

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent for Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and a subset of multicentric Castleman disease (MCD). The KSHV life cycle has two principal gene repertoires, latent and lytic. KSHV viral interleukin-6 (vIL-6), an analog of human IL-6, is usually lytic; production of vIL-6 by involved plasmablasts is a central feature of KSHV-MCD. vIL-6 also plays a role in PEL and KS. We show that a number of plasmablasts from lymph nodes of patients with KSHV-MCD express vIL-6 but not ORF45, a KSHV lytic gene. We further show that vIL-6 is directly induced by the spliced (active) X-box binding protein-1 (XBP-1s), a transcription factor activated by endoplasmic reticulum (ER) stress and differentiation of B cells in lymph nodes. The promoter region of vIL-6 contains several potential XBP-response elements (XREs), and two of these elements in particular mediate the effect of XBP-1s. Mutation of these elements abrogates the response to XBP-1s but not to the KSHV replication and transcription activator (RTA). Also, XBP-1s binds to the vIL-6 promoter in the region of these XREs. Exposure of PEL cells to a chemical inducer of XBP-1s can induce vIL-6. Patient-derived PEL tumor cells that produce vIL-6 frequently coexpress XBP-1, and immunofluorescence staining of involved KSHV-MCD lymph nodes reveals that most plasmablasts expressing vIL-6 also coexpress XBP-1. These results provide evidence that XBP-1s is a direct activator of KSHV vIL-6 and that this is an important step in the pathogenesis of KSHV-MCD and PEL.
IMPORTANCE Kaposi sarcoma herpesvirus (KSHV)-associated multicentric Castleman disease (KSHV-MCD) is characterized by severe inflammatory symptoms caused by an excess of cytokines, particularly KSHV-encoded viral interleukin-6 (vIL-6) produced by lymph node plasmablasts. vIL-6 is usually a lytic gene. We show that a number of KSHV-MCD lymph node plasmablasts express vIL-6 but do not have full lytic KSHV replication. Differentiating lymph node B cells express spliced (active) X-box binding protein-1 (XBP-1s). We show that XBP-1s binds to the promoter of vIL-6 and can directly induce production of vIL-6 through X-box protein response elements on the vIL-6 promoter region. We further show that chemical inducers of XBP-1s can upregulate production of vIL-6. Finally, we show that most vIL-6-producing plasmablasts from lymph nodes of KSHV-MCD patients coexpress XBP-1s. These results demonstrate that XBP-1s can directly induce vIL-6 and provide evidence that this is a key step in the pathogenesis of KSHV-MCD and other KSHV-induced diseases.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent for Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and a subset of multicentric Castleman's disease (KSHV-MCD) (1–3). Like other herpesviruses, the KSHV life cycle includes latent and lytic phases. During the latent phase, gene expression is restricted and focused on promoting cell survival (4). When the virus is activated into the lytic phase through the lytic switch gene, replication and transcription activator (RTA) (5, 6), the full viral genome is expressed, and viral replication ensues. KSHV has coevolved with humans and is finely attuned to respond to the state of infected cells. Several factors that can stimulate lytic replication have been identified, including hypoxia, oxidative stress, and certain cytokines (7–12).
KSHV encodes an analog of human interleukin-6 (hIL-6) called viral IL-6 (vIL-6) (13). vIL-6 is induced by RTA and is produced during lytic KSHV replication (14). However, vIL-6 is also expressed in a subset of otherwise latently infected cells (15–17). Like hIL-6, vIL-6 stimulates proliferation and differentiation of B cells and angiogenesis (18–22). However, vIL-6 can activate additional cell types that do not respond to hIL-6 by binding to the receptor signaling subunit gp130 directly (18, 22, 23). Also, unlike hIL-6, vIL-6 binds to receptors within the endoplasmic reticulum (ER), manifesting effects within the cell in which it is produced (22).
KSHV-MCD is a systemic illness characterized by severe inflammatory flares and is usually fatal if untreated. Local and systemic vIL-6 proteins are important contributors to the pathogenesis and symptomatology of KSHV-MCD (24–27). KSHV-MCD flares are characterized by systemic expression of vIL-6 and/or hIL-6 (24, 25, 28, 29). The key pathological finding is KSHV-infected plasmablasts in affected lymph nodes (21, 29, 30). Previous studies have shown that a subset of these plasmablasts express vIL-6, and there is evidence to suggest that vIL-6 expression in these plasmablasts often occurs without expression of other lytic KSHV genes (21, 28–30). This production of vIL-6 without cell lysis is important in KSHV-MCD pathogenesis, for otherwise the vIL-6 contribution to disease would be self-limited. The factors inducing vIL-6 expression in latent cells are not fully understood though activation by interferon alpha has been reported (17). An understanding of these factors would provide an important insight into the fundamental aspects of KSHV-MCD pathogenesis. There is also evidence that vIL-6 plays a role in PEL and KS, and understanding the regulation of vIL-6 can provide insights into the pathogenesis of these diseases as well (24, 27).
We hypothesized that spliced X-box binding protein 1 (XBP-1) contributes to the activation of vIL-6 in KSHV-MCD lymph node plasmablasts and possibly in PEL (31). XBP-1 is a key protein in the unfolded protein response (UPR) associated with endoplasmic reticulum (ER) stress caused by factors such as an excess of protein production (32, 33). In cells under ER stress, a 26-nucleotide intron is excised from mRNA of unspliced XBP-1 (XBP-1u; inactive) to create a spliced XBP-1 (XBP-1s), which is active (34). XBP-1s enters the nucleus, activating cellular genes of the UPR by binding to XBP-1 response elements (XREs). Interestingly, XBP-1s is expressed during B cell differentiation in lymph nodes, where it is thought to protect against ER stress caused by immunoglobulin production (35). XBP-1s has also been shown to activate KSHV lytic replication, either alone or in combination with hypoxia (36–39). Since vIL-6-producing MCD plasmablasts arise in a setting where many differentiating B cells express XBP-1s, it seemed plausible that XBP-1s might also directly activate vIL-6 in KSHV-infected cells without inducing lytic replication and cell death.
MATERIALS AND METHODS
Cell culture.
Hep3B and HEK-292T cell lines (American Type Culture Collection [ATCC], Manassas, VA) were maintained in Dulbecco's modified Eagle's medium (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (heat inactivated; HyClone, Logan, UT) and penicillin-streptomycin-glutamine (Pen-Strep-glutamine) (Invitrogen Corp., Carlsbad, CA). BCBL-1 (NIH AIDS Research and Reference Reagent Program) and JSC-1 (ATCC) cell lines were maintained in RPMI 1640 medium (Gibco, Grand Island, NY) supplemented with 10% FBS and Pen-Strep-glutamine. BC-3 (ATCC) was maintained in RPMI 1640 medium supplemented with 20% FBS and Pen-Strep-glutamine. Except where indicated, cultures were maintained in 95% air and 5% CO2 at 37°C. In some experiments, cells were treated with brefeldin A (BFA), tunicamycin (TM), or 20 nM 12-O-tetra-dodecanoyl-phorbol-13-acetate (TPA) in dimethyl sulfoxide (DMSO) (all from Sigma, St. Louis, MO) (40). Equivalent final concentrations of DMSO were used as vehicle controls.
Plasmid DNA construction and site-directed mutagenesis.
