

ABSTRACT
Adenovirus 14p1 (Ad14p1) is an emergent variant of Ad serotype 14 (Ad14) that has caused increased severity of respiratory illnesses during globally distributed outbreaks, including cases of acute respiratory distress syndrome and death. We found that human cell infection with Ad14p1 results in markedly decreased expression of the E1B 20-kilodalton (20K) protein compared to that with infection with wild-type (wt) Ad14. This reduced Ad14p1 E1B 20K expression caused a loss-of-function phenotype of Ad-infected cell corpses that, in contrast to cells infected with wt Ad14, either failed to repress or increased NF-κB-dependent, proinflammatory cytokine responses of responder human alveolar macrophages. A small-animal model of Ad14-induced lung infection was used to test the translational relevance of these in vitro observations. Intratracheal infection of Syrian hamsters with Ad14p1 caused a marked, patchy bronchopneumonia, whereas hamster infection with wt Ad14 caused minimal peribronchial inflammation. These results suggest that this difference in E1B 20K gene expression during Ad14p1 infection and its modulating effect on the interactions between Ad14-infected cells and the host innate immune response could explain the increased immunopathogenic potential and associated increase in clinical illness in some people infected with the Ad14p1 outbreak strain.
IMPORTANCE We previously reported that Ad-infected human cells exhibit E1B 19K-dependent repression of virally induced, NF-κB-dependent macrophage cytokine responses (J. R. Radke, F. Grigera, D. S. Ucker, and J. L. Cook, J Virol 88:2658–2669, 2014, http://dx.doi.org/10.1128/JVI.02372-13). The more virulent, emergent strain of Ad14, Ad14p1, causes increased cytopathology in vitro, which suggested a possible E1B 20K defect. Whether there is a linkage between these observations was unknown. We show that there is markedly reduced expression of E1B 20K in Ad14p1-infected human cells and that this causes an increased proinflammatory cytokine response of human alveolar macrophages and more severe inflammatory lung disease in infected hamsters. This is the first evidence of a clinical relevance of differential expression of the small Ad E1B gene product. The results suggest that there is a low, critical threshold of E1B 19/20K expression that is needed for viral replication and infection transmission but that a higher level of E1B 19/20K expression is required for the usual repression and control of the Ad-triggered host innate immune response.
INTRODUCTION
Human adenoviruses (Ads) are nonenveloped double-stranded DNA viruses that consist of at least 52 different serotypes, divided into seven different groups (A to G). Ad infection usually induces mild, self-limited infections in immunocompetent human hosts. However, outbreaks of emergent strains of Ad have resulted in severe and fatal infections in otherwise healthy individuals. Following the discontinuation of Ad vaccination in the U.S. military, there were numerous outbreaks of varying severity in military installations. Ad4 and Ad7 have been the major cause of these infections (1). At the same time, an emergent strain of Ad14 also appeared in U.S. military populations (1). These were the first instances in which Ad14 infection had been documented in the United States.
Ad14 is a member of the B2 subgroup of Ad. Members of the B2 group tend to cause kidney and urinary tract infections but have also been associated with respiratory illness. The emergent Ad14 strain (Ad14p1) was isolated from military personnel at geographically distinct locations. Sequencing of the isolates determined that the strains were 100% identical in the coding regions and differed by just 0.3% from the prototype Ad14 deWit strain (2). Currently, Ad14p1 is circulating worldwide and has been identified as the causative agent in numerous cases of acute respiratory distress syndrome (ARDS); these strains are also highly homologous to the military isolates (3–6). However, it is unclear why Ad14p1 infection can cause more severe forms of respiratory illness than the wild-type (wt) deWit strain of Ad14.
We have previously reported that cells dying as a result of wt Ad5 infection repress the host inflammatory response to the virus (7). Expression of the Ad E1B 19-kilodalton (19K) protein is required for this anti-inflammatory activity since cells infected with an Ad mutant that lacks expression of 19K fail to exhibit anti-inflammatory activity. Ad14 expresses an E1B 20K protein that is the equivalent of Ad5 E1B 19K (19/20K). We postulated that emergent Ad strains that induce ARDS might either lack sufficient expression of 19/20K or express a mutated 19/20K, resulting in an enhanced host proinflammatory response to Ad infection. Adenoviruses that express either reduced levels or a mutant form of 19/20K have a large-plaque (lp) phenotype and increased cytopathic effect (CPE) in cell culture (the so-called cyt phenotype) and generate CPE corpses that either fail to repress or induce increased proinflammatory responses of activated macrophages (7).
In this report we show that CPE corpses from wt Ad14-infected cells, similar to those from wt Ad5-infected cells, repress NF-κB-dependent transcription and cytokine responses, whereas Ad14p1 CPE corpses either fail to repress or induce such proinflammatory responses by human alveolar macrophages (HAM). E1B sequencing and complementation studies revealed that the difference in E1B 20K gene expression is sufficient to explain the observed difference in the innate immune inflammatory responses to wt Ad14- and Ad14p1-infected cells. Studies using a permissive hamster lung infection model revealed an in vivo correlation between the immunomodulatory activities associated with these viral infections and their induction of lung immunopathology. Animals infected with wt Ad14 had minimal, peribronchial inflammatory reactions. In contrast, animals infected with Ad14p1 had a marked, patchy bronchopneumonia, similar to that described in humans infected with this outbreak strain. We discuss the theoretical implications of the expression level dependence of E1B 19/20K for viral replication, transmission of infection, and infected-cell-mediated immunomodulation and virally induced immunopathology.
MATERIALS AND METHODS
Cell lines.
Human A549 and THP-1 cells were obtained from the ATCC; 293 NF-κB (293-κB) luciferase reporter cells were provided by David Ucker. A549 and 293 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% bovine calf serum (BCS). THP-1 cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS). All cells were cultured at 37°C in 5% CO2.
Viruses.
Prototype adenovirus type 14 de Wit (Ad14; VR-15) was obtained from the ATCC. Clinical isolates from U.S. military outbreaks were obtained from Kevin Russell at the U.S. Naval Health Research Center (isolates 1986T and 22039). Separate seed stocks of 1986T and 22039 were provided by Adriana Kajon (Lovelace Respiratory Research Institute). Virus stocks were grown in A549 cells. Adenovirus for hamster infection was purified by CsCl banding as previously described (8). Adenovirus titers were determined either by plaque assays in A549 cells or by quantitative PCR (qPCR) (see below) and expressed as PFU/ml.
