

ABSTRACT
Intrinsic immunity is an aspect of antiviral defense that operates through diverse mechanisms at the intracellular level through a wide range of constitutively expressed cellular proteins. In the case of herpesviruses, intrinsic resistance involves the repression of viral gene expression during the very early stages of infection, a process that is normally overcome by viral tegument and/or immediate-early proteins. Thus, the balance between cellular repressors and virus-counteracting proteins determines whether or not a cell becomes productively infected. One aspect of intrinsic resistance to herpes simplex virus 1 (HSV-1) is conferred by components of promyelocytic leukemia nuclear bodies (PML NBs), which respond to infection by accumulating at sites that are closely associated with the incoming parental HSV-1 genomes. Other cellular proteins, including IFI16, which has been implicated in sensing pathogen DNA and initiating signaling pathways that lead to an interferon response, also respond to viral genomes in this manner. Here, studies of the dynamics of the response of PML NB components and IFI16 to invading HSV-1 genomes demonstrated that this response is extremely rapid, occurring within the first hour after addition of the virus, and that human Daxx (hDaxx) and IFI16 respond more rapidly than PML. In the absence of HSV-1 regulatory protein ICP0, which counteracts the recruitment process, the newly formed, viral-genome-induced PML NB-like foci can fuse with existing PML NBs. These data are consistent with a model involving viral genome sequestration into such structures, thereby contributing to the low probability of initiation of lytic infection in the absence of ICP0.
IMPORTANCE Herpesviruses have intimate interactions with their hosts, with infection leading either to the productive lytic cycle or to a quiescent infection in which viral gene expression is suppressed while the viral genome is maintained in the host cell nucleus. Whether a cell becomes lytically or quiescently infected can be determined through the competing activities of cellular repressors and viral activators, some of which counteract cell-mediated repression. Therefore, the events that occur within the earliest stages of infection can be of crucial importance. This paper describes the extremely rapid response to herpes simplex virus 1 infection of cellular protein IFI16, a sensor of pathogen DNA, and also of the PML nuclear body proteins PML and hDaxx, as revealed by live-cell microscopy. The data imply that these proteins can accumulate on or close to the viral genomes in a sequential manner which may lead to their sequestration and repression.
INTRODUCTION
Whether or not a cell becomes productively infected with herpes simplex virus 1 (HSV-1), as with other herpesviruses, depends on many factors that modulate the initial stages of infection. Among these are cellular proteins that respond in a restrictive manner to repress viral gene expression once the viral genomes have entered the nucleus, while the virus expresses proteins that counteract these repressive effects or stimulate viral gene expression more directly. Over the last decade, it has become clear that one class of restricting cellular factors comprises a number of components of promyelocytic leukemia nuclear bodies (PML NBs, also known as ND10), including PML itself, Sp100, human Daxx (hDaxx), and ATRX (reviewed in references 1, 2, and 3). The HSV-1 immediate-early ICP0 protein is responsible for overcoming restriction mediated by these proteins through mechanisms that require its E3 ubiquitin ligase activity (reviewed in reference 1). HSV-1 mutants that are unable to express active ICP0 have a very low probability of initiating lytic infection in restrictive cell types (4–6) but are able to replicate more efficiently in cells depleted of one or more of these PML NB proteins (7–10).
There is considerable evidence that the restrictive effects of PML NB components depend on their dynamic response to infection. PML, Sp100, and hDaxx are recruited to sites that are closely associated with HSV-1 genomes during the earliest stages of infection (9, 11) by mechanisms that involve sumoylation and/or their ability to interact with sumoylated proteins and which are inhibited by ICP0 (7–9, 12). It is likely that other cellular proteins that accumulate on or near HSV-1 genomes in a SUMO pathway-dependent manner will be identified in the future, and because ICP0 causes a wide-ranging reduction in the levels of sumoylated species during infection (13–15), their recruitment may also be sensitive to ICP0. Interestingly, although PML is required for the assembly of PML NBs in uninfected cells (16, 17), it is not required for recruitment of either hDaxx or Sp100 to viral genomes, and these proteins may indeed be recruited independently (7–9). Recruitment-defective mutants of PML and hDaxx, unlike their wild-type (wt) counterparts, are unable to reverse the stimulatory effects on ICP0-null mutant HSV-1 replication of short hairpin RNA (shRNA)-mediated knockdown of the endogenous proteins (9, 12, 18).
