

ABSTRACT
The cyclic dinucleotide 2′,3′-cGAMP can bind the adaptor protein STING (stimulator of interferon [IFN] genes) to activate the production of type I IFNs and proinflammatory cytokines. We found that cGAMP added to the culture medium could suppress the replication of the hepatitis C virus (HCV) genotype 1b strain Con1 subgenomic replicon in human hepatoma cells. Knockdown of STING expression diminished the inhibitory effect on replicon replication, while overexpression of STING enhanced the inhibitory effects of cGAMP. The addition of cGAMP into 1b/Con1 replicon cells significantly increased the expression of type I IFNs and antiviral interferon-stimulated genes. Unexpectedly, replication of the genotype 2a JFH1 replicon and infectious JFH1 virus was less sensitive to the inhibitory effect of cGAMP than was that of 1b/Con1 replicon. Using chimeric replicons, 2a NS4B was identified to confer resistance to cGAMP. Transient expression of 2a NS4B resulted in a pronounced inhibitory effect on STING-mediated beta IFN (IFN-β) reporter activation compared to that of 1b NS4B. 2a NS4B was found to suppress STING accumulation in a dose-dependent manner. The predicted transmembrane domain of 2a NS4B was required to inhibit STING accumulation. These results demonstrate a novel genotype-specific inhibition of the STING-mediated host antiviral immune response.
IMPORTANCE The cyclic dinucleotide cGAMP was found to potently inhibit the replication of HCV genotype 1b Con1 replicon but was less effective for the 2a/JFH1 replicon and infectious JFH1 virus. The predicted transmembrane domain in 2a NS4B was shown to be responsible for the decreased sensitivity to cGAMP. The N terminus of NS4B has been reported to suppress STING-mediated signaling by disrupting the interaction of STING and TBK1 and/or MAVS. We show that 2a/JFH1 NS4B has an additional mechanism to evade STING signaling through suppressing STING accumulation.
INTRODUCTION
Hepatitis C virus (HCV) infects approximately 170 million people worldwide, leading to liver diseases, such as cirrhosis and hepatocellular carcinoma (1, 2). HCV can be classified into seven major genotypes and over 67 subtypes (3). Intense efforts over the past 20 years to understand the requirements for HCV replication have contributed to the development of several direct-acting antivirals (DAAs) that can cure genotype 1 HCV infection at a significant rate (4). However, differences exist among the various genotypes that could impact therapy (5–8). More research is needed to better understand HCV replication and to effectively inhibit infection by all genotypes.
HCV is an enveloped positive-strand RNA virus in the Flaviviridae family. The HCV genome is of approximate 9.6 kb in length and encodes a single open reading frame flanked by 5′ and 3′ untranslated regions (UTRs). The internal ribosome entry site (IRES) recruits ribosomes to translate viral RNA, producing a single polypeptide that is co- and posttranslationally processed into structural proteins (Core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) by viral and cellular proteases (9, 10). All HCV proteins are likely multifunctional, and Core, NS3, NS4B, NS5A, and NS5B have all been reported to modulate cellular responses to viral infection in addition to acting on viral RNA replication (10, 11).
HCV infection can activate the production of type I interferons (IFNs), which can result in the clearance of viral infection (12, 13). IFNs bind to cognate receptors and activate the transcription of IFN-stimulated genes (ISGs). The ISG-encoded proteins can inhibit virus infection by multiple mechanisms, including suppressing viral mRNA translation and replication and degradation of viral RNA (14, 15). Given the role of the innate immune response activated by IFNs, recombinant alpha IFN (IFN-α) is a part of the standard of care to treat HCV (2).
HCV uses several strategies to counter host antiviral immune response (11). The NS3/4A proteinase can cleave mitochondrial antiviral-signaling protein (MAVS; also known as IPS-1, VISA, and Cardif) and the adaptor protein TRIF (TIR-domain-containing adaptor inducing IFN-β) to abrogate RIG-I- and Toll-like receptor 3 (TLR3)-mediated innate immune signaling pathways (16–18). NS4B can interact with STING (stimulator of interferon gene, also known as MITA, MPYS, ERIS, and TMEM173) and disrupt the interaction of STING and downstream signaling effectors to block host antiviral immune responses (19, 20). The balance between activation and blockade of the immune response could be the key determinant for the outcome of HCV infection.
HCV NS4B can induce membrane rearrangements that serves as a scaffold for the viral replicase complex formation (10). NS4B consists of the N-terminal A domain (amino acids [aa] 1 to 79), a predicted transmembrane B domain (aa 80 to 191), and a C-terminal C domain (aa 192 to 261). The A domain has been reported to inhibit STING-mediated IFN-β activation (19) and is involved in virus production (21). The B domain consists of four predicted transmembrane helices that regulate the replication of HCV genome and virus production (22). The C domain interacts with NS4B itself, NS3, and the viral RNA to regulate RNA replication as well as virus production (23–25).
Cyclic dinucleotides c-di-GMP and c-di-AMP produced by bacteria are strong elicitors of host type I IFN production (26, 27) by binding to the adaptor protein STING (28). Recently, mammalian cells have been reported to express the enzyme cGAS (cyclic GMP-AMP synthase) to produce the 2′,3′-cyclic GMP-AMP (here abbreviated as cGAMP) that can activate type I IFN production (29–34). Although transfection and permeabilization methods are typically used to deliver the cyclic dinucleotides into cells, cGAMP can be added to the cell culture medium to activate IFN-β production in human monocyte THP1 cells (35). In addition, cGAMP can traffic between cells through the gap junctions to transmit antiviral responses between cells (36). These observations suggest that cGAMP could be potentially developed as an innate immune modulator to control viral infection.
We found that cGAMP added to the culture medium of Huh7.5 cells inhibited the genotype 1b Con1 replicon replication via the cGAMP-STING pathway. However, cGAMP was less effective in suppressing the genotype 2a JFH1 replicon replication than that of the 1b/Con1 strain. The 2a/JFH1 NS4B was found to suppress STING accumulation and confer resistance to cGAMP. The inhibitory effect of 2a NS4B on STING accumulation was primarily mapped to the B domain, suggesting that 2a NS4B has multiple mechanisms to abrogate STING-mediated antiviral immune response.
MATERIALS AND METHODS
Cell culture.
The human hepatoma cell line Huh7.5 (37), human embryonic kidney (HEK) 293T cells (ATCC), and 293-MSR cells (Invitrogen) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. Huh7.5 stable cell lines harboring replicons 1b/Con1 (pFKI389/NS3-3′/WT) and 1b/Con1-RLuc were cultured in DMEM-10% FBS containing 0.5 mg/ml G418 (Invitrogen) as described previously (38).
Cyclic dinucleotides.
Cyclic di-GMP (c-di-GMP) was from Invivogen. 2′,3′-Cyclic GMP-AMP (cGAMP) was synthesized with recombinant human cGAS catalytic domain using ATP and GTP substrates in the presence of salmon sperm DNA as described by Li et al. (35). cGAMP was purified by ultrafiltration and Q-Sepharose ion-exchange chromatography and eluted with a gradient of NH4HCO3. Purified cGAMP was lyophilized to remove NH4HCO3. All cyclic dinucleotides were dissolved in double-distilled water (ddH2O) at final concentration of 2 or 5 mg/ml.
Plasmid constructions.