Viral plaque assays.
A549 cells were plated at 5 × 105 cells per 60-mm tissue culture dish overnight and were then inoculated with 0.5 ml of serial, 10-fold dilutions of virus stocks and incubated for 1 h at 37°C, with rocking every 15 min. Three milliliters of complete DMEM containing 1.25% SeaPlaque GTG-agarose (Lonza) was then added to each dish, the viral inoculum was suspended in the DMEM-agarose mix by swirling, and the agarose was allowed to solidify at room temperature (RT). Plaque dishes were incubated at 37°C in 5% CO2 for 14 days, with the addition of 3 ml of DMEM-agarose every 4 to 5 days. Dishes were stained by the addition of 3 ml of 20% ethanol, 2% paraformaldehyde, and 1% crystal violet overnight at RT; plaques were counted, and plaque-forming titers were calculated and expressed as PFU/ml.
qPCR determination of viral titer.
Titers of Ad14 stocks were also determined by qPCR, using the method of Murata and colleagues (9). Ad14 stock at titers of 106 to 100 PFU, as determined by repeated plaque assays, was used to create standard curves. Test samples were serially diluted in diethyl pyrocarbonate (DEPC)-H2O to 10−8. Ad14 and Ad14p1 genomes were detected using the primers CGGAGCTGCCTGGACATG and GCTTTACAGGAATGGGCTTG, which amplify a portion of the E1A genome and are specific for group B adenovirus. PCR was performed with either iQ SYBR green or Sso Advanced SYBR green Supermix (Bio-Rad) on either a MyiQ or CFX 1000 system, respectively (Bio-Rad) (1× final concentration), with primers at a final concentration of 0.5 μM and 5 μl of a standard or test sample in a final reaction volume of 25 μl. The qPCR protocol was as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 0.5 min, 55°C for 1.5 min, and 72°C for 1 min. Data were collected during the 72°C annealing step.
Viral infection of cells and collection of CPE corpses.
A549 cells were infected for 1 h at 37°C in suspension with virus at a multiplicity of infection (MOI) of 10 PFU/cell, unless otherwise indicated, and then plated in 100-mm dishes and allowed to proceed to complete CPE. Floating cells were collected, and any remaining adherent cells were collected by incubation with EDTA at 37°C for 3 min. Cell corpses were washed twice in phosphate-buffered saline (PBS), fixed in 125 mM formaldehyde, and stored at 4°C if not used immediately. Before use in cell culture, fixed CPE corpses were washed twice in warmed complete medium.
Sequence analysis of Ad14p1 20K genes.
E1B genes from the 1986T and 22039 isolates of Ad14p1 were obtained from viral DNA by PCR using the primers ATGGAGGTTTGGGCCATTTTGGAA (forward) and AAACTCCGCCTCCTCCACT (reverse) and cloned into pC4 TOPO Vector for sequencing (Invitrogen). Sequence of the 20K genes was determined with M13 forward and reverse primers. Sequence data were then aligned with published sequences for both wt Ad14 de Wit (GenBank accession number AY803294) and Ad14p1 (GenBank accession number FJ822614) using DNA Workbench (CLC-bio/Qiagen) and were 100% identical to the published sequences.
Analysis of Ad14 E1B 20K protein expression.
A549 cells were infected with either Ad14 or Ad14p1 isolates at an MOI of 50 PFU/cell. At 24 h postinfection, cells were collected and lysed in radioimmunoprecipitation assay (RIPA) buffer. Actin (Sigma-Aldrich antibody) and Ad14 E1B 20K (mouse anti-Ad2 E1B 19K monoclonal antibody, 3D11; Calbiochem) were detected by immunoblotting using enhanced chemiluminescence (ECL).
Analysis of Ad14 E1B 20K transcription.
A549 cells were infected with either an Ad14 or Ad14p1 isolate at an MOI of 10 PFU/cell. At 24 h postinfection, cells were collected, and RNA was isolated with RNeasy kits (Qiagen). PrimeTime Standard qPCR assays from IDT were developed for the detection of Ad14 E1B 20K and beta-actin. Assays for detection of 20K used primers 5′-ATCCCGCAGACTCATTTCAG-3′ and 5′-ATTCCCGCTACACCCAAAG-3′ and probe 5′-56-FAM-ACGTTTTGG-ZEN-ATTTCGTAGCCACAGC-3IABkFQ/-3′ (where FAM is 6-carboxyfluorescein, ZEN is an internal quencher, and 3IABkFQ is a 3′ Iowa Black quencher). For beta-actin, the primers 5′-GGATGCCTCTCTTGCTCTG-3′ and 5′-CGTCTTCCCCTCCATCGTG-3′ and probe 5′-56-FAM-AATCCTTCT-ZEN-GACCCATGCCCACC-3IABkFQ-3′ were used. DNase-treated RNA was added to Brilliant II reverse transcription-qPCR (RT-qPCR) one-step master mix (Agilent), in the presence of either actin or E1B 20K primer/probe mixes. The qPCR protocol was as follows: 50°C for 30 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Data were collected during the third step. All qPCRs were performed in 96-well optical plates with optical adhesive seals (Bio-Rad).
Assay of NF-κB-dependent transcription.
Studies of the effects of cellular corpses on NF-κB-dependent transcription were done with 293 cells, stably transfected with an NF-κB-luciferase reporter, as described previously (10). Plated 293-κB-luciferase cells were stimulated for 18 h with 2 nM phorbol myristate acetate (PMA) in the absence or presence of Ad CPE corpses and were then washed with PBS and lysed for analysis of luciferase activity, which was determined by using a luciferase assay kit (Promega), with activity expressed as the fold induction of stimulated cells versus the level in unstimulated control cells.
Creation of stable Ad5 E1B 19K-expressing A549 cells.