The signals that initiate the recruitment of PML NB components to HSV-1 genomes remain unknown. It is possible that recruitment is related to a DNA repair response, and indeed, several DNA repair proteins respond to infection in similar manners (19), but recruitment of PML still occurs in DNA repair-deficient cells (19). More recently, cellular DNA sensor IFI16 (20, 21) was also implicated in the initiation of pathways that are inhibitory to HSV-1, either through initiating events leading to an interferon response (21, 22) or through more-direct effects on viral gene expression (23–26). ICP0-null mutant HSV-1 has increased plaque-forming potential in cells depleted of IFI16 (23, 25). IFI16 is degraded during HSV-1 infection by a mechanism that was originally thought to be ICP0 dependent (22), although later work showed that ICP0 was neither sufficient nor necessary for this degradation (23, 27). Indeed, cellular factors (and cell type) also contribute to the stability of IFI16 during HSV-1 infection (23, 24, 26). Nonetheless, under normal experimental conditions, IFI16 is degraded more rapidly during a wild-type (wt) infection than during an ICP0-null mutant infection, unless the latter is conducted at a very high multiplicity of infection (MOI). Like the PML NB components, IFI16 also localizes to HSV-1 genomes during the early stages of infection (22–28), and ICP0 inhibits this process (23).
In order to study the dynamic responses of this group of cellular proteins during the very earliest stages of HSV-1 infection, analyses were conducted in live cells expressing near-endogenous levels of PML, hDaxx, or IFI16, or combinations thereof, tagged with enhanced yellow fluorescent protein (EYFP), enhanced cyan fluorescent protein (ECFP), or a dual-fusion protein that expresses red fluorescence constitutively and, in addition, green fluorescence after photoactivation (PA). These studies revealed the extremely rapid and in some cases transient responses of these proteins during HSV-1 infection. Within the first hour after addition of the virus, IFI16 was observed to form distinct but highly transient foci that are almost certainly associated with HSV-1 genomes. This occurred with both wt and ICP0-null mutant viruses, but in the latter case, a second and more stable phase of recruitment of IFI16 was observed. Recruitment of IFI16 to HSV-1 genomes was observed at equal levels of efficiency in PML-depleted cells. Efficient recruitment of hDaxx to these foci also occurred very rapidly, but that of PML took place more slowly. These events occurred in both wt and ICP0-null mutant infections but were very transient in the former, presumably because of the effects of ICP0. Photoactivation experiments illustrated that hDaxx molecules in one part of the cell nucleus could be recruited within seconds to viral genomes in the opposite half of the nucleus. These studies revealed highly dynamic and in some cases sequential or transient events of cellular protein recruitment to HSV-1 genomes that would not be amenable to study by fixed-cell methods. Importantly, the results reveal that the cell responds to the entry of viral genomes into the nucleus certainly within minutes and probably within seconds, long before a cell can be detected as being infected through the production of viral proteins.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 strain 17+ was the wt strain used, from which the ICP0-null mutant dl1403 was derived (5). Virus dl0C4, a derivative of dl1403 that expresses ECFP-linked ICP4, was constructed as described previously (4). All viruses were grown in BHK cells and titrated in U2OS cells, in which ICP0 is not required for efficient HSV-1 replication (6). Human diploid fibroblasts (HFs), PML-depleted HFs (7), telomerase-immortalized HFs (HFTs; a gift from Chris Boutell), and U2OS and HEK-293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). BHK cells were grown in Glasgow modified Eagle's medium supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. HepaRG cells were grown in William's Medium E supplemented with 10% fetal bovine serum Gold (PAA Laboratories Ltd.), 2 mM glutamine, 5 μg/ml insulin, and 500 nM hydrocortisone. All cell growth media were supplemented with 100 units/ml penicillin and 0.1 mg/ml streptomycin. Lentivirus-transduced cells were maintained with continuous antibiotic selection, as appropriate.