Plasmid Luc-2a/JFH1 (22) served as the backbone to construct chimeric replicons using overlapping PCR. J1/C1-NS4B1b was made by replacing the NS4B coding sequence with that of 1b/Con1 NS4B. Chimeric replicons J1/C1-A1b, J1/C1-B1b, and J1/C1-C1b were made by replacement of the sequence coding for the N-terminal 79 codons, predicted transmembrane domain (codons 80 to191), and C-terminal domain (codons 192 to 261) of JFH1 NS4B, respectively, with the sequence from that of 1b/Con1. J1/GND was a replicon construct containing a mutation in the catalytic residues of NS5B (22). Plasmids pcDNA-1bNS4B and pcDNA-2aNS4B with a C-terminal hemagglutinin (HA) tag were constructed by PCR amplification and inserted into NheI/XhoI of pcDNA3.1. The pcDNA-STING with a Flag tag was previously described (39). For transient expression, chimeric NS4Bs, 2a/1b-A, 2a/1b-B, 2a/1b-C, 2a/1b-B(1-2), 2a/1b-B(3-4), and 1b/2a-B, were amplified by fusion PCR and constructed in the pcDNA3.1 vector.
Transient HCV replicon assay.
Plasmids containing the wild type (WT) or the ΔGDD mutant of HCV 1b/Con1 Renilla luciferase (1b/Con1-RLuc) replicon were linearized with ScaI while the 2a/JFH1-Luc replicon, chimeric replicons, and JFH1 full-length infectious cDNA clone were linearized with XbaI and then treated with mung bean nuclease to generate DNA with blunt ends (NEB). In vitro transcription used the AmpliScribe T7-Flash transcription kit (Epicentre Biotechnologies) with 1 μg of linearized DNA as the template. After a 2-h incubation at 37°C, the transcription reaction mixtures were treated with RNase-free DNase I for 30 min, extracted with a 1:1 phenol-chloroform mixture, and precipitated with ethanol. RNA pellets were washed with 75% ethanol, dried, and then dissolved in H2O. The RNA concentration was determined by spectrophotometry. The integrity and concentration were confirmed by agarose gel electrophoresis.
HCV replicon RNA was introduced into Huh7.5 cells by electroporation of trypsinized Huh7.5 cells washed with ice-cold Cytomix (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4-KH2PO4, 25 mM HEPES, 2 mM EGTA, 5 mM MgCl2; pH 7.6) and suspended at 2 × 106 cells/ml. The RNA and cells were added to a cuvette with a 2-mm gap and pulsed at 270 V, 960 μF, with a Gene Pulser (Bio-Rad). Electroporated cells were suspended in complete medium and seeded in a 96-well plate or a 24-well plate at 1.5 × 104 and 5 × 104 cells/well, respectively. The medium was replaced with complete medium containing indicated concentrations of cyclic dinucleotides and incubated for 48 h or 72 h. Luciferase expressed from the 1b/Con1 replicon-harboring Huh7.5 cells was measured using the Dual-Glo luciferase reporter assay (Promega) in a plate reader (BioTek). The WST-1 cell proliferation assay was performed according to the manufacturer's instructions (Clontech).
HCV 2a/JFH1 virus assay and real-time quantitative RT-PCR (qRT-PCR).
2a/JFH1 virus (22) infection of Huh7.5 cells (5 ×104 cells/well) was performed at a multiplicity of infection (MOI) of 0.01. The cells were infected with virus for 4 h and then washed with complete medium and treated with cGAMP for an additional 48 h. Total RNA was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's protocol, and specific RNAs were quantified by real-time reverse transcription-PCR (RT-PCR) using the appropriate primers listed in Table 1. Briefly, 1 μg of total RNA was reverse transcribed into cDNA using Moloney murine leukemia virus (M-MuLV) (NEB) and 4 μM 9-nucleotide (9-nt) random primer mix. Real-time PCR was performed with the Bio-Rad IQ SYBR green kit (Bio-Rad). The samples were heated to 95°C for 10 min, followed by 45 cycles of PCR of 15 s at 95°C, 20 s at 60°C, and 30 s at 72°C. The mRNA levels were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level, and the fold or percent change of RNA was compared to that of mock-treated controls as described by Livak and Schmittgen (40).
TABLE 1.
Primer pairs used for real-time RT-PCR
| Gene | Sequence (5′–3′) |
|
|---|---|---|
| Forward | Reverse | |
| GAPDH | GAGTCAACGGATTTGGTCGT | TGGGATTTCCATTGATGACA |
| Con1 | AGCCATGGCGTTAGTATGAGTGTC | ACAAGGCCTTTCGCGACCCAAC |
| JFH1 | AGCCATGGCGTTAGTATGAGTGTC | ACAAGGCCTTTCGCAACCCAAC |
| STING | GAGAGCCACCAGAGCACAC | CGCACAGTCCTCCAGTAGC |
| IFN-α | GTGAGGAAATACTTCCAAAGAATCAC | TCTCATGATTTCTGCTCTGACAA |
| IFN-β | AGCTGAAGCAGTTCCAGAAG | AGTCTCATTCCAGCCAGTGC |
| TNF-α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| MxA | CCCTTCCCAGAGGCAGCGGG | CTGATTGCCCACAGCCACTC |
| OAS | GGTGGTAAAGGGTGGCTCCTC | TCTGCAGGTAGGTGCACTCC |
| PKR | CCAGTGATGATTCTCTTGAGAG | CCCCAAAGCGTAGAGGTCCA |
Western blot assays.
Huh7.5 cells were seeded onto a 24-well plate at a density of 5 × 104/well and treated with cyclic dinucleotides for 48 or 72 h prior to lysis with radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% NP-40, and a cocktail of proteinase inhibitors (Roche). The lysates were clarified by centrifugation at 13,000 rpm for 30 min, and equal amounts of total protein were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane for Western blot assays. The primary monoclonal antibodies to detect NS5A and β-actin and the goat anti-mouse IgG coupled with horseradish peroxidase (HRP) were from Santa Cruz Biotech. Mouse anti-Flag monoclonal antibody and rat anti-HA antibody were from Sigma and Roche, respectively. Human STING, TBK1, and IRF3 antibodies were from Cell Signaling. Mouse antiubiquitin monoclonal antibody and mouse anti-human α-tubulin antibody were from Santa Cruz Biotech. Western blot signals were detected with ECL prime detection reagent (GE Healthcare) and quantified using the ChemiDoc system and software (Bio-Rad).
Combination of cGAMP with DAAs on HCV 1b/Con1 replicon replication.
Increasing concentrations of direct-acting HCV NS5B inhibitors sofosbuvir (a nucleotide analog; kind gift of Alios Biopharma) and NS5A inhibitor daclatasvir (Selleckchem) were added along with different doses of cGAMP to Huh7.5 cells harboring the 1b/Con1-RLuc replicon, and Renilla luciferase activity was determined at 48 h posttreatment. The 50% and 75% effective concentrations (EC50 and EC75, respectively) of each direct-acting antiviral (DAA) alone or combined with cGAMP were calculated and listed in Table 2. The effect of drug combination was determined using the Chu-Talalay method. The combination index (CI) at EC75 was calculated with CompuSyn software.
TABLE 2.
Inhibitory constants of DAAs alone and with cGAMP
| DAA | EC50 | EC75 |
|---|---|---|
| Sofosbuvir (μM) | ||
| Alone | 0.96 | 1.83 |
| Plus 50 μg/ml cGAMP | 0.13 | 0.60 |
| Fold change | 7.47 | 3.04 |
| Combination index | 1.02 | |
| Daclatasvir (pM) | ||
| Alone | 14.44 | 34.92 |
| Plus 50 μg/ml cGAMP | 1.78 | 10.45 |
| Fold change | 8.08 | 3.34 |
| Combination index | 1.00 |
Knockdown and overexpression of STING.