The expression plasmid encoding degradation-resistant forms of hypoxia-inducible factor-1α (HIF-1α) (pcDNA-HIF-1αm with substitutions P402A and P564A in HIF-1α) has been described previously (10, 41). An expression plasmid encoding KSHV RTA (pcDNA-RTA) was a gift from Keiji Ueda (Osaka University Graduate School of Medicine, Osaka, Japan). Expression plasmids encoding unspliced (pcDNA-XBP1u) and spliced (pcDNA-XBP1s) XBP-1 were gifts from Kazatoshi Mori (Kyoto, Japan) (32). The pcDNA3.1 empty vector was used as a control. All plasmids were purified with a Qiagen Maxiprep kit (Qiagen, Valencia, CA), and inserts were verified by DNA sequencing. Reporter plasmids containing the full-length human vascular endothelial growth factor (VEGF) gene promoter (pVEGF-KpnI) (42) and KSHV-RTA promoter (9) have been described previously.
vIL-6 promoter luciferase reporter constructs (pvIL6-1, pvIL6-2, pvIL6-3, and pvIL6-4) were created spanning regions −1,696 bp, −484 bp, −297 bp, and −198 bp, respectively, upstream of the ATG start site to position +1. DNA fragments were amplified from KSHV in a bacterial artificial chromosome (BAC36) (43) (provided by Joseph Ziegelbauer, National Cancer Institute [NCI]) by PCR with 5′ primers for pvIL6-1 (5′-ACAGGTACCAGATGAGGATGTTCCTGTCTGC-3′), pvIL6-2 (5′-ACAGGTACCCATTGGCGGGTAGAATCAATGT −3′), pvIL6-3 (5′-ACAGGTACCCTGGCCAGTTAGGCTATTTTTA-3′), and pvIL6-4 (5′-ACAGGTACCCAAGCCTGGCGAGCAAGAGAGG-3′), which contained KpnI sites (underlined), and the common 3′ primer 5′-CCGAAGCTTGGCTGCTAACGCGGCATACAC-3′ which contained an HindIII site (underlined). PCR fragments were purified and cloned into the corresponding sites of the reporter vector pGL3-basic (Promega Corporation, Madison, WI). The vIL-6 promoter luciferase reporters pvIL6-M2, pvIL6-M3, and pvIL6-M2/3 containing mutagenized XREs were constructed from pvIL6-2 using a GeneArt site-directed mutagenesis system (Invitrogen Corp., Carlsbad, CA). These mutant plasmids contained 4-nucleotide substitutions (from ACGT to CTAG) in one or two XREs. Primer sequences used to mutate XRE2 were 5′-TTTACATGACTTTGCGTGTGCTAGCTTTCTCTCGCATGATAGCT-3′ and 5′-AGCTATCATGCGAGAGAAACCTAGCACACGCAAAGTCATGTAAA-3′; sequences for mutating XRE3 were 5′-AATGTGGTTCTAAGTCGCCTAGTAGAAACCCCGCCCCCTG −3′ and 5′-CAGGGGGCGGGGTTTCTACTAGCGGACTTAGAACCACATT-3′ (mutated residues are underlined). M2 contained mutated XRE2, M3 contained mutated XRE3, and M2/3 included both mutated XRE2 and XRE3. Reactions and transformation were performed according to the manufacturer's protocol. All constructs were confirmed by DNA sequencing.
Transfection and luciferase reporter assays.
Cells were transfected using Fugene 6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's protocol. For luciferase reporter experiments, 8 × 104 Hep3B cells/well or 1 × 105 HEK-293T cells/well were plated in a 12-well plate and the following day cotransfected with 300 ng/well of reporter plasmid DNA and either an expression plasmid DNA or a control (pcDNA3.1) in the presence of 50 ng of DNA of an internal control plasmid, pSV-β-galactosidase (pSV-β-Gal) (Promega Corporation, Madison, WI) to normalize for transfection efficiency. In experiments to assess the response of the vIL-6 promoter to HIF-1α or HIF-2α, a PTPRZ-1 reporter plasmid that was previously shown to be responsive to HIF-2α and, to a lesser extent, to HIF-1α (41) was used as a control. The HIF plasmids utilized encode HIF with mutagenized prolines that are not resistant to hydroxylation and degradation under normoxic conditions (41); the degradation-resistant HIF-1α plasmid was a gift of L. Eric Huang, previously of the NCI (44). The amount of each expression plasmid DNA used was as follows: pcDNA-XBP1u, 100 ng/well; pcDNA-XBP1s, 100 ng/well; pcDNA-RTA, 250 ng/well; pcDNA degradation-resistant HIF-1α or HIF-2α, 250 ng/well. The amount of control plasmid pCDNA3.1 used was equal to the amount of expression plasmid. Cells were incubated for 48 h, washed with phosphate-buffered saline (PBS), lysed with 250 μl per well of 1× reporter lysis buffer (Promega Corporation, Madison, WI), and freeze-thawed once. After centrifugation at 13,000 × g for 8 min, 20 μl and 50 μl of cell lysates were used to determine luciferase and β-Gal in the extracts. Means and standard deviations were calculated, and conditions were compared using a two-tailed Student t test for paired values.
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed using a SimpleChIP Enzymatic Chromatin IP kit (no. 9003; Cell Signaling Technologies). BCBL-1 cells were treated with 2 μg/ml TM or the DMSO control for 48 h at 37°C and then cross-linked with 37% formaldehyde at a final concentration of 1% at room temperature for 10 min. Fragmented chromatin was treated with nuclease and subjected to sonication. Chromatin immunoprecipitation was performed with rabbit anti-XBP-1 antibody (M-186; 5 μg) (sc7160; Santa Cruz Biotechnology), rabbit anti-histone H3 (a technical positive control; 1:50) (catalog no. 4620; Cell Signaling Technologies), and normal rabbit IgG (a negative control; 5 μg) (catalog no. 2729; Cell Signaling Technologies). After reverse cross-linking and DNA purification, immunoprecipitated DNA was quantified by real-time PCR using power SYBR green (catalog no. 4367659; Applied Biosystems) with primers for XBP-1 binding sites in the vIL-6 promoter (XRE2 forward primer 5′-AGAAGCCCAGAGCTAGCACA-3′ and reverse primer 5′-CATACGCAGCCAAGCTATCA-3′; XRE3 forward primer 5′-CAGCTGACTACCGACTGTGC-3′ and reverse primer 5′-GAACTCGCCAAAAAGTGAGC-3′) and RPL30 exon 3 (catalog no. 7014; Cell Signaling Technologies). Fold enrichment was calculated based on the threshold cycle (CT) value of the IgG control using the comparative CT method.
IHC.
Lymph nodes from MCD biopsy specimens and PEL pleural effusions were obtained from patients on protocols of the HIV and AIDS Malignancy Branch; these protocols were approved by the National Cancer Institutional Review Board, and all patients gave written informed consent. For latency-associated nuclear antigen (LANA) and for vIL-6 immunohistochemistry (IHC), formalin-fixed, paraffin-embedded (FFPE) sections were deparaffinized in xylene, rehydrated in graded alcohol, placed in hot, 1× low-pH antigen retrieval solution (Dako, CA), microwaved for 6 min (for LANA) or 10 min (for vIL-6), and blocked with Tris-goat serum buffer for 15 min. For LANA staining, sections were incubated with anti-LANA rat monoclonal antibody ([MAb] 1:400 dilution; Advanced Biotechnologies, Inc.) for 2 h at room temperature and detected with Ventana BenchMark XT system using an ultraView universal diaminobenzidine (DAB) detection kit (catalog number 760-500; Ventana, Tucson, AZ). For vIL-6 staining, sections were incubated with anti-vIL-6 rabbit polyclonal antibody (1:500 dilution) (catalog number 13-214-100; Advanced Biotechnologies, Inc.) for 2 h at room temperature and detected with a Dako Autostainer universal staining system. Images were collected using Zen software controlling a Zeiss AxioObserver Z1 widefield microscope equipped with a 10× Plan Apochromat (numerical aperture [NA], 0.45) objective lens and Axiocam MRc5 color charge-coupled-device (CCD) camera. Individual images were acquired using the tiling module of the Zen software and were stitched together to provide one large resultant image with an extended field of view. The images were exported as tiff files.