Lentiviral vectors.
Lentiviral vectors expressing EYFP-linked PML isoform I or hDaxx from the weak HSV-1 glycoprotein D promoter and including G418 resistance have been described previously (9, 12, 18). Derivatives of these were constructed in which the G418 resistance marker was replaced by puromycin resistance, while versions were also constructed in which ECFP was used in place of EYFP. The same backbone was used to express EYFP-linked IFI16 using a cDNA purchased from Origene. IFI16 mutant m3 (S27A/L28A/D50A [29]) was constructed by replacement of the wt cDNA with the complete m3 cDNA made by PCR from a plasmid supplied by Jungsan Sohn. A derivative ΔHIN2 mutant which lacks IFI16 codons 518 to 729 was constructed by PCR splicing and replacement of the wt sequence. Lentiviral vectors expressing PML isoform I or hDaxx linked to a fusion of photoactivatable EGFP and constitutively fluorescent mCherry were constructed by replacement of the EYFP coding region of vectors pLNGY-PML.I and pLNGY-hDaxx with the dual-fluorescence G(PA)C (denoting green [photoactivation] Cherry) cassette (30). Lentivirus transduction, selection of transduced cells, and maintenance of cell lines were performed as described previously (7). Sequential transduction was used to prepare cells expressing EYFP-IFI16 with either ECFP-hDaxx or ECFP-PML or with EYFP-hDaxx and ECFP-PML. Selection during routine culture used G418 at 0.5 mg/ml and/or puromycin at 500 ng/ml as appropriate. The antibiotic was omitted from cells seeded for and during experimentation.
Abs.
The following antibodies (Abs) were used: anti-actin monoclonal Ab (MAb) AC-40 (Sigma-Aldrich), anti-PML rabbit polyclonal Ab (rAb) ABD030 (Jena Bioscience) or MAb 5E10 (31), anti-IFI16 MAb ab55328 (Abcam), anti-hDaxx rAb 07-471 (Upstate), anti-ICP4 MAb 58S (32), and anti-EGFP rAb ab290 (Abcam).
Immunofluorescence.
Cells on 13-mm-diameter glass coverslips were fixed with formaldehyde and prepared for immunofluorescence using standard methods. The secondary antibodies used were fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse IgG (Sigma), Alexa 555-conjugated goat anti-mouse or anti-rabbit IgG, and Alexa 633-conjugated goat anti-rabbit IgG (Invitrogen). The samples were examined using a Zeiss LSM 710 confocal microscope, with 488-nm, 561-nm, and 633-nm-wavelength laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were exported as tif files, minimally adjusted using Photoshop, and then assembled into the figures using Illustrator.
Live-cell microscopy.
Cells were seeded into Nunc Lab-Tec chambered coverglass live-cell chambers at 1 × 105 cells per well (4 chambered units) and then infected or left uninfected as relevant the following day. Immediately prior to infection, the cells were washed with DMEM without phenol red, and after virus adsorption, they were overlaid with the same medium containing 2% FCS and antibiotics as described above. A range of MOIs varying between 25 and 100 was used for high-MOI experiments. The nature of the results was not essentially influenced by the choice of the MOI except that events occurred more commonly and were thus easier to detect as the MOI was increased. If plaques were to be examined later, the medium in the overlay also included 1% human serum. If the cells were to be examined immediately, the virus absorption period was 15 min, after which the cells were placed into the microscope incubation chamber (preheated to 37°C) and imaging was commenced as soon as possible. In the latter case, the times stated in the figures relate to the times after addition of the virus. The microscope was a Zeiss Axio Observer Z1 equipped with definite focus control to maintain the correct focal plane during a time course. The S1 environmental system was used to maintain CO2 at 5% and temperature and humidity. Filter sets 47 HE and 46 HE were used for detecting ECFP and EYFP, respectively, using illumination from an HXP 120-V unit. The samples were examined using either a 40× numerical aperture (NA) 1.3 or a 63× NA 1.4 oil immersion lenses. Image capture conditions generally utilized 70% or 50% HXP power with 2× gain and 4-by-4 binning, with exposure times on the order of 100 to 200 ms for both channels. The time intervals between images are noted in each figure as relevant. Imaging conditions were adjusted so that the cells did not suffer detectably from light poisoning