STING knockdown was performed by transfection of STING-specific small interfering RNA (siRNA) (Santa Cruz Biotechnology) into Huh7.5 cells harboring 1b/Con1-RLuc replicon using Lipofectamine RNAiMax (Invitrogen) at a final concentration of 40 nM. At 48 h posttransfection, the mRNA level of STING was analyzed using quantitative RT-PCR (qRT-PCR), and protein level was determined by Western blotting. The siRNA-transfected cells were then trypsinized and seeded in 96-well plates. cGAMP was added at indicated concentrations, and the Renilla luciferase activity was determined at 48 h posttreatment. For cells overexpressing human STING, 1b/Con1 replicon cells were transfected with pcDNA-STING for 48 h and then treated with cGAMP. After an additional 48 h, luciferase activity was measured.
IFN-β luciferase reporter assays.
STING-mediated activation of the IFN-β promoter was performed as described in the work of Yi et al. (39). 293T cells were seeded at 4 × 104 cells/well for 24 h in 96-well plates prior to transfection with a mixture of plasmids that can express firefly luciferase regulated by the IFN-β or interferon-stimulated response element (ISRE) promoter elements (pIFN-FFLuc or pISRE-FFLuc; 30 ng), Renilla luciferase driven by the constitutive thymidine kinase (TK) promoter (phRL-TKRen; 2 ng), STING (pcDNA-STING; 10 ng), and various amounts of pcDNA-NS4B to express NS4B. All transfections were amended with the empty pcDNA3.1 plasmid to allow equal amounts of plasmid to be transfected. Transfections were performed with Lipofectamine 2000 (Invitrogen). Firefly and Renilla luciferase activities were determined using the Dual-Glo luciferase reporter assay (Promega). The ratio of firefly luciferase to Renilla luciferase activities was normalized to the level of the vector control and plotted as relative fold induction.
Indirect immunofluorescence assay (IFA).
The 1b/Con1-RLuc-harboring Huh7.5 cells were seeded on glass coverslips in 6-well plates overnight and then treated with cGAMP for 48 h. The cells were washed with phosphate-buffered saline (PBS) and fixed for 15 min in 4% paraformaldehyde (in PBS) at room temperature followed by 0.5% Triton X-100 treatment at 4°C for 15 min to permeabilize the cells. The fixed cells were stained with monoclonal NS5A antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:200; Molecular Probes). After three washes with PBS-Tween (PBST), the coverslips were mounted onto glass slides with Vectashield (Vector Laboratories) and sealed with clear nail polish. For the NS4B and STING colocalization experiment, Huh7.5 cells were seeded in glass coverslips in a 6-well plate overnight and then were transfected with indicated amounts of pcDNA-NS4B or chimeras along with pcDNA-STING for 24 h. The cells were fixed and stained with rat anti-HA and mouse anti-Flag antibodies following by Alexa 647-conjugated goat anti-rat IgG antibody and Alexa 488-conjugated goat anti-mouse IgG antibody. Immunostained samples were visualized with a Leica SP5 confocal microscope.
Immunoprecipitation assay.
293T cells were seeded onto 6-well plates and transfected with pcDNA-STING-Flag (1 μg) and pcDNA-1bNS4B-HA (1 μg) by Lipofectamine 2000. After 24 h, the cells were washed and lysed in RIPA buffer with a cocktail of proteinase inhibitors (Roche). Cell lysates were clarified by centrifugation at 13,000 rpm for 30 min at 4°C. Immunoprecipitation used either a rat-anti-HA monoclonal antibody or a mouse anti-Flag monoclonal antibody and protein A/G magnetic beads (Thermo Scientific) according to the manufacturer's protocol. The immunoprecipitated materials were dissolved in 1× Laemmli buffer and heated at 95°C for 5 min prior to separation by SDS-PAGE and detection via immunoblotting.
Statistical analysis.
The data were analyzed using the Student t test. Data were expressed as an average of triplicates with standard deviation (error bar), unless stated otherwise. P values of <0.05 were considered statistically significant.
RESULTS
cGAMP inhibits HCV 1b/Con1 replicon replication.
Huh7.5 cells harboring a 1b/Con1 replicon that expresses the Renilla luciferase (1b/Con1-RLuc) were used to examine whether cGAMP could inhibit HCV replication. cGAMP was added into the culture medium in increasing concentrations, and Renilla luciferase activity was determined at 48 h posttreatment. cGAMP, but not c-di-GMP, inhibited luciferase activity from 1b/Con1 in a concentration-dependent manner. The effective concentration for 50% reduction (EC50) by cGAMP was ∼50 μg/ml (Fig. 1A). cGAMP had a minor impact on cell proliferation and viability when added up to 80 μg/ml, as determined by measuring the mitochondrial dehydrogenase activity using the WST-1 reagent (Fig. 1A). The 50% cytotoxicity concentration (CC50) for cGAMP was in excess of 400 μg/ml (data not shown).
FIG 1.

cGAMP can inhibit 1b/Con1 replicon replication in Huh7.5 cells. (A) cGAMP suppresses 1b/Con1-RLuc replication. Renilla luciferase, expressed from the replicon, was measured at 48 h after the addition of either cGAMP or c-di-GMP to the medium. The data were plotted as percent inhibition relative to mock-treated sample. Cell viability data (WST-1) were added to the same graph by determining the optical density at 450 nm according to the manufacturer's protocol. The cells were treated with cGAMP in parallel with those used in the luciferase assay. The data were expressed as an average of triplicates with standard deviation (error bar). (B) Viral RNA level in replicon-harboring cells in the presence of increasing cGAMP concentrations. The viral RNA was quantified by real-time RT-PCR. The results were normalized to the level of GAPDH mRNA. (C) Western blot analysis of NS5A level in cGAMP-treated 1b/Con1-RLuc replicon cells. The NS5A was detected with mouse monoclonal anti-NS5A antibody and HRP-conjugated goat anti-mouse IgG. β-Actin served as the loading control. (D) Indirect immunofluorescence analysis of NS5A expression in replicon cells. The replicon cells were treated with cGAMP for 48 h and stained with mouse anti-NS5A monoclonal antibody and Alexa Fluor 488-conjugated goat anti-mouse antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue).
To confirm that cGAMP has inhibitory effects on HCV replication, we determined the level of 1b/Con1 viral RNA using quantitative RT-PCR. The viral RNA level decreased with the increasing concentrations of cGAMP added to the medium (Fig. 1B). A reduction in HCV NS5A protein accumulation was also detected via Western blotting and immunofluorescence assays (Fig. 1C and D).
cGAMP and direct-acting antivirals additively inhibit 1b/Con1 replicon replication.
To examine whether the antiviral activity of cGAMP could act in concert with direct-acting antivirals (DAAs) to inhibit HCV RNA replication, the NS5B polymerase inhibitor sofosbuvir, a nucleotide analog (41), and daclatasvir, an NS5A inhibitor (42), were tested in combination with different concentrations of cGAMP for 48 h. In the presence of 50 μg/ml of cGAMP, cells treated with daclatasvir or sofosbuvir decreased the EC50 by more than 7-fold (Table 2). The combination index (CI) for cGAMP with sofosbuvir or daclatasvir was 1.02 or 1 (Table 2), respectively, suggesting that the inhibitor combinations had an additive effect. These results demonstrate that cGAMP could enhance the inhibitory effects of DAAs on 1b/Con1 replicon replication.