Dual IHC staining for KSHV ORF45 and vIL-6 was done on KSHV-MCD FFPE tissue sections prepared as described above, except that they were steamed for 20 min instead of being microwaved and were blocked with Dako Protein Block, Serum-Free, for 15 min two times and then with 5% bovine serum albumin (BSA; Sigma-Aldrich) for 15 min two times. Sections were then incubated with anti-ORF45 mouse monoclonal antibody (1:1,000 dilution) (2D4A5; Abcam) overnight at room temperature. Horseradish peroxide (HRP)-conjugated anti-rabbit IgG (Dako) was used as a secondary antibody for a 30-min incubation at room temperature, and DAB-chromogen (Dako) was used for detection after a 5-min incubation at room temperature. ORF45-stained sections were reretrieved in hot Dako 1× low-pH antigen retrieval solution, steamed for 20 min, and blocked with Dako Protein Block, Serum-Free, for 15 min two times and with 5% BSA for 15 min two times; sections were then incubated with vIL-6 rabbit polyclonal antibody (1:400 dilution; made under contract with Epitomics, Burlingame, CA) for 2 h at room temperature and detected with a Ventana BenchMark XT system using a Ventana (Tucson, AZ) ultraView Universal Alkaline Phosphatase Red Detection Kit (catalog number 760-501). Images were collected using Zen software controlling a Zeiss AxioObserver Z1 widefield microscope equipped with a 10× Plan Apochromat (NA, 0.45) objective lens and Axiocam MRc5 color CCD camera. Individual images were acquired using the tiling module of the Zen software and were stitched together to provide one large resultant image with an extended field of view. The images were exported as tiff files.
IF.
Dual immunofluorescence IF for ORF45 and vIL-6 was performed on FFPE slides of MCD lymph node biopsy specimens that were deparaffinized and rehydrated in Histo-Clear (National Diagnostics, Atlanta, GA) and an ethanol series. Antigen retrieval was performed by boiling samples for 20 min in citric acid-based Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Slides were blocked with 1% bovine serum albumin (BSA) (Sigma, St. Louis, MO) in PBS with 0.05% Tween 20 (PBST) (Abcam, Cambridge, MA) for 30 min and incubated with primary antibodies overnight at 4°C. Primary antibodies used were monoclonal mouse anti-ORF45 (1:200 dilution; Abcam, Cambridge, MA) and anti-vIL-6 rabbit monoclonal antibody (1:400 dilution; made under contract with Epitomics, Burlingame, CA). After three washes with PBS for 10 min, Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen Corp., Carlsbad, CA) and Alexa Fluor 555 goat anti-mouse IgG (Invitrogen Corp., Carlsbad, CA), at a 1:200 dilution, were applied as secondary antibodies and incubated for 30 min at room temperature. After three 10-min washes with PBST, the slides were mounted in ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen Corp., Carlsbad, CA) and incubated in the dark for 24 h. Confocal fluorescence images were acquired using a Zeiss LSM 510 Meta laser scanning confocal microscope equipped with a 63× Plan Apochromat (NA, 1.4) oil immersion objective lens (Carl Zeiss Microscopy, LLC, Thornwood, NY). Consistent detector settings were used for collecting images from all samples, including 0.14-μm x-y pixel size, 0.9-μm optical slice thickness, and 4× frame averaging. The final images were exported as tiff files and arranged into figures using PowerPoint 2007 (Microsoft, Redland, OR).
For IF in PEL cell lines, cells were washed in PBS, centrifuged at 1,000 × g for 5 min, and placed on slides precoated with poly-l-lysine to allow adherence. Cells were fixed in fresh 4% paraformaldehyde at room temperature for 15 min and then permeabilized with 0.25% Triton X-100 for 10 min. For PEL pleural effusions, mononuclear cells were enriched by Ficoll-Paque Plus (GE Healthcare Bio-Sciences, Pittsburgh, PA) according to the manufacturer's protocol, and fixed as described for cell lines. Slides were blocked with 1% bovine serum albumin (BSA) (Sigma, St. Louis, MO) in PBS with 0.05% Tween 20 (PBST) (Abcam, Cambridge, MA) for 30 min and incubated with primary antibodies overnight at 4°C. Primary antibodies used for PEL cell cultures included anti-vIL-6 rabbit monoclonal antibody (1:400 dilution; made under contract with Epitomics, Burlingame, CA) and anti-XBP-1s mouse monoclonal antibody (1:200 dilution; BioLegend, San Diego, CA). Primary antibodies used for primary PEL effusions included anti-vIL-6 mouse monoclonal antibody (clone 12.1.1; 1:200 dilution) (18) and anti-XBP-1 rabbit polyclonal antibody (M-186, 1:100 dilution; Santa Cruz Biotechnology). After three washes with PBS for 10 min, Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen Corp., Carlsbad, CA) and Alexa Fluor 555 goat anti-mouse IgG (Invitrogen Corp., Carlsbad, CA), at 1:200 dilutions, were applied as secondary antibodies and incubated for 30 min at room temperature. Subsequent processing and acquisition of confocal images were as described above for ORF45 and vIL-6 IF of MCD FFPE specimens
Dual IF for vIL-6 and XBP-1 was performed in FFPE MCD sections that were deparaffinized in xylene and rehydrated in ethanol (100% to 50%). Tissue sections were refixed in 4% PFA-PBS (20 min, room temperature). After samples were washed with PBS, antigen retrieval was performed with Uni-Trieve solution (Inovex Bioscience) at 70°C for 30 min. After being washed with PBS, tissues were blocked (1 h at room temperature) with PBS–0.05% Tween 20 containing 1% BSA (Jackson ImmunoResearch Laboratories, West Grove, PA) and 5% normal human serum. Rabbit monoclonal anti-XBP-1 antibody (EPR4086) (ab109221; Abcam) was used at 1:200 in blocking buffer; mouse monoclonal antibody (MAb) to vIL-6 (clone 12.1.1) was used at 2 μg/ml in blocking buffer. Tissue slides were incubated with the primary antibodies (anti-XBP-1 and anti-vIL-6 primary antibodies) or with control rabbit IgG and mouse IgG (both from Vector Laboratories) for 18 h at 4°C. After three 10-min washes (PBS–0.05% Tween 20), the secondary antibodies Alexa 594-conjugated donkey anti-rabbit IgG (Invitrogen) and Alexa 488-conjugated goat anti-mouse IgG (Life Technologies, Grand Island, NY) were added at a 1:2,000 dilution in blocking buffer for 1 h at room temperature. After three 10-min washes (PBS–0.05% Tween 20), the slides were mounted in DAPI Fluoromount-G (Southern Biotech, Birmingham, AL). Images were obtained with an LSM 710NLO microscope using ZEN software (Carl Zeiss) and exported as tiff files.
RNA isolation, RT-PCR, real-time qPCR, and Western blot analysis.
Total cellular RNA was isolated from cells using an RNeasy minikit (Qiagen, Hilden, Germany). Reverse transcription-PCR (RT-PCR) was performed using a Superscript III One-step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen Corp., Carlsbad, CA). Primers used are listed in Table 1; RT-PCR was performed according to the manufacturer's protocol. Unspliced XBP-1 product contained a PstI (New England BioLabs, Ipswich, MA) cutting site; after digestion, it degraded into 147-bp and 102-bp fragments, which could be separated from XBP-1s by electrophoresis in a 2% agarose gel. For real-time quantitative PCR (qPCR), cDNA was synthesized using the reverse transcriptase enzyme SuperScript II (Invitrogen). Real-time quantitative PCR was performed with SYBR green (catalog no. 4367659; Applied Biosystems) and the gene-specific PCR primers listed in Table 1. Samples were run in triplicate, and PCR was performed using an ABI Step One Plus thermocycler (Applied Biosystems). 18S ribosomal RNA was used as a housekeeping gene, and qPCR data (CT values) were analyzed using the 2−ΔΔCT method. The qPCR data are presented as the fold change in target gene expression ± standard error of mean.
TABLE 1.