Ad5 E1B 19K was cloned from the viral genome by PCR using the primers CACCATGGAGGCTTGGGAGTGTTTGG (forward) and AAATTCCCGAGGGTCCAGGCC (reverse). The PCR product was cloned in pENTR vector by TOPO cloning (Invitrogen) and then cloned into pcDNA DEST 40 vector (Invitrogen) by LR cloning, which introduced a C-terminal V5 tag, and the resulting vector (DEST-40 19K) was sequenced. A total of 2 × 106 A549 cells were electroplated with 2 μg of DEST-40 19K with a Gene Pulser II (Bio-Rad) in 0.4-cm gap cuvettes at 0.3 kV and 950 μF. Stable cell lines were selected in 1 mg/ml G418-containing DMEM with 5% BCS. Individual clones were selected and screened for 19K expression by Western blotting for the V5 tag (anti-V5; Invitrogen).
Differentiation of THP-1 monocytes into macrophages.
THP-1 monocytes were plated at a density of 105 cells per well in 24-well plates in complete RPMI medium containing 100 nM PMA and incubated for 3 days at 37°C. After 3 days, the medium was changed, and the differentiated THP-1 cells were used as described below for cytokine analysis.
Isolation of HAM.
Human alveolar macrophages (HAM) were obtained with Institutional Review Board (IRB) approval from excess bronchial alveolar lavage samples from HIV-negative patients without an infectious lung process. Cells from lavage samples were pelleted by centrifugation and washed twice with RPMI medium. Cell counts were determined on a TC20 cell counter (Bio-Rad), and the total numbers of macrophages were determined by gating on the cells between 9 and 20 μm in diameter. Cells were plated at a density of 2.5 × 104 per well of 24-well plates in RPMI medium–10% FBS. Macrophages were adhered overnight, and nonadherent cells were removed by repeated washing with fresh complete RPMI medium. HAM were allowed to rest for 3 days before use in experiments.
Cytokine/chemokine analysis.
Cytokine and chemokine (here referred to as proinflammatory mediator) production by HAM or PMA-differentiated THP-1 cells was determined using a Bio-Plex Suspension Array System (Bio-Rad). Adherent macrophages were tested either without or with stimulation with lipopolysaccharide (LPS; 1 μg/ml for 18 h) in the absence or presence of cell corpses at a ratio of 10 CPE corpses per macrophage. Medium was collected, cleared of debris by centrifugation, and frozen at −20°C for later proinflammatory mediator analysis.
Syrian hamster model of lung infection and immunopathogenesis.
Female Syrian hamsters, 4 to 5 weeks old, were obtained from Harlan Laboratories (Indianapolis, IN). After 1 week of rest, hamsters were mildly sedated with isoflurane to slow breathing and calm animals for infection. Infections were carried out with 50 μl of Ad14 or Ad14p1 stock (which corresponded to 5 × 109 PFU per animal) via intratracheal inoculation of panting hamsters. Hamsters were held in an upright, slightly recumbent position for 30 breaths to enhance inhalation of the viral inoculum. At 7 days postinfection, animals were sacrificed by CO2 narcosis, and lungs were removed, inflated and fixed with formalin, and embedded in paraffin. Lung sections were stained with hematoxylin and eosin (H&E). Slides were scored in a blinded manner by a board-certified clinical pathologist, with expertise in microbiology and infectious diseases. A second consecutive microtome slice from the paraffin block was stained for Ad-infected cells with a pan-anti-hexon antibody mix (adenovirus 20/11 and 2/6; Cell Marque). This hamster protocol was approved by both the Edward Hines, Jr., Veterans Administration (VA) Hospital and the Loyola-Stritch School of Medicine IACUC review committees.
Statistical analysis.
The significance of differences in comparative data was estimated with SigmaPlot software. Normal distribution was first assessed using the Kolmogorov-Smirnov test, and all sets of data displayed normal distribution. Student's t test was used to determine the significance of differences between two groups of data. For analysis of data with three groups, one-way analysis of variance (ANOVA) was first performed, followed by post hoc analysis by the Holm-Sidak method to determine significance of differences between two specific data sets. For all analyses, a P value of <0.05 was considered a significant difference.
RESULTS
Ad14p1 isolates exhibit large-plaque and cytocidal phenotypes.
Isolates of Ad14p1 were obtained from the U.S. Naval Health Research Center. For initial studies, we used Ad14p1 isolate 1986T from a fatal infection at Lackland Air Force Base in Texas and Ad14p1 isolate NHRC22039 from an outbreak at the Marine Corps Recruit Depot in California (2, 11). The isolates were propagated, and titers were determined by plaque assay in A549 cells. These Ad14p1 isolates induced heterogeneous but generally larger plaques in human cell monolayers than those induced by wt Ad14 (Fig. 1A). The heterogeneous nature of the plaque sizes was consistent through three rounds of plaque purification regardless of the size of the original plaque (data not shown). It is not uncommon for Ad variants to exhibit heterogeneous plaque sizes (12). The large-plaque (lp) phenotype is characteristic of loss of E1B 19K function (13). Ad14p1 isolates also induced a more rapid onset of cytopathic effect (CPE; cell rounding and loss of adherence) in human cells than wt Ad14 (Fig. 1B). This cytocidal (cyt) phenotype is another characteristic of reduced E1B 19/20K function (14). Mutations within the E1B 19/20K protein or decreased expression of E1B 19/20K is generally responsible for most lp Ad strains.
FIG 1.