STING is required for the inhibitory effect of cGAMP.
Cyclic dinucleotides can trigger type I IFNs and proinflammatory responses through several proteins, including STING, DDX41, and NLRP3 (26, 43, 44). To determine whether STING is required for the inhibitory effect of cGAMP, we used siRNA to knock down the endogenous STING in 1b/Con1 replicon cells and evaluated the effect of cGAMP on HCV replication. The STING protein level was only 5% of that of U937 human leukemic monocyte lymphoma cells (data not shown) but could be detected in the 1b/Con1 replicon-harboring Huh7.5 cells (Fig. 2A). The knockdown by siRNA decreased the STING mRNA and protein levels by 75% and 50%, respectively (Fig. 2A). Knockdown of STING increased 1b/Con1 replicon replication by ∼1.7-fold even without the exogenous cGAMP (Fig. 2B), suggesting that STING is involved in restricting 1b/Con1 replication in Huh7.5 cells. The knockdown of STING dramatically diminished the inhibitory effect of cGAMP on 1b/Con1 replicon replication (Fig. 2C).
FIG 2.

Effects of knockdown and overexpression of STING on cGAMP-mediated inhibition of HCV 1b replicon replication. (A) Knockdown of endogenous STING. The replicon-harboring Huh7.5 cells were transfected with 40 nM human STING siRNA or control siRNA. Forty-eight hours later, the mRNA level of STING or GAPDH was determined by real-time RT-PCR, and the protein level of STING or α-tubulin was determined by Western blotting. (B) Impact of STING knockdown on HCV 1b/Con1 replicon replication. The Renilla luciferase level in the siRNA-transfected replicon cells was determined at 48 h posttransfection and plotted as the percentage of control siRNA. The data were plotted as an average of triplicates with standard deviation (error bar). The asterisk denotes a P value of <0.05. (C) Effects of STING knockdown on the inhibitory effect of 1b/Con1 replicon replication by cGAMP. The replicon cells were treated with siRNA for 48 h and then treated with cGAMP. Renilla luciferase activity expressed from the replicon was measured at 48 h posttreatment and plotted relative to the mock-treated sample. The black circles and white squares denote cells treated with control siRNA and STING siRNA, respectively. (D) Overexpression of human STING in replicon-harboring Huh7.5 cells. The replicon cells were transfected with pcDNA-STING plasmid for 48 h. The mRNA level and protein level of STING were determined by quantitative RT-PCR and Western blotting. (E) Effect of STING overexpression on HCV 1b/Con1 replicon replication. The asterisk denotes a P value of <0.05. (F) Effect of STING overexpression on the inhibitory effect of 1b/Con1 replicon replication by cGAMP. The black circles and white squares denote cells transfected with pcDNA vector and pcDNA-STING plasmid, respectively.
The effect of STING overexpression on HCV Con1 replicon replication was also examined. Transfection of pcDNA-STING plasmid into replicon cells reduced 1b/Con1 RNA level by 35 to 40% even in the absence of cGAMP (Fig. 2D and E). In the presence of cGAMP, the EC50 was ca. 20 μg/ml for replicon cells overexpressing STING, while it was 50 μg/ml for cells with endogenous levels of STING (Fig. 2F and 1A). Results from both knockdown and overexpression of STING demonstrate that STING mediates the inhibition of HCV 1b/Con1 replicon replication in Huh7.5 cells.
cGAMP triggers type I IFN and antiviral ISG expression.
STING interacts with TBK1 and IRF3 to promote the phosphorylation of IRF3 that leads to type I interferon production (45). To examine whether cGAMP could activate the antiviral innate immune response in the Huh7.5 cells harboring the 1b/Con1 replicon, the mRNA levels for various IFNs and interferon-stimulated genes (ISGs) were determined by real-time RT-PCR 48 h after cGAMP treatment. Relative to the mock-treated cells, the mRNA levels for IFN-α, IFN-β, and tumor necrosis factor alpha (TNF-α) were increased by 6-, 12-, and 26-fold, respectively, in cells treated with 80 μg/ml of cGAMP (Fig. 3A). The mRNA levels of antiviral ISGs MxA and OAS were increased by 6- and 5-fold, respectively, compared to that of mock-treated samples (Fig. 3B). Notably, the increase of the cytokines and ISG mRNA levels correlated with a decrease in HCV 1b/Con1 RNA levels (Fig. 1B and data not shown). These results suggest that cGAMP activates the host antiviral innate immune response that likely results in the inhibition of 1b/Con1 replication.
FIG 3.
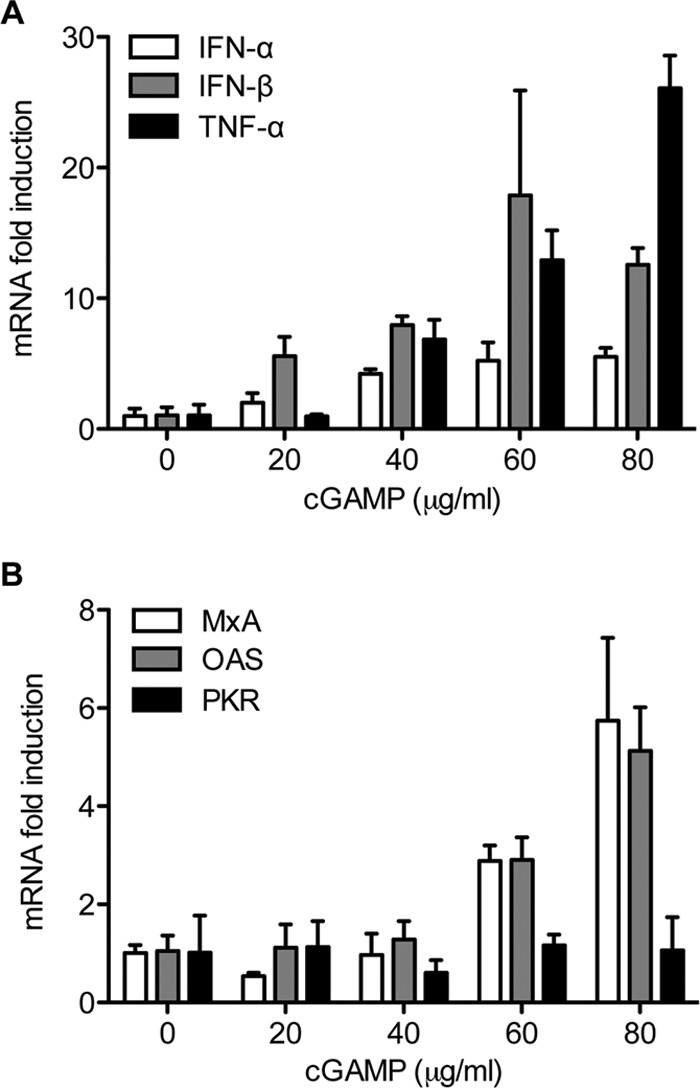
cGAMP triggers the expression of type I IFNs and ISGs in 1b/Con1 replicon-harboring Huh7.5 cells. (A) Effects of cGAMP on IFN-α, IFN-β, and TNF-α mRNA expression. Approximately 5 × 104 replicon cells were seeded on a 24-well plate and treated with cGAMP for 48 h. Total RNA was isolated and reverse transcribed into cDNA with random primers. cDNA was quantified by real-time PCR with the specific primer pairs listed in Table 1. The data were plotted as fold change from triplicate compared to that of mock-treated sample. (B) Effects of cGAMP on ISG MxA, OAS, and PKR mRNA levels.
cGAMP is less effective in reducing 2a/JFH1 replicon replication.