Primers used in RT-PCR
| Target | Size(s) (bp) | Primer |
|
|---|---|---|---|
| Direction | Sequence (5′–3′) | ||
| vIL-6 | 295 | Forward | 5′-CTGTTACCGTACCGGCATCT-3′ |
| Reverse | 5′-GGGTGGACTGTAGTGCGTCT-3′ | ||
| RTA | 343 | Forward | 5′-GTCATGTCACCCTTGCGATC-3′ |
| Reverse | 5′-ACGCTTCTTTGAGCTCCTCT-3′ | ||
| 18S | 479 | Forward | 5′-AAACGGCTACCACATCCAAG-3′ |
| Reverse | 5′-CCCTCTTAATCATGGCCTCA-3′ | ||
| XBP-1 | 223 (spliced), 249 (unspliced) | Forward | 5′-GGAGTTAAGAAGCGCTTGG-3′ |
| Reverse | 5′-ACTGGGTCCAAGTTGTCCAG-3′ | ||
For Western blotting, nuclear extracts and whole-cell lysate were prepared using NE-PER nuclear and cytoplasmic protein extraction reagent and M-PER mammalian protein extraction reagent (Pierce, Rockford, IL). Thirty micrograms of nuclear protein or whole-cell lysate was electrophoresed on 4 to 12% NuPAGE gels (Invitrogen Corp., Carlsbad, CA). Proteins were transferred to a nitrocellulose membrane and blocked with 50% Odyssey blocking buffer (Li-Cor Biosciences) diluted in washing buffer (1× Tris-buffered saline containing 0.05% Tween 20 [TBST]). The membrane was incubated with anti-XBP-1s rabbit IgG (BioLegend, San Diego, CA) at a 1:500 dilution, rabbit anti-RTA (custom synthesized from Alpha Diagnostic International, Inc., San Antonio, TX) at a 1:500 dilution, rabbit anti-vIL-6 monoclonal antibody (custom made by Epitomics, Inc. Burlingame, CA) at a 1:10,000 dilution, and mouse-anti-β-actin (Sigma) at a 1:1,000,000 dilution. After the membrane was washed with washing buffer, it was incubated with appropriate anti-rabbit, or anti-mouse IRDye 800 CW secondary antibody (Li-Cor Biosciences). Western blots (WB) were visualized using an Odyssey scanner and analyzed using ImageStudio (Li-Cor Biosciences).
RESULTS
Expression of vIL-6 and KSHV ORF45 by KSHV-MCD plasmablasts.
KSHV-MCD is characterized by KSHV-infected plasmacytoid B cells in affected lymph nodes. As can be seen in a KSHV-MCD node, there are a number of MCD plasmablasts expressing the latent KSHV protein LANA; these are predominantly in the mantle zone of the germinal center (Fig. 1A). Figure 1B shows a different slice of the same node, stained for KSHV vIL-6. Consistent with previous reports (21, 30, 45), a number of plasmablasts express vIL-6 although the amount is less than the number that express LANA. Since previous studies have suggested that some cells may produce vIL-6 but not other lytic genes in KSHV-MCD (15, 16), we costained KSHV-MCD lymph node biopsy specimens for vIL-6 and ORF45, an immediate early KSHV lytic gene, both by IHC (Fig. 1C and D) and IF (Fig. 1E). Counts of IHC MCD lymph node slides from two patients (one shown in Fig. 1C and D) showed that of 233 cells in total expressing vIL-6 and/or ORF45, 117 (50.2%) expressed vIL-6 alone, 65 (27.9%) expressed vIL-6 and ORF45, and 51 (21.9%) expressed ORF45 but not vIL-6. Thus, while some cells expressed both proteins, a number of cells expressed vIL-6 but not ORF45, showing that vIL-6 is expressed in a substantial percentage of cells not manifesting the full lytic repertoire. A smaller number of cells expressed ORF45 and not vIL-6, perhaps because the level of vIL-6 was below the level of detection of the stain in these patients.
FIG 1.

vIL-6 and ORF45 in KSHV-MCD Patients. (A) LANA expression in a germinal center involved with KSHV-MCD. LANA, stained in brown with DAB, appears as nuclear speckles in KSHV-infected cell nuclei. The majority of LANA-expressing plasmablasts are in the mantle zone. (B) vIL-6 expression in an adjacent cut from the same germinal center. There were fewer vIL-6-expressing cells, and these were less focused in the mantle zone. (C and D) ORF45 and vIL-6 coexpression in a germinal center involved with KSHV-MCD. ORF45 (a KSHV lytic gene) was stained in brown, while vIL-6 was stained in red. Panel C shows the whole germinal center while panel D shows a section at higher magnification. ORF45 and vIL-6 are colocalized in many cells (solid arrows). Some cells express only vIL-6 and not ORF45 (dashed arrows). (E) ORF45 and vIL-6 expression in a KSHV-MCD lymph node observed by IF and confocal microscopy. Each horizontal row shows a separate field from the same section in one of two patients examined. ORF45 was stained in red, and vIL-6 was stained in green. Some cells expressed only vIL-6 and not ORF45.
The vIL-6 promoter contains potential XREs and responds to XBP-1s.
Since B cells maturing in germinal centers express XBP-1s (33, 46), we hypothesized that XBP-1s might drive vIL-6 production in cells latently infected with KSHV. We analyzed the promoter sequence of vIL-6 for potential XREs (Fig. 2A). Ten XRE core sequences, ACGT (47), were found within 1.7 kb upstream of the vIL-6 start codon (positions 17871 to 19567 of KSHV-BAC36; GenBank accession number HQ404500.1). By further analysis using MATCH (public version 1.0), four of these potential XREs (XRE2, XRE3, XRE8, and XRE9) were found to have consensus XRE sequences. XRE2 and XRE9 are encoded in the 5′-to-3′ direction on the sense strand, and XRE3 and XRE8 are encoded in the 5′-to-3′ direction on the antisense strand.
FIG 2.

Schematic of vIL-6 luciferase (LUC) promoter and constructs showing the location of the potential XBP response elements (XRE) and activation of the vIL-6 promoter constructs by spliced XBP-1. (A) The sequence of the vIL-6 promoter contains two TATA boxes. Ten XRE core sequences, ACGT (47), are found within 1.7 kb upstream of the vIL-6 start codon (positions 17871 to 19567 of KSHV-BAC36; GenBank accession number HQ404500.1): XRE1, −163 to −160; XRE2, −245 to −242; XRE3, −447 to −444; XRE4, −769 to −766; XRE5, −991 to −988; XRE6, −1262 to −1259; XRE7, −1331 to −1328; XRE8, −1352 to −1349; XRE9, −1401 to −1398; and XRE10, −1607 to −1604. Each potential XRE sequence is denoted as a square. Consensus XREs are indicated in black, and other (core-only) XREs are shown in gray. The direction of each consensus XRE is indicated with an arrow (core XRE sequence only, 5′-ACGT-3′; the core sequence is underlined in the consensus XRE, 5′-NNGNTGACGTGKNNNWT-3′). Constructs pvIL6-2, pvIL6-3, and pvIL6-4 were made by sequential deletions, as shown. (B) Comparison of the activation of the vIL-6 promoter luciferase reporter by the XBP-1 unspliced (XBP-1u) or spliced (XBP-1s) form. Hep3B cells were cotransfected with 300 ng of a vIL-6 promoter luciferase construct and 50 ng of an internal β-Gal control plasmid (pGL3-basic [pGL3b]) in the presence of 100 ng of an expression plasmid encoding XBP-1u, XBP-1s, or the pcDNA3.1 expression plasmid control. Values are expressed as fold increase over the value for the pGL3-basic reporter transfected with an empty expression vector (pcDNA3.1) and represent the mean of three independent experiments. Error bars denote the standard deviations (*, P ≤ 0.01; **, P ≤ 0.005, for the comparisons shown, normalized in each case for the results with the pGL3-basic control). The comparison between results for pvIL6-1/XBP-1u and pvIL6-1/pcDNA was not significant (P > 0.05). (C) Comparison of the activation of truncated forms of the vIL-6 luciferase reporter by XBP-1s or the pcDNA3.1 plasmid control. Hep3B cells were cotransfected with 300 ng of each vIL-6 promoter and 50 ng of an internal β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding XBP-1s or the pcDNA3.1 control. Values are expressed as fold increase over the value for pvIL6-1 transfected with an empty expression vector (pcDNA3.1) and represent the means of three independent experiments. Error bars denote the standard deviations.