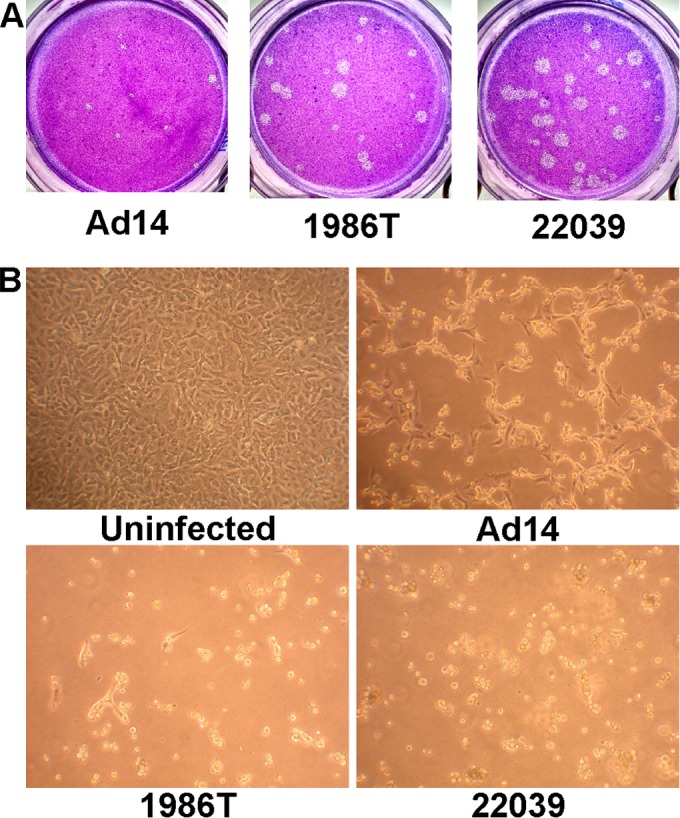
Ad14p1 isolates display an lp and slight cyt phenotype. (A) Plaque sizes of wt Ad14, and 1986T and 22039 Ad14p1 isolates on day 14 postinfection of A549 cells. (B) CPE phenotype of A549 cells 48 h after infection with wt Ad14 and 1986T and 22039 Ad14p1 isolates at an MOI of 10 PFU/cell.
The Ad14p1 E1B 20K gene contains a single, silent point mutation.
The DNA sequences of Ad14 de Wit and Ad14p1 have been determined previously (2, 15). Houng et al. have shown that the isolates 1986T (fatal isolate) and 22039 (mild outbreak) are nearly 100% identical, except for 5 bp (2). Comparison of the published sequences of Ad14 de Wit (GenBank Accession number AY803294) and Ad14p1 (GenBank Accession number FJ822614) showed that, within the E1B 20K coding region, there is one silent point mutation in Ad14p1 (Fig. 2). Sequences of the E1B 20K genes from Ad14p1 isolates 1986T and 22039 were 100% identical to the published sequence of Ad14p1 (GenBank accession number FJ822614), which is in agreement with the work of Houng et al. (2). Therefore, the observed lp and cyt traits observed in the Ad14p1 isolates could not be explained by a mutation that alters the function of the E1B 20K protein in cells infected with the Ad14p1 isolates.
FIG 2.

Ad14p1 contains a silent point mutation within the E1B 20K gene. The DNA sequences of wt Ad14 (GenBank Accession number AY803294) and Ad14p1 (GenBank Accession number FJ822614) were aligned for the E1B 20K gene. A single silent point mutation is shown in gray.
E1B 20K expression is reduced during Ad14p1 infection.
It has been demonstrated previously that mutations within the E1B 19K gene can alter 19K expression (12). Based on these observations and our results suggesting the possibility of altered E1B 20K function in Ad14p1-infected cells, we compared the expression of 20K in A549 cells infected with Ad14p1 isolate 1986T with that in prototype Ad14 de Wit and prototype Ad5 infection. Infection of A549 cells with the Ad14p1 isolate resulted in a marked decrease in 20K protein expression compared to 20K protein expression in A549 cells infected with Ad14 de Wit or Ad 5 (Fig. 3A). The observed reduction in 20K expression is similar to that reported for Ad5 lp mutants (12). To determine if decreased 20K expression in Ad14p1-infected cells was caused by decreased transcription of the E1B 20K gene, we developed a real-time PCR assay to quantitate E1B 20K mRNA expression in infected A549 cells. As shown in Fig. 3B, 20K mRNA levels in Ad14p1-infected cells were decreased by 80% compared to the 20K mRNA expression level in Ad14 de Wit-infected cells. The mechanism(s) of this marked reduction in E1B 20K transcription in Ad14p1-infected cells is unknown and could theoretically include any of the multifactorial explanations for reduced E1B promoter activity that have been postulated for other Ad serotypes (16).
FIG 3.
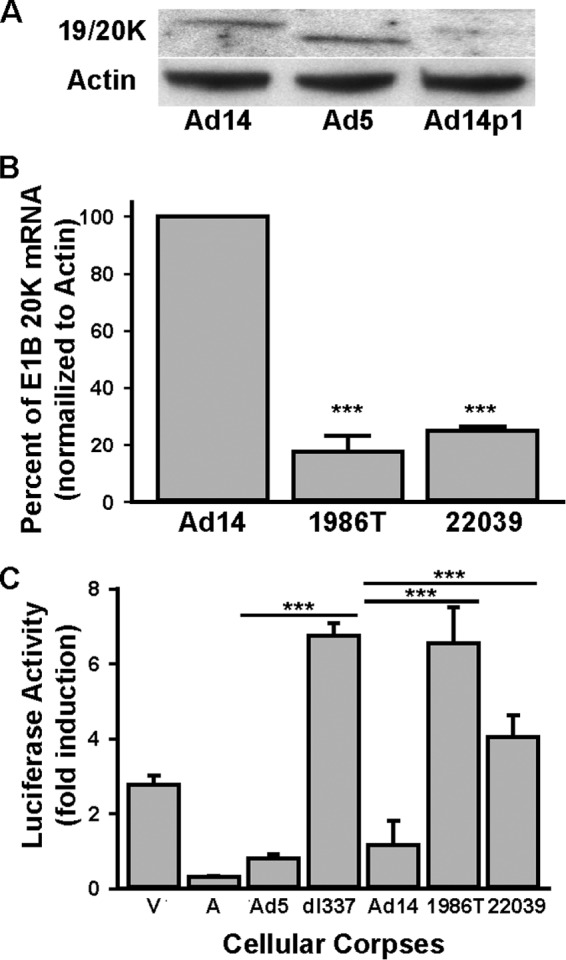
Reduced E1B 20K expression correlated with loss of Ad14p1 CPE corpse repression of NF-κB activation. (A) Immunoblot analysis of E1B 19/20K expression in A549 cells infected with wt Ad14, wt Ad5, or Ad14p1 clinical isolate 1986T. (B) RT-qPCR analysis of E1B 20K transcripts in A549 cells infected with either Ad14, 1986T, or 22039. Values are means ± standard errors of the means (n = 3; ***, P < 0.001, by a t test). (C) PMA-induced 293-κB luciferase reporter cell responses when cells were cocultured at a 1:10 ratio with viable (V) or staurosporine-treated, apoptotic (A) A549 cells or with Ad-CPE A549 corpses infected with the indicated viruses: wt Ad5, E1B 19K-negative Ad5 (dl337), wt Ad14, or the two Ad14p1 clinical isolates, 1986T and 22039 Values are means ± standard errors of the means (n = 3; ***, P < 0.001, by a t test).