Different genotypes of HCV are known to be differentially sensitive to direct-acting antivirals (6–8). Therefore, we examined whether cGAMP could inhibit the replication of the genotype 2a JFH1 replicon. Huh7.5 cells electroporated with 1b/Con1-RLuc or 2a/JFH1-Luc replicon RNA were treated with increasing concentrations of either c-di-GMP or cGAMP for 72 h, and the replication efficiency was determined. The replication of the 1b/Con1-RLuc was inhibited by cGAMP with an EC50 of 50 μg/ml. However, cGAMP was not effective in reducing replication of the 2a/JFH1 replicon (Fig. 4A).
FIG 4.

Effects of cyclic dinucleotides on genotype 2a JFH1 replicon and infectious virus replication. (A) Effects of cyclic dinucleotides on 2a/JFH1 replicon replication. Huh7.5 cells were electroporated with in vitro-transcribed replicon RNA and seeded onto a 96-well plate. After 24 h, the medium was changed and the cells were treated with indicated concentrations of cyclic dinucleotides for an additional 48 h. Luciferase activity was measured, and the data were plotted relative to the mock-treated control. The data represent two independent experiments, each with triplicate samples. The open circle and triangle represent c-di-GMP and cGAMP, respectively. The dashed line represents the result from 1b/Con1 replicon, which served as the control. (B) Effects of cyclic dinucleotides on JFH1 infectious virus replication. Huh7.5 cells were infected with JFH1 virus at an MOI of 0.01 FFU/cell for 4 h and then treated with indicated concentrations of cyclic dinucleotides. Viral RNA was quantified by real-time RT-PCR 48 h posttransfection and normalized to GAPDH. The data represent the percentage of virus replication compared to mock-treated sample.
We also examined the effects of cyclic dinucleotides on Huh7.5 cells infected with the genotype 2a JFH1 virus. Huh7.5 cells were infected with the virus at a multiplicity of infectivity (MOI) of 0.01 and treated with indicated concentrations of c-di-GMP or cGAMP for 48 h. The JFH1 viral RNA was only modestly inhibited in cells treated with 100 μg/ml of cGAMP as determined by qRT-PCR (Fig. 4B). In agreement with the results from 2a/JFH1 replicon, these data indicate that the replication of 2a/JFH1 is less sensitive to the effects of cGAMP than that of 1b/Con1.
The 2a/JFH1 NS4B suppresses STING accumulation.
HCV has several strategies to modulate the innate immune responses (11). For example, NS4B was reported to inhibit the STING-mediated type I IFN response (19, 20). We therefore determined the subcellular distribution of NS4B and STING in cells coexpressing the two proteins. As shown in Fig. 5A, both 1b and 2a NS4Bs colocalized with STING in Huh7.5 cells. Furthermore, coimmunoprecipitation experiments showed that 1b/Con1 and 2a/JFH1 NS4Bs associated with STING in 293T cells (Fig. 5B). This has led us to postulate that NS4Bs from 1b/Con1 and 2a/JFH1 have distinct abilities to inhibit STING-mediated innate immune responses. To test this hypothesis, the 293T cells were transfected to express increasing amounts of either 1b or 2a NS4B along with a fixed amount of STING, IFN-β–firefly luciferase reporter, and TK-Renilla luciferase reporter. While both NS4B proteins reduced firefly luciferase expressed from the IFN-β promoter, 2a NS4B exhibited a more pronounced inhibitory effect than did 1b NS4B (Fig. 5C). 2a NS4B expressed from 20 ng of transfected plasmid reduced luciferase activity to background, while the comparable amount of 1b NS4B decreased luciferase readout by only 15% (Fig. 5C). A similar result was obtained with a luciferase reporter driven by an interferon-stimulated response element (ISRE) (Fig. 5D).
FIG 5.

2a NS4B can downregulate STING accumulation. (A) Colocalization of NS4B with STING in Huh7.5 cells. The cells were transfected with STING-Flag plasmid along with equal amounts of either 1b NS4B-HA or 2a NS4B-HA plasmid. After 30 h, the cells were processed for fluorescence microscopy with anti-HA antibody (NS4B; red) and anti-Flag antibody (STING; green). The fluorescence was visualized with a Leica SP5 confocal microscope. (B) Immunoprecipitation of NS4B and STING in 293T cells. The cells were transfected with equal amounts of the empty vector or STING-Flag and either 1b NS4B-HA or 2a NS4B-HA plasmid. The cell lysate was immunoprecipitated with mouse anti-Flag monoclonal antibody followed by immunoblotting with rat anti-HA antibody. The STING and NS4B protein levels in cell lysates were probed with anti-Flag and anti-HA antibodies. (C) Effects of transient expression of NS4B proteins from 1b and 2a on STING-mediated IFN-β reporter expression in 293T cells. The cells were transfected with constant amounts of STING-Flag, IFN-β firefly luciferase (IFN-β-FLuc) reporter, and TK-Renilla luciferase (TK-RLuc) reporter together with indicated amounts of either 1b NS4B-HA or 2a NS4B-HA. The FLuc and RLuc activities were measured, and the ratio of FLuc to RLuc was determined. The data were plotted as fold induction for the IFN promoter activity in the presence of STING with or without NS4B protein. (D) Effects of NS4B expression on ISRE reporter expression. The experiment was performed as in panel C except that the transfection mixture included ISRE-Luc reporter. (E) Western blot analysis of STING accumulation in the presence of NS4B in 293T cells. The cells were transfected with fixed amount of STING-Flag along with increasing concentrations of either 1b NS4B-HA or 2a NS4B-HA. Cell lysate was subjected to SDS-PAGE followed by Western blotting with Flag- and HA-specific antibodies. α-Tubulin served as a loading control. The STING level was quantified and normalized to α-tubulin using ImageQuant software. (F) Effects of NS4B expression on STING accumulation in Huh7.5 cells. The experiment was performed as in panel E except that Huh7.5 cells were transfected. (G) Effects of 2a NS4B/fs mutant expression on STING and IRF3 accumulation in HEK293T cells. (H) Effects of NS4B expression on endogenous STING accumulation in 293 cells. The cells were transfected with increasing concentrations of NS4B, and the endogenous levels of STING, TBK1, and IRF3 were examined by Western blotting. (I) Effects of NS4B on endogenous STING accumulation in Huh7.5 cells.
To examine whether NS4B proteins could differentially affect STING accumulation, 293T cells were transfected with a fixed amount of STING plasmid along with increasing concentrations of either 1b or 2a NS4B. As seen in Fig. 5E (right), 2a NS4B suppressed STING accumulation in a dose-dependent manner. In contrast, 1b NS4B had only a modest effect on the STING accumulation (Fig. 5E, left). Similar results were obtained from Huh7.5 cells (Fig. 5F). To exclude the possibility that the mRNA of 2a NS4B might mediate the inhibitory effect, we introduced a stop codon at amino acid position 22 of NS4B, named 2a NS4B/fs. The 2a NS4B/fs mutant dramatically diminished the inhibitory effect (Fig. 5G). Both WT and 2a NS4B/fs had only modest effects on IRF3 accumulation (Fig. 5G). This result suggests that 2a NS4B protein, instead of mRNA, is required to suppress STING accumulation.