To explore whether the vIL-6 promoter responds to XBP-1s, a 1,696-bp region of the vIL-6 promoter region containing the 10 possible XREs was cloned into a luciferase reporter construct to generate pGL3-pvIL6-1 Luc(1,696 bp) (Fig. 2). This construct was transiently transfected into Hep3B cells along with a plasmid encoding XBP-1u, a plasmid containing XBP-1s, or a control plasmid (pcDNA). When cotransfected with plasmids encoding XBP-1u, there was little or no increase in promoter activity compared to the level of the pcDNA control (Fig. 2B). In contrast, cotransfection with XBP-1s increased the promoter activity approximately 3-fold compared to transfection with XBP-1u and 3.4-fold compared to the level with the pcDNA3.1 control (P value at least of ≤0.01 for each comparison) (Fig. 2B). Similar results were also observed with HEK-293T cells (results not shown). The results provide evidence that XBP-1s activates the vIL-6 promoter.
The responsiveness of the IL-6 promoter to XBP-1s is mediated at least in part by XRE2 and XRE3.
To further investigate which response elements were responsible, we made reporter constructs with sequential deletions: pvIL6-2 (484 bp), pvIL6-3 (297 bp), and pvIL6-4 (198 bp) (Fig. 2C). When cotransfected with XBP-1s, pvIL6-2, containing XRE-1 through XRE3, induced essentially the same degree of activation as pvIL6-1, while pvIL6-3 and pvIL6-4 had lower levels of absolute activity with XBP-1s (Fig. 2C). Since pvIL6-3 lacks XRE3 and pvIL6-4 lacks both XRE2 and XRE3 sequences, these findings suggested that XRE2 and/or XRE3 might be functional XREs.
To determine this, a 4-bp mutation of the core XRE sequence was introduced into XRE2 and/or XRE3 in pvIL6-2 to produce pvIL6-M2, pvIL6-M3, and pvIL6-M2/3, respectively (Fig. 3A). Mutation of XRE2 and XRE3 (pvIL6-M2/3) significantly decreased the XBP-1s-induced activation of pvIL6-2 (P ≤ 0.05), and there was a suggestion that mutation of XRE2 by itself also had some effect although it was not significant (Fig. 3B). These results show that at least a substantial portion of the responsiveness of the vIL-6 promoter to XBP-1s is mediated by XRE2 plus XRE3.
FIG 3.

Site-directed mutagenesis of XRE2 and XRE3 of the pvIL6-2 promoter and response to XBP-1s, RTA, and HIF-1. (A) Constructs of the wild-type pvIL6-2 and mutant reporter plasmids. Three different mutant reporters (pvIL6-M2, pvIL6-M3, and pvIL6-M2/3) were constructed, each containing a 4-base substitution within the core XRE sequence or sequences. All mutations were made in the pvIL6-2 promoter (HRE core sequence, 5′-RCGTG-3′; RTA response element [RRE] sequence in the vIL-6 promoter, 5′-GTGGTTCTAAGTCGCACGTTAGAAACCCCGCCCCCTGGTGCTCACTTT-3′; the core XRE sequence is underlined). Consensus XREs including XRE2 and XRE3 are indicated in black, and XRE1 is indicated in gray. (B) Comparison of the activation of a wild-type pvIL6-2 luciferase reporter with that of the three mutant luciferase reporters by XBP-1s. Hep3B cells were cotransfected with 300 ng of pvIL6-2 or mutant promoters and 50 ng of an internal β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding XBP-1s or a pcDNA3.1 expression plasmid control. Values are expressed as fold increase over the value for the pvIL6-2 reporter transfected with an empty expression vector (pcDNA3.1) and represent the means of three independent experiments. Error bars denote the standard deviations. After values were normalized to the results with the pcDNA3.1 expression plasmid control for each promoter, the activity of the pvIL6-M2/3 reporter was significantly lower than that of pvIL6-2 (P ≤ 0.05), while comparisons of results of the other reporters were not significant. (C) Comparison of the activation by RTA of the wild-type pvIL6-2 luciferase reporter with that of the three mutant luciferase reporters. Hep3B cells were cotransfected with 300 ng of a pvIL6-2 promoter luciferase reporter or the mutant promoters and 50 ng of an internal β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding RTA or a pcDNA3.1 expression plasmid control. Values are expressed as the fold increase over the value for the pvIL6-2 reporter transfected with an empty expression vector (pcDNA3.1) and represent the means of three independent experiments. Error bars denote the standard deviations. After normalization to the results with the pcDNA3.1 expression plasmid control for each promoter, the activity of the pvIL6-M2 reporter was significantly greater than that of pvIL6-2 (P ≤ 0.05), while the results with the other reporters in comparison to those with pvIL6-2 were not significant. (D) Comparison of the activation of the wild-type vIL6-1 (1,696 bp) luciferase reporter or the HIF-responsive PTPRZ-1 reporter in response to degradation-resistant HIF-1α or HIF-2α. Hep3B cells were cotransfected with 300 ng of the pvIL6-1 luciferase reporter or PTPRZ-1 promoter and 50 ng of an internal β-Gal control plasmid in the presence of 250 ng of an expression plasmid encoding degradation-resistant HIF-1α, HIF-2α, or the pcDNA3.1 expression plasmid control. Values are expressed as the fold increase over the value of the pcDNA3.1 expression vector-treated control for each reporter construct in a representative experiment; error bars denote the standard deviations of triplicate determinations.
RTA, the KSHV lytic switch gene, activates other lytic genes by binding to RTA response elements (RRE) in their promoter regions. We asked if the vIL-6 promoter could respond to RTA with the XRE mutations. The response of pvIL6-M3 was of particular interest in this regard since XRE3 is embedded within an RRE sequence (5′-GTGGTTCTAAGTCGCACGTTAGAAACCCCGCCCCCTGGTGCTCACTTT-3′; XRE core consensus sequence is underlined) in the vIL-6 promoter (48). We cotransfected Hep3B cells with an RTA-expressing plasmid and the pvIL6-2 wild-type and mutant reporter constructs. RTA provided >125-fold activation of the pvIL6-2 reporter construct (Fig. 3C). Essentially no decrease in RTA-induced activation was seen with any of the mutants; in fact, there was some increase in the responsiveness of pvIL6-M2. This showed that the decreased responsiveness to XBP-1s of the pvIL6 promoter with mutated XRE2 and XRE3 was not due to a global loss of promoter function.
The vIL-6 promoter contains potential HRE but does not respond to HIF.
Cells exposed to hypoxia increase their levels of HIF, which activates hypoxia-responsive genes by binding to hypoxia response elements (HRE). Hypoxia can cause an upregulation of vIL-6 in KSHV-infected cells (11), and the XRE core consensus sequence ACGT is contained within the HRE consensus sequence (RCGTG), where R is A or G (49). Also, the promoter region of vIL-6 contains 13 core consensus HRE, and we wondered if HIF could directly activate vIL-6. To explore whether HIF could directly activate vIL-6, we cotransfected degradation-resistant HIF-1α or HIF-2α with the vIL-6 reporter constructs pvIL6-1 (1,696 bp) or the mutant constructs. As a control, we utilized a PTPRZ-1 reporter construct that has previously been shown to respond to HIF-2α and, to a lesser extent, to HIF-1α (41). As seen in the results shown in Fig. 3D, neither HIF-1α nor HIF-2α had any substantial effect on the vIL6-2 promoter, while HIF-2α and, to a lesser extent, HIF-1α were able to activate PTPRZ-1. These experiments suggest that upregulation of vIL-6 by hypoxia is an indirect effect, perhaps mediated through RTA.