Ad14p1 CPE corpses fail to repress NF-κB-dependent transcription in responder cells.
We reported that Ad5 E1B 19K function is required for Ad-infected, dying cells (CPE corpses) to repress stimulus-induced, NF-κB-dependent transcription and related cytokine production by responder macrophages, an E1B 19K function termed apoptotic mimicry (7). As observed with wt Ad5 infection, wt Ad14-infected human A549 (lung epithelial cell) CPE corpses repressed PMA-induced NF-κB-dependent transcription in reporter cells, whereas CPE corpses infected with either Ad14p1 clinical isolate failed to repress this response, in a pattern similar to that of infection with the 19K-deleted Ad mutant, H5 dl337 (Fig. 3C). All future studies of Ad14p1 were done using the 1986T isolate.
Expression of Ad5 E1B 19K in trans complements the reduction of Ad14p1 E1B 20K.
E1B 19K expressed in trans complements the loss of Ad5 E1B 19K for both lp and immunorepressive phenotypes (7, 14, 17). We therefore tested the complementation activity of Ad5 E1B 19K, stably expressed in human A549 cells, for reversal of the loss-of-function phenotypes of Ad14p1 (Fig. 4A). As seen in Fig. 4B and C, E1B 19K expressed in trans eliminated the lp phenotype of Ad14p1 and restored the immunorepressive activity of Ad14p1-infected CPE corpses. These data demonstrated cross-complementation of Ad5 E1B 19K for Ad14 E1B 20K function and showed that reduced E1B 20K expression of Ad14p1 in infected human cells is sufficient to explain the loss of immunorepressive activity of Ad14p1 CPE corpses.
FIG 4.
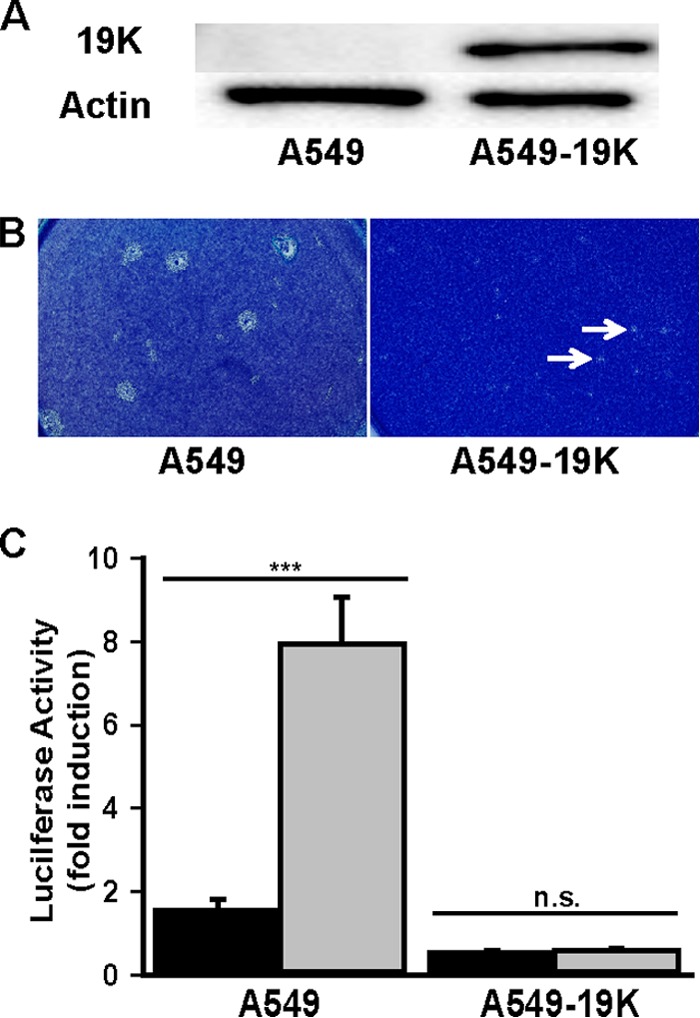
Restoration of NF-κB-repressive activity of Ad14p1 CPE corpses by E1B 19K complementation in trans. (A) Immunoblot of Ad5 E1B 19K expression in stably transfected A549 cells (A549-19K). (B) Plaque size of Ad14p1 in either A549 cells or A549 cells expressing Ad5 E1B 19K (A549-19K). Arrows indicate small plaques. (C) PMA-induced 293-κB luciferase reporter cell responses when cells were cocultured for 18 h at a 1:10 ratio with wt Ad14 (black bars) or Ad14p1 (gray bars) CPE corpses, using infected A549 or infected E1B 19K-expressing A549 (A549-19K) cells. Values are means ± standard errors of the means (n = 3; ***, P < 0.001; n.s., not significant, by a t test).
Ad14p1 CPE corpses have a net proinflammatory effect on human alveolar macrophages.