Next, we sought to examine whether 2a NS4B could inhibit the endogenous STING accumulation using HEK293 cells, which express relatively lower levels of endogenous STING (46). Increasing expression of 2a NS4B, but not 1b NS4B, suppressed the endogenous STING accumulation, while TBK1 and IRF3 levels were only modestly affected (Fig. 5H). A similar result was observed in Huh7.5 cells (Fig. 5I). Since STING is required for the inhibition of 1b/Con1 replicon replication by cGAMP, the downregulation of STING accumulation by 2a NS4B could account for the decreased sensitivity to cGAMP by the 2a/JFH1 replicon.
2a/JFH1 NS4B confers resistance to the inhibitory effect of cGAMP.
We used chimeric replicons to determine whether 2a/JFH1 NS4B could confer resistance to cGAMP. The J1/C1-NS4B1b chimeric replicon had the 2a/JFH1 NS4B coding sequence replaced with the comparable sequence from 1b/Con1 NS4B (Fig. 6A). The replication of J1/C1-NS4B1b at 72 h postelectroporation was examined relative to the replication-competent JFH1 replicon and the J1/GND replicon, which encodes a catalytically inactive NS5B (Fig. 6B). J1/C1-NS4B1b replicated at levels about 5- to 10-fold lower than those of the 2a/JFH1 replicon, consistent with a previous report (22). In the presence of 75 μg/ml cGAMP, the luciferase expression from the J1/C1-NS4B1b chimera decreased by 68% to the level of the mock-treated control (Fig. 6C). The replication of the 1b/Con1 replicon was decreased to near background. A similar inhibitory effect was observed at 72 h posttreatment (data not shown). This result demonstrates that the NS4B proteins from the two different HCV genotypes can affect the inhibition by cGAMP.
FIG 6.
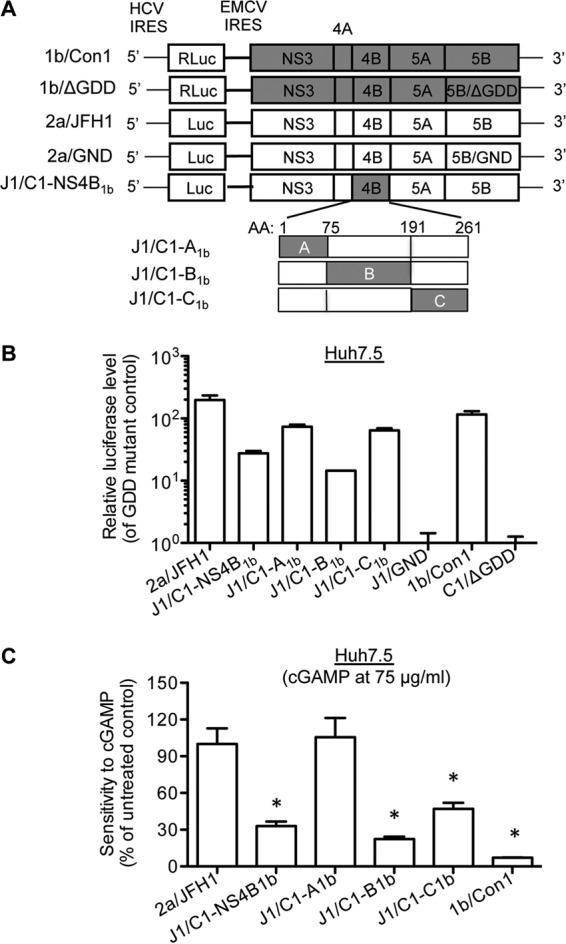
NS4B contributes to the distinct sensitivities of 1b/Con1 and 2a/JFH1 replicons to cGAMP. (A) Schematic diagram of chimeric replicons. 2a/JFH1-Luc replicon contains HCV 5′ UTR with its IRES directing firefly luciferase expression and the NS3-5B protein-coding region under the control of the encephalomyocarditis virus (EMCV) IRES. J1/C1-NS4B1b is a chimeric replicon that contains full-length 1b/Con1 NS4B in the 2a/JFH1-Luc backbone. J1/C1-A1b, J1/C1-B1b, and J1/C1-C1b represent the chimeric replicons in which the A, B, and C domains of 2a NS4B were replaced with those of 1b NS4B. (B) Replication of parental and chimeric replicons in Huh7.5 cells. Huh7.5 cells were electroporated with the indicated replicon RNAs. At 48 h posttransfection, the luciferase activity was measured and plotted as fold induction relative to the catalytic site mutant GND or ΔGDD. (C) Effects of cGAMP on the chimeric replicon replication in Huh7.5 cells. Huh7.5 cells were seeded onto a 96-well plate and electroporated with replicon RNA for 24 h. The cells were then mock treated or treated with 75 μg/ml of cGAMP for 48 h, and the luciferase activity was determined. The data were expressed as an average from triplicates and plotted as a percentage of the mock-treated samples. The statistical analysis of the inhibitory effects relative to the 2a/JFH1 replicon was calculated using the Student t test. The asterisk denotes a P value of <0.05.
NS4B contains three domains (Fig. 6A). To determine the domains in NS4B that confer the resistance to cGAMP, we used chimeric replicons in 2a/JFH1 backbone that had exchanges of the three 2a NS4B domains with those of 1b NS4B (Fig. 6A). In the absence of cGAMP, the replication level of J1/C1-A1b, which has the A domain of 2a NS4B replaced with that of 1b NS4B, was similar to that of J1/C1-C1b, which has the C domain of 2a NS4B replaced with that of 1b NS4B (Fig. 6A and B). J1/C1-B1b, which has the B domain from 1b/Con1 NS4B, replicated to levels about 10-fold lower than those of 2a/JFH1 (Fig. 6B). In the presence of 75 μg/ml of cGAMP, replication of J1/C1-B1b was significantly inhibited, similarly to that of J1/C1-NS4B1b, while J1/C1-A1b was unaffected compared to the mock-treated control (Fig. 6C). The replication of J1/C1-C1b was also inhibited by 53%, suggesting that the B and C domains of 2a NS4B contribute to the resistance to cGAMP.
The B domain of 2a NS4B is required to inhibit STING accumulation.
To identify the domain(s) in 2a NS4B required to inhibit STING accumulation, the chimeric NS4Bs were constructed in the pcDNA vector (Fig. 7A). Expression of the chimeric 2a/1b-B protein (2a NS4B with the B domain from 1b) abrogated the inhibitory effect of 2a NS4B on STING, while 2a/1b-A protein (2a NS4B with the A domain from 1b) decreased STING accumulation to a level similar to that of WT 2a NS4B (Fig. 7B), suggesting that the B domain primarily contributes to the inhibition of STING accumulation. 2a/1b-C also partially suppressed STING accumulation but less efficiently than did WT 2a NS4B. Comparable results were observed in 293T cells (Fig. 7C).
FIG 7.