Evidence of direct binding of XBP-1s to the vIL-6 promoter by ChIP.
To further explore the activation of vIL-6 by XBP-1s, we assessed the binding of XBP-1s to the promoter DNA by ChIP. BCBL-1 cells were treated with 2 μg/ml TM to induce XBP-1s and cross-linked with 37% formaldehyde, and the DNA was fragmented. Immunoprecipitation was performed with rabbit anti-XBP-1 antibody, and after reverse cross-linking immunoprecipitated DNA was purified by PCR using primers surrounding the XRE2 and the XRE3 sites. As seen in Fig. 4, anti-XBP-1 antibody produced enrichment of the vIL-6 promoter DNA sequences, especially as amplified by primers surrounding XRE3. These results provide evidence that XBP-1s can bind to the vIL-6 promoter in the region of XRE-2 and XRE3. As the degree of binding to a given XRE may not directly correlate with XRE activity and as the DNA fragments in ChIP assays can extend for 900 bp or so, the relative contributions of XRE2 and XRE3 cannot be further established from these results.
FIG 4.

CHIP assay showing binding of XBP-1s to the vIL-6 promoter. BCBL-1 endogenous XBP-1s was induced by TM treatment (2 μg/ml) for 48 h and then cross-linked. Chromatin IP of fragmented DNA was performed with XBP-1 antibody or control IgG. Precipitated DNA was assayed by qPCR with specific primers for amplification of XRE2 or XRE3 of the vIL-6 promoter, and the data were quantitated as described in Materials and Methods. Results shown are the means ± standard deviations of triplicate determinations from a typical experiment of two experiments performed. In controls performed at the same time, DNA immunoprecipitated with histone H3 antibody, but not with XBP-1 antibody or a control IgG, was enriched for RPL30 exon 3.
Chemical ER stress induction of XBP-1s upregulates vIL-6 mRNA and protein in PEL lines.
We next asked if induction of XBP-1s in PEL cells would lead to increased production of vIL-6 mRNA and vIL-6. BCBL-1 PEL cells were exposed to tunicamycin (TM), an ER stress inducer known to increase levels of XBP-1s by inhibiting N-linked glycosylation and causing protein misfolding (50). RT-PCR showed that TM induces XBP-1s mRNA in BCBL-1 PEL cells (Fig. 5A). XBP-1s was induced even at the lowest dose tested (0.25 μg/ml). As determined by real-time quantitative PCR, TM also induced production of vIL-6 mRNA in a dose-dependent manner at both 8 h and 24 h (Fig. 5B and C). Also, TM induced production of vIL-6 protein at 24 h (Fig. 5D); the appearance of two vIL-6 bands in TM-treated cells is likely due to the effects of TM on vIL-6 glycosylation.
FIG 5.

vIL-6 upregulation mediated by chemical inducers of XBP-1s in the BCBL-1 PEL line. BCBL-1 cells were treated with increasing doses of tunicamycin (TM) to induce ER stress; cells were also treated with 15 nM 12-O-tetra-dodecanoyl-phorbol-13-acetate (TPA) as an inducer of RTA. (A) RT-PCR showing expression at of total XBP-1, XBP-1s, XBP-1u, vIL-6, RTA, and 18S ribosomal RNA as a loading control in BCBL-1 cells treated with various doses of TM, a DMSO control, or 15 nM TPA. Cells treated with TM or the DMSO control were harvested at 24 h, while those treated with TPA were harvested at 48 h. (B) Real-time quantitative PCR showing expression of vIL-6 and RTA mRNA in BCBL-1 cells cultured in the same way as described for panel A and harvested at 8 h and 24 h, as well as a TPA control harvested at 48 h. Shown are the means ± the standard deviations of triplicate determinations from one representative experiment expressed as the fold change compared to the level of the DMSO control. (C) Time course (24 h) of the fold increase of vIL-6 and RTA mRNA over baseline in cells exposed to 0.25 μg/ml TM. Shown are the means ± the standard deviations of triplicate determinations from one representative experiment expressed as the fold change compared to the level of the DMSO control. (D) Western blot showing XBP-1s (55 kDa), vIL-6 (22 kDa and 28 kDa), and RTA (85 kDa) expression in BCBL-1 cells 24 h after treatment with 0.5 μg/ml TM, 2 μg/ml TM, or 15 nM TPA. DMSO was used as the control.
XBP-1s alone or in combination with hypoxia has previously been reported to upregulate RTA (36, 38, 39), and we wondered whether the effects of XBP-1s on vIL-6 were mediated through RTA. In these same BCBL-1 cells, TM induced an increase of RTA mRNA at 8 h and 24 h (Fig. 5B) although little or no RTA protein as detected by Western blotting was observed in response to 0.5 μg/ml or 2 μg/ml at 24 h (Fig. 5D). Also, a time course experiment showed that the increase in vIL-6 mRNA in response to a low dose (0.25 μg/ml) of TM occurred at the same time as the increase in RTA mRNA (Fig. 5C). At the higher doses, however, the increase in vIL-6 mRNA lagged behind the increase in RTA mRNA (Fig. 5B and results not shown). Taken together with the previous findings, these results provide evidence that XBP-1 can directly induce vIL-6 and that this may be particularly important in the early hours after XBP-1 induction. The results also provide evidence that TM can induce vIL-6 indirectly through upregulation of RTA.
Consistent with these results, XBP-1s was detected in the nucleus in a number of BCBL-1 cells treated with TM or with BFA, another inducer of XBP-1s which acts by blocking ER-to-Golgi compartment transport (51) (Fig. 6A). However, no XBP-1s was detected in cells exposed to DMSO as a control or to TPA (Fig. 6A). Some BFA-exposed or TM-exposed cells that expressed XBP-1s also stained for vIL-6. Similar results were also seen in PEL cell lines BC-3 and JSC-1 (data not shown).
FIG 6.

Expression of XBP-1 and vIL-6 in BCBL-1 cells exposed to chemical inducers of ER stress and in primary PEL cells. (A) BCBL-1 cells were treated with BFA for 24 h or TM for 48 h to induce ER stress or with TPA for 48 h to induce KSHV lytic replication. Cells expressing XBP-1s were stained in red, and cells expressing vIL-6 were stained in green. BFA-treated and TM-treated cells showed coexpression of XBP-1s and vIL-6. TPA-treated cells expressed vIL-6 but not XBP-1s. (B) Staining of primary PEL cells from a plural effusion for XBP and vIL-6. Cells expressing vIL-6 were stained in red while cells expressing XBP-1 were stained in green. A second patient tested gave similar results.
XBP-1 and vIL-6 proteins are associated in PEL and MCD.
To determine if induction of vIL-6 by XBP-1s may occur in infected patients, we looked for XBP-1 protein in primary PEL cells and lymph node biopsy specimens from MCD patients. Pleural effusion PEL cells were isolated, fixed, and stained with vIL-6 and XBP-1 (Fig. 6B). Many tumor cells displayed both XBP-1 and vIL-6. We also examined lymph node biopsy specimens from two patients with KSHV-MCD (one lymph node is shown in Fig. 7). A number of the cells contained XBP-1, and in 30% of these cells, it was located in the nucleus. A smaller number expressed vIL-6, and most of these cells coexpressed nuclear XBP-1. In scans of seven representative fields, out of 46 cells expressing vIL-6, 37 coexpressed nuclear XBP-1; in the same fields, an additional 363 cells expressed XBP-1 alone. It is quite likely that many of these cells expressing XBP-1 alone were not infected with KSHV; in KSHV-MCD lymph nodes, only a small percentage of cells are infected with KSHV (Fig. 1A) (21, 29, 30). With respect to the cells expressing vIL-6 but not XBP, it is possible that the level of XBP was too low to be detected by IF. Alternatively, another mechanism might be causing the vIL-6 expression in these cells.