The human ARDS response to viral infection is associated with increased expression of the NF-κB-dependent cytokines, interleukin-1β (IL-1β), IL-6, IL-8, and tumor necrosis factor alpha (TNF-α) (18). We predicted that Ad14p1 CPE corpses would be relatively ineffective at repressing stimulus-induced macrophage production of these cytokines compared with the effect of wt Ad14 CPE corpses. To test this prediction, we isolated primary human alveolar macrophages (HAM) from bronchoalveolar lavage fluid and incubated the HAM with unfixed Ad CPE corpses to simulate in vivo conditions that would occur during viral infection. We have shown that Ad CPE corpse stimulation of macrophages is caused by Ad virions associated with (and contaminating) Ad CPE corpse preparations and that it is the balance between this virion stimulation and E1B-19K-dependent, Ad CPE corpse repression of macrophages that determines the net effect on NF-κB-dependent cytokine production (7; also unpublished data). In the current studies, Ad14p1 CPE corpses induced significantly increased HAM production of IL-6, IL-8, and TNF-α but had no effect on production of IL-β (Fig. 5A). In contrast, wt Ad14 CPE corpses had no net stimulatory effect on IL-6, IL-8, and TNF-α production by HAM and repressed IL-1β production. These differences in the immunomodulatory effects of Ad14p1 versus those of wt Ad14 CPE corpses were not explained by differences in the amounts of virus contaminating the CPE preparations. There were no differences in the particle counts of the infecting viruses or of the viral PFU associated with these cellular preparations (data not shown). To further test the macrophage immunomodulatory effects of Ad14p1 versus those of wt Ad14 CPE corpses, we contrasted their effects on HAM stimulated with LPS (Fig. 5B), a known inducer of macrophage, NF-κB-dependent, cytokine responses (19). Ad14p1 CPE corpses enhanced the LPS-induced IL-1β macrophage response, but wt Ad14 CPE corpses had no such stimulatory effect. wt Ad14 CPE corpses markedly repressed LPS-induced macrophage production of IL-6, IL-8, and TNF-α. For each of these three cytokines, Ad14p1 CPE cells were either significantly less repressive than wt Ad CPE corpses (IL-6 and IL-8) or did not repress LPS-induced production of the cytokine (TNF-α). These same relative effects of Ad14p1 versus wt Ad14 CPE corpses on macrophage cytokine production were observed with HAM from three human donors and with LPS-stimulated human THP-1 monocyte-derived macrophages (Fig. 5C). In summary, these results indicate that there is a difference in the interactions of Ad14p1 CPE corpses with human alveolar macrophages (versus that of wt Ad14 CPE corpses) that results from a combination of an increase of the basic macrophage cytokine production response to viral stimulation and a loss of repression of macrophage responses to other stimuli (here represented by LPS). Macrophages are the key innate immune effector cells that determine the intensity and regulation of the early lung inflammatory responses to viral infection (20). These in vitro studies of alveolar macrophages suggested that Ad14p1 lung infection would generate a greater cellular inflammatory response than infection with wt Ad14. A small-animal model of Ad14 lung infection was developed to test this hypothesis.
FIG 5.

Loss of macrophage-immunorepressive activity of Ad14p1 CPE corpses compared with that of wt Ad14 CPE corpses. (A and B) Supernatant cytokine concentrations of human alveolar macrophages cocultured with unfixed wt Ad14 or Ad14p1 A549 CPE corpses in the absence (A) or presence (B) of 1 μg/ml LPS for 18 h. Data from a single macrophage donor, representative of similar data from three individual donors, are shown. Values are means of triplicate cultures ± standard deviations. One-way ANOVA was performed, and P values were determined by a post hoc Holm-Sidak test. (C) Supernatant cytokine concentrations of PMA-differentiated human THP-1 cells cocultured with unfixed wt Ad14 or Ad14p1 CPE A549 corpses in the presence of 1 μg/ml LPS for 18 h. Values are means ± standard errors of the means (n = 4; P ≤ 0.002 for both comparisons, as determined by a t test).
Ad14p1 infection induces increased lung immunopathogenesis in infected Syrian hamsters.
Small-animal models of productive human Ad infection are limited to Syrian hamsters and cotton rats, both of which are permissive for Ad replication (21–26). We developed a hamster model of comparative, viral lung pathology to contrast the effects of Ad14p1 and wt Ad14 by infecting 4- to 5-week-old hamsters intratracheally with 5 × 109 PFU of each virus. Seven days later, lungs were fixed in formalin, sectioned, and stained with hematoxylin and eosin (H&E) or tested for Ad infection by immunohistochemistry. Gross inspection of Ad14p1-infected lungs revealed patchy, multifocal infiltrates, whereas wt Ad14 infection resulted in minimal, peribronchial reaction (Fig. 6A to C). Microscopic examination of Ad14p1-infected lungs revealed marked alveolar, peribronchial, perivascular, and interstitial mononuclear cell infiltrates, whereas wt Ad14-infected lungs showed limited peribronchial, mononuclear cell infiltrates (Fig. 6D to F). Adenovirus-positive cells were detected at sites of inflammation with both infections (Fig. 6G to I). Quantitation of virus in the lungs of infected hamsters revealed that there was no significant difference in the in vivo replication levels of Ad14 (2.2 × 106 ± 0.29 × 106 PFU/g) and Ad14p1 (1.78 × 106 ± 0.86 × 106 PFU/g). Preliminary data with earlier and later time points after hamster infection did not show any difference in the kinetics of viral infections or inflammatory responses between Ad14- and Ad14p1-infected animals. Specifically, there were no time points along the continuum through 2 weeks after infection where the inflammatory responses to Ad14 infection ever approached the intensity of the responses observed to Ad14p1 infection (unpublished data). The patchy bronchopneumonia observed in Ad14p1-infected hamsters is similar to the pattern of lung pathology described for human infection (4). These data provided the first link between altered viral gene expression during infection with Ad14p1 and increased lung immunopathology of this emergent Ad strain. The results indicate that reduced E1B 20K expression during Ad14p1 infection of human cells results in both loss of the immunomodulatory activity that is a general trait of human cells infected with wild-type (wt) adenovirus (7) and enhancement of virally induced immunopathogenesis.
FIG 6.

Comparative lung pathology induced by infection with wt Ad14 versus that with Ad14p1. Lungs were harvested from infected hamsters at 7 days after intratracheal viral inoculation (5 × 109 PFU). (A to C) H&E staining for macroscopic comparison. (D to F) Comparison of H&E staining results at a magnification of ×10. Green, yellow, and red arrows indicate perivascular, alveolar, and peribronchial inflammation, respectively. (G to I) Immunohistochemistry staining of Ad-infected cells with an anti-hexon antibody mix (adenovirus 20/11 and 2/6; Cell Marque). Black arrows point to brown, Ad-infected cells. Images are representative of similar findings in eight hamsters per condition.
DISCUSSION
We previously discovered a new function of the E1B 19K protein—apoptotic mimicry—that causes repression of the innate immune response to Ad infection, mediated by interactions between cells dying from Ad infection and responder, host macrophages (7). In that report, we postulated that Ad variants that induce increased inflammatory responses in vivo (like ARDS observed during clinical outbreaks) would express either insufficient E1B 19K or a mutant E1B 19K, resulting in a loss of the immunorepressive, apoptotic mimicry phenotype of infected cells.