The inhibitory effect of NS4B on STING accumulation is primarily mapped to the B domain. (A) Schematic representation of parental and chimeric NS4B constructs. A, B, and C denote the three domains of the NS4B protein. (B) Effects of NS4B chimeric constructs on STING accumulation. Huh7.5 cells were transfected with fixed amounts of STING and increasing concentrations of NS4B chimeras for 30 h. The cell lysate was separated by SDS-PAGE followed by Western blotting. Tubulin level served as a loading control. The relative level of STING was quantified and normalized to α-tubulin with ChemiDoc software. (C) Effects of NS4B subdomain chimeras on STING accumulation. The experiment was performed as in panel B, except that 293T cells were transfected. (D) Effects of NS4B domain B helices on STING accumulation in 293T cells. (E) Effect of 1b/2a-B NS4B expression on STING accumulation. (F) Effects of NS4Bs and chimera 2a/1b-B on the endogenous STING accumulation in 293 cells. 293 cells were transfected with increasing concentrations of either 2a NS4B, 1b NS4B, or 2a/1b-B plasmid. The endogenous STING, TBK1, and IRF3 levels were detected by Western blotting. (G) Effects of NS4B chimera on the inhibition of IFN-β-Luc reporter expression. 293T cells were transfected with fixed amounts of STING, IFN-β-Luc, and TK-Luc along with increasing concentrations of either NS4B or its chimeras. After 24 h, luciferase activity was determined and the data were plotted as IFN-β fold induction relative to the cells treated with 1b/Con1 NS4b. Data were expressed as an average of triplicates with standard deviation of the mean. (H) Effects of 1b/2a-B on the IFN-β reporter expression. The asterisk denotes a P value of <0.05.
The NS4B B domain consists of four predicted transmembrane helices. To further map the elements required for the inhibition of STING accumulation, the sequences that form helices 1 and 2 and helices 3 and 4 of 2a/1b-B NS4B were replaced with those from 2a NS4B and named 2a/1b-B(1-2) or 2a/1b-B(3-4) NS4B, respectively (Fig. 7A). While 2a/1b-B NS4B did not inhibit STING accumulation, 2a/1b-B(1-2) and 2a/1b-B(3-4) proteins could partially restore the ability to suppress STING accumulation (Fig. 7D). This result suggests that transmembrane domain helices 1 and 2 and helices 3 and 4 of 2a NS4B both contribute to the inhibition of STING, presumably through the helix-helix interactions in the transmembrane domain (47). Next, we replaced the B domain of 1b NS4B with the 2a counterpart (named 1b/2a-B [Fig. 7A]) to test whether the B domain of 2a NS4B was sufficient to enable 1b NS4B to suppress STING accumulation. The 1b NS4B protein had minor effects on STING accumulation as expected, while the chimeric 1b/2a-B protein inhibited STING accumulation to a level similar to that of 2a NS4B (Fig. 7E), indicating that the B domain of NS4B is primarily responsible for the distinct regulation of STING accumulation.
To further determine whether the B domain of 2a NS4B could affect the accumulation of endogenous STING, 293 cells were transfected with increasing concentrations of either 2a NS4B, 2a/1b-B, or 1b NS4B. The endogenous levels of STING and downstream signaling molecules TBK1 and IRF3 were examined by Western blotting. 2a NS4B could suppress STING but had only modest effects on TBK1 and IRF3 accumulation, while 2a/1b-B dramatically decreased the inhibitory effect on STING accumulation (Fig. 7F).
To confirm these results, chimeric constructs were tested in the STING-mediated IFN-β luciferase reporter assay in 293T cells. As expected, 2a NS4B inhibited IFN-β promoter activation more efficiently than 1b NS4B (Fig. 7F). The chimeric protein 2a/1b-B dramatically decreased the ability of 2a NS4B to inhibit IFN-β reporter expression (Fig. 7F) and exhibited an inhibitory effect similar to that of 1b NS4B. In addition, the 1b/2a-B chimera exhibited the same inhibitory effect as that of 2a NS4B (Fig. 7G). Altogether, these results demonstrate that the B domain of 2a NS4B primarily contributes to the genotype-specific suppression of STING accumulation.
DISCUSSION
In this work, we found that cGAMP could inhibit HCV genotype 1b/Con1 replicon replication in Huh7.5 cells through activation of STING-mediated type I IFNs and cytokine production. cGAMP was less effective in inhibiting the replication of the genotype 2a JFH1 replicon or the infectious JFH1 virus. The distinct effects of cGAMP are at least partially due to the different abilities of 1b and 2a NS4Bs to inhibit STING signaling through suppressing STING accumulation. The predicted transmembrane domain of 2a NS4B, the B domain, was primarily responsible for the resistance to cGAMP and suppression of STING accumulation.
cGAS can bind DNA from pathogens such as herpes simplex virus and HIV (cDNA) and produce cGAMP to trigger the host innate immune response (48, 49). However, cGAS cannot sense and bind RNA, and the antiviral role of cGAS and its product cGAMP for RNA viruses is still not clear. Nevertheless, overexpression of cGAS could regulate the replication of an RNA virus, West Nile virus (50). Our results demonstrate that the cGAS product cGAMP could directly inhibit HCV genotype 1b Con1 replicon replication when it was added into the cell medium. The inhibitory effect was not observed with bacterial second messenger c-di-GMP, likely due to c-di-GMP not being able to enter cells. cGAMP has been reported to bind the recombinant STING protein with a Kd (dissociation constant) of 5 to 60 nM while c-di-GMP bound to STING with a Kd of 1 to 5 μM (34, 35). The distinct phosphodiester linkages between the dinucleotides could also affect binding to STING as well as their sensitivities to phosphodiesterases (31, 32, 34).
STING is a key player in the response to the DNA and RNA virus-activated signaling pathway (51, 52). Similarly to cGAS, STING cannot directly bind RNA in vitro, raising questions as to how STING can recognize an RNA virus. Since STING can bind MAVS, TBK1, and IRF3, the downstream molecules of the RNA sensors RIG-I and MDA5 (45, 51, 53), it is possible that STING triggers the innate immune response through cross talk with RNA sensors. STING has been reported to restrict the replication of a number of RNA viruses, including Japanese encephalitis virus, dengue virus, hepatitis C virus, and coronavirus (19, 54–56). Our results confirm that STING can restrict HCV replication in Huh7.5 cells. Knockdown of STING enhanced HCV 1b/Con1 replicon replication, while its overexpression inhibited HCV replication in Huh7.5 cells (Fig. 2B and E). Huh7.5 cells contain a mutant RIG-I protein and a defective RIG-I signaling pathway (57); therefore, the restriction of HCV replication by STING is likely independent of RIG-I.
The inhibition of HCV 2a/JFH1 replication by cGAMP was not as pronounced as that of 1b/Con1. Results with chimeric replicons suggest that the NS4B protein contributes to the distinct inhibition by cGAMP through suppressing STING accumulation. NS4Bs from different genotypes thus have different capacities to evade STING-mediated innate immune responses. The ability of NS4B to inhibit STING accumulation and cGAMP-mediated immune responses could potentially influence the outcome of HCV infection. Indeed, knockdown of STING expression enhanced 1b/Con1 replicon replication by 60 to 70% (Fig. 2B). The chimeric replicons J1/C1-NS4B1b and J1/C1-B1b, which contain full-length 1b/Con1 NS4B and the predicted transmembrane domain of 1b NS4B, respectively, were more susceptible to the inhibitory effect of cGAMP and replicated at levels 5- to 10-fold lower than that of the WT 2a/JFH1 replicon. However, not only is NS4B involved in membranous web formation, associated with other nonstructural proteins, NS3, NS5A, and NS5B, to form the replicase complex, but also it affects virion assembly (21, 25, 58). Therefore, we cannot conclude that the defect of replication by NS4B chimeric replicon J1/C1-NS4B1b was merely due to the different regulation of STING accumulation.