and so that any photobleaching was minimized during the course of the experiment. This was generally not a problem for EYFP-IFI16 or ECFP-PML, but ECFP-hDaxx gave weaker fluorescence and was subject to photobleaching if the image capture frequency or illumination intensity was too high. Cells were selected for imaging on the basis of signal intensities to ensure sufficient image quality. Signal intensities were fairly uniform for IFI16, but cells expressing sufficient ECFP-hDaxx were in a minority. Relevant segments of the image series were cropped and exported as avi files (for the videos in the supplemental material) or saved separately for excision of individual frames for presentation in the figures, after adjustment of minimum and maximum thresholds for each channel for ease of visualization. Due to space constraints, only a small number of examples of each phenomenon can be presented in the figures. In some cases, additional events of the same nature can be seen in the accompanying videos in the supplemental material (which may cover a longer time frame). In all cases, the data presented are representative of the results of several independent experiments.
For the photoactivation experiments, cells were examined in a live-cell-adapted Zeiss LSM 510 Meta microscope with incubation at 37°C and 5% CO2. Selected cells were first imaged for mCherry, and then regions of interest were illuminated with the 405-nm laser at 50% power for 10 reiterations using the bleach program in the LSM 510 software. Subsequent imaging was performed at timed intervals thereafter for both EGFP and mCherry using the 488-nm and 543-nm lasers, respectively. For G(PA)C-hDaxx, images were acquired at 2-s intervals after bleaching, while 15-s intervals were used for G(PA)C-PML.I.
RESULTS
Description of the basic experimental system and underlying assumptions and extrapolations.
Gerd Maul was first to observe that the genomes of many DNA viruses, particularly HSV-1 and human cytomegalovirus (HCMV), could be observed to be in association with PML NBs during the early stages of infection (33, 34). These studies used the difficult technique of fluorescence in situ hybridization (FISH) to detect the viral genomes, a method which, although providing direct evidence, is limited because the antibody staining techniques required to detect viral or cellular proteins are not always compatible with the required harshness of the hybridization procedure. Later, it was discovered that, in cells at the edges of developing plaques, viral genomes could be detected frequently in large numbers of foci that formed characteristic arcs just inside the nuclear envelope. These genomes could be detected by FISH but also, importantly, by simple fluorescence detection of the viral transcriptional activator protein ICP4 (11). This occurs because ICP4 binds avidly to viral DNA. Thus, an asymmetric pattern of ICP4 foci near the nuclear periphery in such cells could be taken to identify viral genome sites, without the need for FISH. Because this highly asymmetric staining pattern is completely distinct from the normal situation in uninfected cells, it can be safely deduced that PML NB protein foci in this arrangement are also associated with the mutant viral genomes, even if neither FISH nor ICP4 staining is included in the protocol. Several of the experiments described in this report were dependent on this deduction. Analogous recruitment can also be observed in wt HSV-1 infections, but this is difficult to detect because the effects of ICP0 render it weak and transient (11). Having established these facts, it becomes possible to infer that small novel foci of relevant cellular proteins that can be detected only after a cell is infected are also highly likely to be associated with viral genomes. This is an important extrapolation in a number of the following experiments, but it is also supported by additional data where possible. These reasonable deductions and extrapolations are necessary because it is not possible to utilize FISH in live cells and because it is sometimes difficult to combine an experimental protocol with the use of a virus expressing fluorescently tagged ICP4. As examples of these phenomena, various phenotypes of PML and hDaxx recruited to ICP4 foci in cells at the edge of a developing ICP0-null mutant plaque are presented in Fig. 1 and for IFI16 in Fig. 2.
FIG 1.