FIG 7.
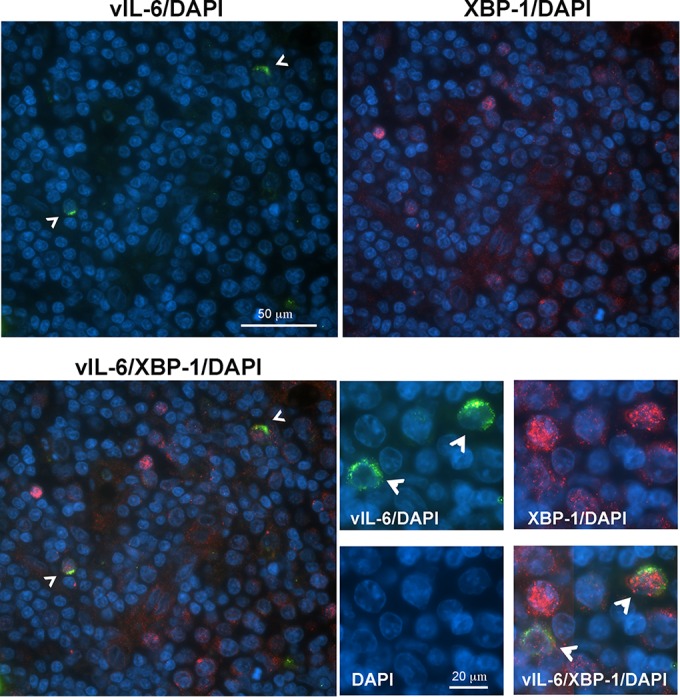
Detection of XBP-1- and vIL-6-positive cells in an MCD lymph node. Fluorescence immunostaining of a KSHV-MCD lymph node shows the distribution of vIL-6 (green) and XBP-1 (red) proteins in nucleated cells (marked by DAPI, blue. Arrowheads in images stained for vIL-6 point to XBP-1 and vIL-6 double-positive cells. Lower-magnification images (upper and left lower quadrants) show that the vIL-6-positive cells are also XBP-1 positive; higher-magnification images (lower right panels) show the predominantly nuclear localization of XBP-1 and cytoplasmic localization of vIL-6.
DISCUSSION
It was previously shown that XBP-1s can induce lytic activation of KSHV through RTA (36–39) We extend these findings and provide evidence that XBP-1s can directly bind to and activate the promoter of KSHV vIL-6. Much of this activation is mediated through two XREs, labeled here XRE2 and XRE3, possibly acting in tandem. We further show that a vIL-6 promoter with mutated XRE2 and XRE3 that has a decreased response to XBP-1s is still activated by RTA. TM, a chemical inducer of XBP-1s, also upregulates vIL-6, and KSHV-MCD plasmablasts and PEL cells in which vIL-6 is expressed frequently have coexpressed nuclear XBP-1. Taken together, these results provide evidence that XBP-1s can activate vIL-6 in latently infected KSHV cells and that this process may play a role in the pathogenesis of KSHV-induced lymphoproliferative disorders.
KSHV has developed a number of mechanisms to optimize its gene expression under various conditions. vIL-6 enhances the survival and proliferation of KSHV infection, and it is not surprising that KSHV may have developed strategies to fine-tune its expression. XBP-1s is produced in cells with ER stress, and activation of vIL-6 in response to XBP-1s may serve to increase survival of these cells and their passenger virus. The results shown here provide evidence that XBP-1s can activate vIL-6 directly; this mechanism may be particularly important in cells where RTA is tightly controlled. In addition, as seen in the PEL lines studied, XBP-1 activation can also be associated with increased expression of RTA, as previously reported (36–39) (Fig. 5); activation of vIL-6 can also occur through this mechanism. The binding of vIL-6 to the intracellular ER (22) would then serve to specifically increase survival of the KSHV-infected cells that may lack surface IL-6 receptors. Induction of RTA by XBP-1s may also lead to KSHV lytic replication, allowing the virus to spread. The prosurvival and proliferative mechanisms may be especially useful in tonsils, which are a potential portal of entry for KSHV and which contain a particularly high percentage of cells expressing XBP-1 (31). It should be noted that vIL-6 may also be constitutively formed in a subset of cells with latent KSHV infection and that interferon alpha has been reported to upregulate production of vIL-6 (17).
Activation of vIL-6 by XBP-1s may be important in the pathogenesis of KSHV-MCD and PEL. Although most PEL cells are latently infected, vIL-6 plays a role in PEL pathogenesis (27), and as shown here, a subset of PEL cells from a patient express both XBP-1 and vIL-6; activation of vIL-6 in these cells likely helps promote growth or survival of PEL by autocrine or paracrine activation. B cells undergoing terminal differentiation within lymph node germinal centers express XBP-1s (33, 46), and when certain of these cells are infected with KSHV, direct activation of vIL-6 may be a key step in the development and proliferation of KSHV-MCD plasmablasts, many of which express vIL-6 but not other lytic genes. Some of these cells, perhaps those with higher levels of XBP-1s, may then go on to complete lytic replication and die. This cycle, played out systemically, may help explain the spontaneous waxing and waning of symptoms in untreated KSHV-MCD (52).
Interactions among XBP-1, vIL-6, and human IL-6 may also contribute to KSHV-MCD pathogenesis. XBP-1s can stimulate human IL-6 expression in some cells (33). Also, human IL-6 (and plausibly also vIL-6) can induce XBP-1 (53), thus forming positive-feedback loops between XBP-1 and either human IL-6 or vIL-6. In addition, vIL-6 can induce the expression of hIL-6 (26), further augmenting the effect of vIL-6 since, unlike hIL-6, vIL-6 is produced only by the relatively small number of KSHV-infected cells. Thus, XBP-1s-induced activation of vIL-6 in a small subset of cells has the potential to initiate KSHV-MCD flares through the paracrine recruitment of neighbor IL-6-producing cells.
XBP-1s activation of vIL-6 is likely to be particularly important in KSHV-infected B cells and associated malignancies. It has recently been reported by Leung et al. that 2-deoxy-d-glucose, a stimulator of ER stress, suppressed KSHV replication and expression of vIL-6 in butyrate-stimulated endothelial or epithelial cells (54). At first glance, these results seem at odds with the current study. However, Leung et al. were looking at a modulation of KSHV induction by butyrate in KSHV-infected endothelial cells and cell lines and note that their findings would probably not apply to B cells, given the substantial expression of XBP-1 during B cell differentiation (54). Thus, these results are not inconsistent although it is possible that the upregulation of RTA and vIL-6 in our PEL cultures exposed to chemical inducers of ER stress was reduced somewhat by that mechanism.
In summary, this study shows that XBP-1s can directly activate production of KSHV vIL-6. This effect may be an important contributor to the pathogenesis of KSHV-MCD as well as PEL. These results also add to our appreciation of the physiologic activation of specific KSHV lytic genes outside the general activation of lytic replication.
