Le Gall S, Mothe B, Brander C, Rolland M, Mullins JI, Pavlakis GN, Felber BK. 2014. Altered response hierarchy and increased T-cell breadth upon HIV-1 conserved element DNA vaccination in macaques. PLoS One 9:e86254. doi: 10.1371/journal.pone.0086254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayton EJ, Rose A, Ibrahimsa U, Del Sorbo M, Capone S, Crook A, Black AP, Dorrell L, Hanke T. 2014. Safety and tolerability of conserved region vaccines vectored by plasmid DNA, simian adenovirus and modified vaccinia virus ankara administered to human immunodeficiency virus type 1-uninfected adults in a randomized, single-blind phase I trial. PLoS One 9:e101591. doi: 10.1371/journal.pone.0101591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, Hayton EJ, Black A, Bridgeman A, Rosario M, Hill AV, Berrie E, Moyle S, Frahm N, Cox J, Colloca S, Nicosia A, Gilmour J, McMichael AJ, Dorrell L, Hanke T. 2014. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther 22:464–475. doi: 10.1038/mt.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, et al. . 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Connor SL, Becker EA, Weinfurter JT, Chin EN, Budde ML, Gostick E, Correll M, Gleicher M, Hughes AL, Price DA, Friedrich TC, O'Connor DH. 2012. Conditional CD8+ T cell escape during acute simian immunodeficiency virus infection. J Virol 86:605–609. doi: 10.1128/JVI.05511-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu H, Wang X, Lackner AA, Veazey RS. 2013. CD8 down-regulation and functional impairment of SIV-specific cytotoxic T lymphocytes in lymphoid and mucosal tissues during SIV infection. J Leukoc Biol 93:943–950. doi: 10.1189/jlb.1112580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Connor SL, Lhost JJ, Becker EA, Detmer AM, Johnson RC, Macnair CE, Wiseman RW, Karl JA, Greene JM, Burwitz BJ, Bimber BN, Lank SM, Tuscher JJ, Mee ET, Rose NJ, Desrosiers RC, Hughes AL, Friedrich TC, Carrington M, O'Connor DH. 2010. MHC heterozygote advantage in simian immunodeficiency virus-infected Mauritian cynomolgus macaques. Sci Transl Med 2:22ra18. doi: 10.1126/scitranslmed.3000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Budde ML, Greene JM, Chin EN, Ericsen AJ, Scarlotta M, Cain BT, Pham NH, Becker EA, Harris M, Weinfurter JT, O'Connor SL, Piatak MJ, Lifson JD, Gostick E, Price DA, Friedrich TC, O'Connor DH. 2012. Specific CD8+ T cell responses correlate with control of simian immunodeficiency virus replication in Mauritian cynomolgus macaques. J Virol 86:7596–7604. doi: 10.1128/JVI.00716-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adnan S, Colantonio AD, Yu Y, Gillis J, Wong FE, Becker EA, Piatak MJ, Reeves RK, Lifson JD, O'Connor SL, Johnson RP. 2015. CD8 T cell response maturation defined by anentropic specificity and repertoire depth correlates with SIVΔnef-induced protection. PLoS Pathog 11:e1004633. doi: 10.1371/journal.ppat.1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson CW, Moncla LH, Hughes AL. 29 July 2015. SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. [DOI] [PubMed] [Google Scholar]
- 16.Bailey AL, Lauck M, Weiler A, Sibley SD, Dinis JM, Bergman Z, Nelson CW, Correll M, Gleicher M, Hyeroba D, Tumukunde A, Weny G, Chapman C, Kuhn JH, Hughes AL, Friedrich TC, Goldberg TL, O'Connor DH. 2014. High genetic diversity and adaptive potential of two simian hemorrhagic fever viruses in a wild primate population. PLoS One 9:e90714. doi: 10.1371/journal.pone.0090714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson CW, Hughes AL. 2015. Within-host nucleotide diversity of virus populations: insights from next-generation sequencing. Infect Genet Evol 30:1–7. doi: 10.1016/j.meegid.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris M, Burns CM, Becker EA, Braasch AT, Gostick E, Johnson RC, Broman KW, Price DA, Friedrich TC, O'Connor SL. 2013. Acute-phase CD8 T cell responses that select for escape variants are needed to control live attenuated simian immunodeficiency virus. J Virol 87:9353–9364. doi: 10.1128/JVI.00909-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AL, Becker EA, Lauck M, Karl JA, Braasch AT, O'Connor DH, O'Connor SL. 2012. SIV genome-wide pyrosequencing provides a comprehensive and unbiased view of variation within and outside CD8 T lymphocyte epitopes. PLoS One 7:e47818. doi: 10.1371/journal.pone.0047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budde ML, Lhost JJ, Burwitz BJ, Becker EA, Burns CM, O'Connor SL, Karl JA, Wiseman RW, Bimber BN, Zhang GL, Hildebrand W, Brusic V, O'Connor DH. 2011. Transcriptionally abundant major histocompatibility complex class I alleles are fundamental to nonhuman primate simian immunodeficiency virus-specific CD8+ T cell responses. J Virol 85:3250–3261. doi: 10.1128/JVI.02355-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burwitz BJ, Pendley CJ, Greene JM, Detmer AM, Lhost JJ, Karl JA, Piaskowski SM, Rudersdorf RA, Wallace LT, Bimber BN, Loffredo JT, Cox DG, Bardet W, Hildebrand W, Wiseman RW, O'Connor SL, O'Connor DH. 2009. Mauritian cynomolgus macaques share two exceptionally common major histocompatibility complex class I alleles that restrict simian immunodeficiency virus-specific CD8+ T cells. J Virol 83:6011–6019. doi: 10.1128/JVI.00199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cain BT, Pham NH, Budde ML, Greene JM, Weinfurter JT, Scarlotta M, Harris M, Chin E, O'Connor SL, Friedrich TC, O'Connor DH. 2013. T cell response specificity and magnitude against SIVmac239 are not concordant in major histocompatibility complex-matched animals. Retrovirology 10:116. doi: 10.1186/1742-4690-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohns MS, Greene JM, Cain BT, Pham NH, Gostick E, Price DA, O'Connor DH. 2015. Expansion of simian immunodeficiency virus (SIV)-specific CD8 T cell lines from SIV-naive Mauritian cynomolgus macaques for adoptive transfer. J Virol 89:9748–9757. doi: 10.1128/JVI.00993-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Negri DR, Borghi M, Baroncelli S, Macchia I, Buffa V, Sernicola L, Leone P, Titti F, Cara A. 2006. Identification of a cytotoxic T-lymphocyte (CTL) epitope recognized by Gag-specific CTLs in cynomolgus monkeys infected with simian/human immunodeficiency virus. J Gen Virol 87:3385–3392. doi: 10.1099/vir.0.81934-0. [DOI] [PubMed] [Google Scholar]
- 25.Durrett R, Schmidt D. 2008. Waiting for two mutations: with applications to regulatory sequence evolution and the limits of Darwinian evolution. Genetics 180:1501–1509. doi: 10.1534/genetics.107.082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durrett R, Schmidt D, Schweinsberg J. 2009. A waiting time problem arising from the study of multi-stage carcinogenesis. Ann Appl Probab 19:676–718. doi: 10.1214/08-AAP559. [DOI] [Google Scholar]