In this report, we show that Ad14p1, an ARDS-inducing variant of wt Ad14, has lp and cyt phenotypes, indicative of reduced E1B 20K function (Fig. 1), and reduced expression of E1B 20K at the level of transcription in infected human cells (Fig. 2). This reduced E1B 20K expression caused a loss-of-function phenotype of Ad14p1 CPE corpses, manifested by their loss of immunorepressive activity for virion-stimulated and LPS-stimulated human alveolar macrophages, in contrast to the activity with wt Ad14 CPE corpses (Fig. 3 and 5). Complementation studies using Ad5 E1B 19K expressed in trans in human A549 cells eliminated the lp phenotype and restored the immunorepressive phenotype to Ad14p1 CPE corpses (Fig. 4), thereby indicating that no other genetic change in Ad14p1 was required to explain the loss Ad CPE corpse immunorepressive activity. Further studies are in progress of the effects on immunomodulatory activity in vitro and in vivo of restoring E1B 20K expression to wild-type levels in genetically engineered Ad14p1.
We propose that there is a link between the increased appearance of clinical outbreaks and virulence of Ad14p1 infections and the loss-of-function phenotypes caused by low-level E1B 20K expression. This could result from yet to be defined Ad mutations that result in a reduced, “critical threshold” of E1B 20K expression that is sufficient to sustain normal viral replication (and therefore infection transmission) but insufficient to convey immunomodulatory activity to infected cells. Complete loss of Ad5 E1B 19K expression results in a log reduction in viral replication (27). In contrast, the low but critical threshold of Ad14p1 E1B 20K expression does not reduce viral fitness (28) or viral replication in vivo (Fig. 6H and I) and can cause increased lung pathology (Fig. 6) and more severe clinical illness (1, 4, 6, 29–32).
We speculate that the ongoing molecular evolution of human adenoviral strains, such as Ad14, might result in variable levels of E1B 19/20K expression that would usually be “silent” clinically and would continue undetected during infection in the human population. We propose that mutant viruses that would express either no or subthreshold E1B 19/20K would be defective for viral replication and would not cause infections that could be sustained in the population. Conversely, mutant viruses that expressed only slightly lower than normal levels of E1B 19/20K might be indistinguishable clinically from wild-type virus because of their normal viral replication and infection transmission and the retained immunomodulatory effect of virally infected cells. An E1B 19/20K critical-threshold mutant virus in this scenario would be represented by Ad14p1, where E1B 20K expression is sufficient to sustain normal viral replication and infectivity but insufficient to convey immunorepressive activity to infected human cells, resulting in the potential for increased immunopathogenicity and therefore greater clinical illness and outbreaks of infection that would be more severe than usual. The model systems in this report provide a basis for studies of this hypothesis using other Ad outbreak strains by comparing E1B 19/20K gene expression with that of respective, wild-type Ad strains and by comparing the macrophage immunomodulatory activity of Ad CPE corpses, the cytokine-mediated respiratory tract inflammation in animal models, and the differences in the spectrum of clinical illnesses.
ACKNOWLEDGMENTS
Author contributions are as follows: J.R.R. conceived the experiments, performed all experiments, and wrote the paper; S.L.Y. developed the lung pathology scoring system and did all lung pathology interpretations; J.L.C. conceived the experiments, assisted with hamster experiments, and wrote the paper.
We thank Amit Goyal from Loyola University Medical Center for BAL fluid samples and David Ucker and Andrew M. Lewis, Jr., for their support and encouragement during this project.
The views expressed here are those of the authors and do not reflect the opinion or policy of the VA or the Government of the United States.
Funding Statement
Infectious Diseases and Immunology Research Institute—Loyola University Chicago (LUC) provided funding to Jay R. Radke through a Falk Foundation grant. LUC Stritch School of Medicine (SSOM) provided funding to James L. Cook through a Laboratory Development fund. Funding was also provided by a James and Marion Grant Fellowship in Immunology and Infectious Diseases.
REFERENCES
- 1.Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, Russell KL. 2007. Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. J Infect Dis 196:1465–1473. doi: 10.1086/522970. [DOI] [PubMed] [Google Scholar]
- 2.Houng H-SH, Gong H, Kajon AE, Jones MS, Kuschner RA, Lyons A, Lott L, Lin K-H, Metzgar D. 2010. Genome sequences of human adenovirus 14 isolates from mild respiratory cases and a fatal pneumonia, isolated during 2006-2007 epidemics in North America. Respir Res 11:116. doi: 10.1186/1465-9921-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang L, An J, Xie Z, Dehghan S, Seto D, Xu W, Ji Y. 2013. Genome and bioinformatic analysis of a HAdV-B14p1 virus isolated from a baby with pneumonia in Beijing, China. PLoS One 8:e60345. doi: 10.1371/journal.pone.0060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girouard G, Garceau R, Thibault L, Oussedik Y, Bastien N, Li Y. 2013. Adenovirus serotype 14 infection, New Brunswick, Canada, 2011. Emerging Infect Dis 19:119–122. doi: 10.3201/eid1901.120423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Flanagan D, O'Donnell J, Domegan L, Fitzpatrick F, Connell J, Coughlan S, De Gascun C, Carr MJ. 2011. First reported cases of human adenovirus serotype 14p1 infection, Ireland, October 2009 to July 2010. Euro Surveill 16(8):pii=19801. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19801. [PubMed] [Google Scholar]
- 6.Huang G, Yu D, Zhu Z, Zhao H, Wang P, Gray GC, Meng L, Xu W. 2013. Outbreak of febrile respiratory illness associated with human adenovirus type 14p1 in Gansu Province, China. Influenza Other Respir Viruses 7:1048–1054. doi: 10.1111/irv.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radke JR, Grigera F, Ucker DS, Cook JL. 2014. Adenovirus E1B 19-kilodalton protein modulates innate immunity through apoptotic mimicry. J Virol 88:2658–2669. doi: 10.1128/JVI.02372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tollefson AE, Kuppuswamy M, Shashkova EV, Doronin K, Wold WS. 2007. Preparation and titration of CsCl-banded adenovirus stocks. Methods Mol Med 130:223–235. [DOI] [PubMed] [Google Scholar]