ACKNOWLEDGMENTS
We thank the research volunteers who contributed to this study through their participation on NCI protocols. We also thank the medical and nursing staffs of the Medical Oncology Service of the NCI Center for Cancer Research and the NIH Clinical Center and the NCI Laboratory of Pathology. We appreciate the help of Michael L. Kruhlak in the Experimental Immunology Branch for microscope platform support.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson PG, Boshoff C. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406–1412. [PubMed] [Google Scholar]
- 4.Sarid R, Wiezorek JS, Moore PS, Chang Y. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J Virol 73:1438–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 6.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milligan S, Robinson M, O'Donnell E, Blackbourn DJ. 2004. Inflammatory cytokines inhibit Kaposi's sarcoma-associated herpesvirus lytic gene transcription in in vitro-infected endothelial cells. J Virol 78:2591–2596. doi: 10.1128/JVI.78.5.2591-2596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Feng J, Sun R. 2011. Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J Virol 85:715–724. doi: 10.1128/JVI.01742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haque M, Davis DA, Wang V, Widmer I, Yarchoan R. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J Virol 77:6761–6768. doi: 10.1128/JVI.77.12.6761-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. 2006. Genetic organization and hypoxic activation of the Kaposi's sarcoma-associated herpesvirus ORF34-37 gene cluster. J Virol 80:7037–7051. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250. doi: 10.1182/blood.V97.10.3244. [DOI] [PubMed] [Google Scholar]
- 12.Cai Q, Lan K, Verma SC, Si H, Lin D, Robertson ES. 2006. Kaposi's sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J Virol 80:7965–7975. doi: 10.1128/JVI.00689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore PS, Boshoff C, Weiss RA, Chang Y. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 14.Bu W, Palmeri D, Krishnan R, Marin R, Aris VM, Soteropoulos P, Lukac DM. 2008. Identification of direct transcriptional targets of the Kaposi's sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J Virol 82:10709–10723. doi: 10.1128/JVI.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brousset P, Cesarman E, Meggetto F, Lamant L, Delsol G. 2001. Colocalization of the viral interleukin-6 with latent nuclear antigen-1 of human herpesvirus-8 in endothelial spindle cells of Kaposi's sarcoma and lymphoid cells of multicentric Castleman's disease. Hum Pathol 32:95–100. doi: 10.1053/hupa.2001.21131. [DOI] [PubMed] [Google Scholar]
- 16.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Am J Pathol 156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. 2002. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298:1432–1435. doi: 10.1126/science.1074883. [DOI] [PubMed] [Google Scholar]
- 18.Aoki Y, Narazaki M, Kishimoto T, Tosato G. 2001. Receptor engagement by viral interleukin-6 encoded by Kaposi sarcoma-associated herpesvirus. Blood 98:3042–3049. doi: 10.1182/blood.V98.10.3042. [DOI] [PubMed] [Google Scholar]
- 19.Sakakibara S, Tosato G. 2011. Viral interleukin-6: role in Kaposi's sarcoma-associated herpesvirus: associated malignancies. J Interferon Cytokine Res 31:791–801. doi: 10.1089/jir.2011.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suthaus J, Adam N, Grotzinger J, Scheller J, Rose-John S. 2011. Viral interleukin-6: structure, pathophysiology and strategies of neutralization. Eur J Cell Biol 90:495–504. doi: 10.1016/j.ejcb.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. 1997. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am J Pathol 151:1517–1522. [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Sandford G, Nicholas J. 2009. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J Virol 83:722–733. doi: 10.1128/JVI.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. 1997. A Kaposi's sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J Biol Chem 272:19625–19631. doi: 10.1074/jbc.272.31.19625. [DOI] [PubMed] [Google Scholar]
- 24.Aoki Y, Yarchoan R, Wyvill K, Okamoto S, Little RF, Tosato G. 2001. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97:2173–2176. doi: 10.1182/blood.V97.7.2173. [DOI] [PubMed] [Google Scholar]
- 25.Polizzotto MN, Uldrick TS, Wang V, Aleman K, Wyvill KM, Marshall V, Pittaluga S, O'Mahony D, Whitby D, Tosato G, Steinberg SM, Little RF, Yarchoan R. 2013. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 122:4189–4198. doi: 10.1182/blood-2013-08-519959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G, Janz S, Scheller J, Rose-John S. 2012. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood 119:5173–5181. doi: 10.1182/blood-2011-09-377705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, Tosato G. 1999. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi's sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 94:2871–2879. [PubMed] [Google Scholar]
- 28.Aoki Y, Tosato G, Fonville TW, Pittaluga S. 2001. Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood 97:2526–2527. doi: 10.1182/blood.V97.8.2526. [DOI] [PubMed] [Google Scholar]
- 29.Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, Agbalika F. 2000. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric Castleman disease in HIV-infected patients. Blood 96:2069–2073. [PubMed] [Google Scholar]
- 30.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J Virol 73:4181–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balague O, Mozos A, Martinez D, Hernandez L, Colomo L, Mate JL, Teruya-Feldstein J, Lin O, Campo E, Lopez-Guillermo A, Martinez A. 2009. Activation of the endoplasmic reticulum stress-associated transcription factor X box-binding protein-1 occurs in a subset of normal germinal-center B cells and in aggressive B-cell lymphomas with prognostic implications. Am J Pathol 174:2337–2346. doi: 10.2353/ajpath.2009.080848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 33.Iwakoshi NN, Lee AH, Glimcher LH. 2003. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev 194:29–38. doi: 10.1034/j.1600-065X.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 34.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 35.Gass JN, Jiang HY, Wek RC, Brewer JW. 2008. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol Immunol 45:1035–1043. doi: 10.1016/j.molimm.2007.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett 581:3485–3488. doi: 10.1016/j.febslet.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 37.Lai IY, Farrell PJ, Kellam P. 2011. X-box binding protein 1 induces the expression of the lytic cycle transactivator of Kaposi's sarcoma-associated herpesvirus but not Epstein-Barr virus in co-infected primary effusion lymphoma. J Gen Virol 92:421–431. doi: 10.1099/vir.0.025494-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalton-Griffin L, Wilson SJ, Kellam P. 2009. X-box binding protein 1 contributes to induction of the Kaposi's sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J Virol 83:7202–7209. doi: 10.1128/JVI.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. 2010. Characterization of the activation of protein tyrosine phosphatase, receptor-type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor-2 alpha. PLoS One 5:e9641. doi: 10.1371/journal.pone.0009641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. 1996. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16:4604–4613. doi: 10.1128/MCB.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol 76:6185–6196. doi: 10.1128/JVI.76.12.6185-6196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang LE, Gu J, Schau M, Bunn HF. 1998. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A 95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aoki Y, Tosato G. 2003. Pathogenesis and manifestations of human herpesvirus-8-associated disorders. Semin Hematol 40:143–153. doi: 10.1016/S0037-1963(03)70006-7. [DOI] [PubMed] [Google Scholar]
- 46.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 47.Clauss IM, Chu M, Zhao JL, Glimcher LH. 1996. The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res 24:1855–1864. doi: 10.1093/nar/24.10.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song MJ, Deng H, Sun R. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol 77:9451–9462. doi: 10.1128/JVI.77.17.9451-9462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wenger RH, Stiehl DP, Camenisch G. 2005. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005:re12. [DOI] [PubMed] [Google Scholar]
- 50.Duksin D, Mahoney WC. 1982. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem 257:3105–3109. [PubMed] [Google Scholar]
- 51.Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. 1986. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem 261:11398–11403. [PubMed] [Google Scholar]
- 52.Oksenhendler E, Duarte M, Soulier J, Cacoub P, Welker Y, Cadranel J, Cazals-Hatem D, Autran B, Clauvel JP, Raphael M. 1996. Multicentric Castleman's disease in HIV infection: a clinical and pathological study of 20 patients. AIDS 10:61–67. doi: 10.1097/00002030-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 53.Wen XY, Stewart AK, Sooknanan RR, Henderson G, Hawley TS, Reimold AM, Glimcher LH, Baumann H, Malek LT, Hawley RG. 1999. Identification of c-myc promoter-binding protein and X-box binding protein 1 as interleukin-6 target genes in human multiple myeloma cells. Int J Oncol 15:173–178. [DOI] [PubMed] [Google Scholar]
- 54.Leung HJ, Duran EM, Kurtoglu M, Andreansky S, Lampidis TJ, Mesri EA. 2012. Activation of the unfolded protein response by 2-deoxy-d-glucose inhibits Kaposi's sarcoma-associated herpesvirus replication and gene expression. Antimicrob Agents Chemother 56:5794–5803. doi: 10.1128/AAC.01126-12. [DOI] [PMC free article] [PubMed] [Google Scholar]