RNA viruses have evolved different mechanisms to antagonize the innate immune response by STING. Dengue virus NS2B/3 can bind and cleave human STING (54, 59). Nitta et al. (19) and Ding et al. (20) reported that HCV NS4B can suppress the type I interferon response through disruption of STING interaction with downstream signaling molecule TBK1 or MAVS (19, 20). However, they did not observe a decrease of STING accumulation. Our results with HCV 1b NS4B are consistent with those of Nitta et al. (19) and Ding et al. (20). However, we observed that 2a NS4B could antagonize STING-mediated signaling through decreasing STING accumulation (Fig. 5E and F). Furthermore, we showed that 2a NS4B suppression of STING accumulation is strictly dependent on the level of STING and NS4B expression (Fig. 5E and F). Ding et al. (20) have not detected the decrease of STING accumulation by 2a NS4B; this could be due to the large amount of STING protein expressed or an insufficient amount of NS4B plasmid transfected in cells. We note that NS4B from 2a/JFH1 could account for a significant portion of resistance to cGAMP; however, it is possible that other HCV molecules could also contribute to resistance through association with NS4B. While HCV NS4B has different strategies to suppress STING-mediated antiviral signaling, whether STING signaling is activated and whether STING contributes to the interferon response during HCV infection in patients remain to be determined.
The inhibitory effect of 2a NS4B was mapped to the B and C domains that contain, respectively, the predicted transmembrane domain and the RNA binding domain. The B domain is primarily responsible for the inhibitory effect. Sequence alignment shows that NS4Bs of the 7 major genotypes contain 44% identity. However, NS4Bs from 1b/Con1 and 2a/JFH1 share 72% amino acid identity. The B domains and C domains of 1b/Con1 and 2a/JFH1 share 77% and 73% amino acid identity, respectively. The arginine residues in the C domain of NS4B have been shown to be involved in RNA binding (60); however, they are conserved in different genotypes of HCV, suggesting that RNA binding might not be primarily responsible for the distinct inhibition of STING accumulation by NS4Bs. The B domain contains four predicted transmembrane helices; how this domain contributes to the inhibition of STING accumulation remains to be determined.
Last, we demonstrated that cGAMP added to the cell medium could enhance antiviral activity through activating IFNs and antiviral ISG production. The use of cGAMP to treat HCV infection could have advantages from the use of recombinant IFN. First, cGAMP could trigger the production of multiple subtypes of IFNs, ISGs, and cytokines, such as TNF-α expression (Fig. 3A). Second, cGAMP can traffic from cell to cell to expand the innate immune response to neighbor cells (36). Third, cGAMP contains distinct 2′-5′ phosphodiester linkages between the dinucleotides, which could increase the stability in cells. Patients with genotype 1 HCV infection are the most difficult to cure, and a combination of cGAMP with DAAs can enhance the antiviral effect in vitro (Table 2). It may be worthwhile to examine the antiviral effect of cGAMP and the combined effects of cGAMP and DAAs in treatment of other viral diseases.
ACKNOWLEDGMENTS
We thank members of the Kao lab for helpful discussions during this work.
Funding Statement
This work was partially supported by NIAID.
REFERENCES
- 1.Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 2.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 3.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment Web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiser JJ, Flexner C. 2013. Direct-acting antiviral agents for hepatitis C virus infection. Annu Rev Pharmacol Toxicol 53:427–449. doi: 10.1146/annurev-pharmtox-011112-140254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng L, Hirata Y, Arai M, Kohara M, Wakita T, Watashi K, Shimotohno K, He Y, Zhong J, Toyoda T. 2010. Sphingomyelin activates hepatitis C virus RNA polymerase in a genotype-specific manner. J Virol 84:11761–11770. doi: 10.1128/JVI.00638-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li YP, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J. 2014. Differential sensitivity of 5′UTR-NS5A recombinants of hepatitis C virus genotypes 1-6 to protease and NS5A inhibitors. Gastroenterology 146:812–821. doi: 10.1053/j.gastro.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Clark PJ, Thompson AJ, Vock DM, Kratz LE, Tolun AA, Muir AJ, McHutchison JG, Subramanian M, Millington DM, Kelley RI, Patel K. 2012. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology 56:49–56. doi: 10.1002/hep.25631. [DOI] [PubMed] [Google Scholar]
- 8.Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079. doi: 10.1053/j.gastro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Lindenbach BD, Rice CM. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 10.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 11.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredericksen B, Akkaraju GR, Foy E, Wang C, Pflugheber J, Chen ZJ, Gale M Jr. 2002. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol 15:29–40. doi: 10.1089/088282402317340215. [DOI] [PubMed] [Google Scholar]
- 13.Kanda T, Steele R, Ray R, Ray RB. 2007. Hepatitis C virus infection induces the beta interferon signaling pathway in immortalized human hepatocytes. J Virol 81:12375–12381. doi: 10.1128/JVI.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foy E, Li K, Wang C, Sumpter R Jr, Ikeda M, Lemon SM, Gale M Jr. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 17.Foy E, Li K, Sumpter R Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A 102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, Kiyohashi K, Murakawa M, Nishimura-Sakurai Y, Azuma S, Tasaka-Fujita M, Asahina Y, Yoneyama M, Fujita T, Watanabe M. 2013. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57:46–58. doi: 10.1002/hep.26017. [DOI] [PubMed] [Google Scholar]
- 20.Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J. 2013. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 59:52–58. doi: 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Gouttenoire J, Montserret R, Paul D, Castillo R, Meister S, Bartenschlager R, Penin F, Moradpour D. 2014. Aminoterminal amphipathic alpha-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog 10:e1004501. doi: 10.1371/journal.ppat.1004501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han Q, Aligo J, Manna D, Belton K, Chintapalli SV, Hong Y, Patterson RL, van Rossum DB, Konan KV. 2011. Conserved GXXXG- and S/T-like motifs in the transmembrane domains of NS4B protein are required for hepatitis C virus replication. J Virol 85:6464–6479. doi: 10.1128/JVI.02298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paredes AM, Blight KJ. 2008. A genetic interaction between hepatitis C virus NS4B and NS3 is important for RNA replication. J Virol 82:10671–10683. doi: 10.1128/JVI.00875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Q, Manna D, Belton K, Cole R, Konan KV. 2013. Modulation of hepatitis C virus genome encapsidation by nonstructural protein 4B. J Virol 87:7409–7422. doi: 10.1128/JVI.03523-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burdette DL, Vance RE. 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. 2013. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. 2013. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi G, Deval J, Fan B, Cai H, Soulard C, Ranjith-Kumar CT, Smith DB, Blatt L, Beigelman L, Kao CC. 2012. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob Agents Chemother 56:830–837. doi: 10.1128/AAC.05438-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. 2013. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One 8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 42.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, Liu ZJ, Modlin RL, Liu YJ, Cheng G. 2012. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 13:1155–1161. doi: 10.1038/ni.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abdul-Sater AA, Tattoli I, Jin L, Grajkowski A, Levi A, Koller BH, Allen IC, Beaucage SL, Fitzgerald KA, Ting JP, Cambier JC, Girardin SE, Schindler C. 2013. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep 14:900–906. doi: 10.1038/embor.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. 2009. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A 106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li E, Wimley WC, Hristova K. 2012. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim Biophys Acta 1818:183–193. doi: 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A. 2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nazmi A, Mukhopadhyay R, Dutta K, Basu A. 2012. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci Rep 2:347. doi: 10.1038/srep00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, Clementz MA, Banach BS, Li K, Baker SC, Chen Z. 2012. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20:117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 59.Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL. 2012. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SR. 2008. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat Biotechnol 26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]