- 9.Murata H, Teferedegne B, Lewis AM, Peden K. 2009. A quantitative PCR assay for SV40 neutralization adaptable for high-throughput applications. J Virol Methods 162:236–244. doi: 10.1016/j.jviromet.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. 2006. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J Biol Chem 281:20055–20067. doi: 10.1074/jbc.M603920200. [DOI] [PubMed] [Google Scholar]
- 11.Tate JE, Bunning ML, Lott L, Lu X, Metzgar D, Brosch L, Panozzo CA, Marconi VC, Faix DJ, Prill M, Johnson B, Erdman DD, Fonseca V, Anderson LJ, Widdowson MA. 2009. Outbreak of severe respiratory disease associated with emergent human adenovirus serotype 14 at a US Air Force training facility in 2007. J Infect Dis 199:1419–1426. doi: 10.1086/598520. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian T, Vijayalingam S, Chinnadurai G. 2006. Genetic identification of adenovirus type 5 genes that influence viral spread. J Virol 80:2000–2012. doi: 10.1128/JVI.80.4.2000-2012.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chinnadurai G. 1983. Adenovirus 2 Ip+ locus codes for a 19 kd tumor antigen that plays an essential role in cell transformation. Cell 33:759–766. doi: 10.1016/0092-8674(83)90018-1. [DOI] [PubMed] [Google Scholar]
- 14.White E, Grodzicker T, Stillman BW. 1984. Mutations in the gene encoding the adenovirus early region 1B 19,000-molecular-weight tumor antigen cause the degradation of chromosomal DNA. J Virol 52:410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seto J, Walsh MP, Mahadevan P, Purkayastha A, Clark JM, Tibbetts C, Seto D. 2009. Genomic and bioinformatics analyses of HAdV-14p, reference strain of a re-emerging respiratory pathogen and analysis of B1/B2. Virus Res 143:94–105. doi: 10.1016/j.virusres.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Mautner V, Bailey A, Steinthorsdottir V, Ullah R, Rinaldi A. 1999. Properties of the adenovirus type 40 E1B promoter that contribute to its low transcriptional activity. Virology 265:10–19. doi: 10.1006/viro.1999.0014. [DOI] [PubMed] [Google Scholar]
- 17.Chiou SK, Tseng CC, Rao L, White E. 1994. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol 68:6553–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aggarwal NR, King LS, D'Alessio FR. 2014. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 306:L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bastarache JA, Blackwell TS. 2009. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech 2:218–223. doi: 10.1242/dmm.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pribul PK, Harker J, Wang B, Wang H, Tregoning JS, Schwarze J, Openshaw PJM. 2008. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol 82:4441–4448. doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hjorth RN, Bonde GM, Pierzchala WA, Vernon SK, Wiener FP, Levner MH, Lubeck MD, Hung PP. 1988. A new hamster model for adenoviral vaccination. Arch Virol 100:279–283. doi: 10.1007/BF01487691. [DOI] [PubMed] [Google Scholar]
- 22.Prince GA, Porter DD, Jenson AB, Horswood RL, Chanock RM, Ginsberg HS. 1993. Pathogenesis of adenovirus type 5 pneumonia in cotton rats (Sigmodon hispidus). J Virol 67:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas MA, Spencer JF, La Regina MC, Dhar D, Tollefson AE, Toth K, Wold WSM. 2006. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res 66:1270–1276. doi: 10.1158/0008-5472.CAN-05-3497. [DOI] [PubMed] [Google Scholar]
- 24.Tollefson AE, Spencer JF, Ying B, Buller RML, Wold WSM, Toth K. 2014. Cidofovir and brincidofovir reduce the pathology caused by systemic infection with human type 5 adenovirus in immunosuppressed Syrian hamsters, while ribavirin is largely ineffective in this model. Antiviral Res 112:38–46. doi: 10.1016/j.antiviral.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Toth K, Lee SR, Ying B, Spencer JF, Tollefson AE, Sagartz JE, Kong I-K, Wang Z, Wold WSM. 2015. STAT2 knockout Syrian hamsters support enhanced replication and pathogenicity of human adenovirus, revealing an important role of type I interferon response in viral control. PLoS Pathog 11:e1005084. doi: 10.1371/journal.ppat.1005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toth K, Ying B, Tollefson AE, Spencer JF, Balakrishnan L, Sagartz JE, Buller RML, Wold WSM. 2015. Valganciclovir inhibits human adenovirus replication and pathology in permissive immunosuppressed female and male Syrian hamsters. Viruses 7:1409–1428. doi: 10.3390/v7031409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilder S, Logan J, Shenk T. 1984. Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular-weight polypeptide leads to degradation of viral and host cell DNA. J Virol 52:664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson BD, Barr KL, Heil GL, Friary JA, Gray GC. 2012. A comparison of viral fitness and virulence between emergent adenovirus 14p1 and prototype adenovirus 14p strains. J Clin Virol 54:265–268. doi: 10.1016/j.jcv.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter RN, Cantrell JA, Mallak CT, Gaydos JC. 2012. Adenovirus-associated deaths in US military during postvaccination period, 1999–2010. Emerg Infect Dis 18:507–509. doi: 10.3201/eid1803.111238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carr MJ, Kajon AE, Lu X, Dunford L, O'Reilly P, Holder P, De Gascun CF, Coughlan S, Connell J, Erdman DD, Hall WW. 2011. Deaths associated with human adenovirus-14p1 infections, Europe, 2009–2010. Emerg Infect Dis 17:1402–1408. doi: 10.3201/1708.101760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vento TJ, Prakash V, Murray CK, Brosch LC, Tchandja JB, Cogburn C, Yun HC. 2011. Pneumonia in military trainees: a comparison study based on adenovirus serotype 14 infection. J Infect Dis 203:1388–1395. doi: 10.1093/infdis/jir040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis PF, Schmidt MA, Lu X, Erdman DD, Campbell M, Thomas A, Cieslak PR, Grenz LD, Tsaknardis L, Gleaves C, Kendall B, Gilbert D. 2009. A community-based outbreak of severe respiratory illness caused by human adenovirus serotype 14. J Infect Dis 199:1427–1434. doi: 10.1086/598521. [DOI] [PubMed] [Google Scholar]