Examples of recruitment of PML NB proteins to sites associated with HSV-1 genomes, as detected by staining for ICP4. The panels show views of cells close to developing ICP0-null mutant HSV-1 plaques in HFs, stained for ICP4 (green) and PML (red) (A) or ICP4 (green) and hDaxx (red) (B). Cells indicated by “a” show the pattern of PML or hDaxx expected of uninfected cells. Cells labeled “b” show various typical asymmetric patterns of ICP4 and PML NB protein staining, showing recruitment of PML or hDaxx to sites associated with HSV-1 DNA. The cell labeled “c” in panel B exhibits some faint foci of ICP4, each of which is associated with hDaxx staining. The cell labeled “d” in panel B shows a less common phenotype, in which hDaxx foci are distributed in a highly asymmetric pattern, very likely associated with parental HSV-1 genomes, but before ICP4 expression has reached a detectable level.
FIG 2.

The PYD domain is required for the recruitment of IFI16 to HSV-1 genomes. (A) A map of a lentivirus vector that expresses EYFP-linked IFI16. (B) Western blot analysis of cell extracts from a cell line transduced with the vector, analyzed for IFI16 (left) and EGFP (right). There are three endogenous IFI16 isoforms, which differ in the number of “S” regions in the hinge region between Hin-200A and Hin-200B. Only a single EYFP-linked isoform is expressed in these cells. (C) A map of the coding sequence of IFI16, indicating the PYD and the two HIN domains, with two linker sequences (S1 and S2). Also marked are the locations of the triple point mutations in mutant m3 and the region deleted in the mutant ΔHIN2. (D) The nuclear distribution of the wild-type (wt) and mutant m3 and ΔHIN2 mutant forms of IFI16, detected by autofluorescence in transduced cell lines. (E) The distributions of wt and mutant m3 and ΔHIN2 mutant forms of IFI16 in cells at the edge of developing ICP0-null mutant plaques, indicating prominent recruitment of the wt and ΔHIN2 IFI16 strains, but not the m3 mutant, to sites that are closely associated with HSV-1 genomes (detected by staining for ICP4). (F) Depletion of PML does not compromise efficient recruitment of IFI16 to HSV-1 genomes. The panels show either uninfected HFs or examples of cells at the edge of ICP0-null mutant HSV-1 plaques stained for hDaxx and IFI16, in control and PML-depleted cells (left and right pairs of images, respectively).
The pyrin domain of IFI16 is required for its recruitment to HSV-1 genomes.
Previous publications have noted that IFI16 is also recruited efficiently to HSV-1 genomes at the early stages of infection (22–28). In order to study this in more detail, a lentiviral vector system was used to express EYFP-tagged IFI16 (Fig. 2A). Transduced human fibroblasts were selected with G418, and then EYFP-positive cells were enriched by fluorescence-activated cell sorter (FACS) analysis. Analysis of the resultant cell line by Western blotting showed that the cells expressed EYFP-linked IFI16 at levels on the same order as those seen with the three endogenous isoforms (Fig. 2B). The same system was used to express two mutant forms of IFI16, one with three point mutations in the pyrin domain (mutant m3) (29) and one with a deletion of the second DNA binding HIN domain (ΔHIN2) (Fig. 2C). The EYFP-IFI16 proteins expressed in these cell lines were distributed similarly in the nucleus, with a general diffuse signal throughout the nucleoplasm and a concentration in the nucleoli (Fig. 2D). This is very similar to the distribution of endogenous IFI16 in HFs (23, 25). As with the endogenous protein (23), wt EYFP-IFI16 was clearly recruited to sites associated with HSV-1 genomes (detected by ICP4) in cells at the edge of developing ICP0-null mutant plaques (Fig. 2E, upper row). This recruitment is not detectable in this way in wt-infected cells because of ICP0 activity (23). The recruitment of EYFP-IFI16 also occurred normally in the case of the ΔHIN2 mutant (Fig. 2E, bottom row), showing that one DNA binding HIN domain is sufficient for this activity, but the PYD m3 mutant was not efficiently recruited (Fig. 2E, middle row). The pyrin domain is required for oligomerization of IFI16 when bound to DNA (29); therefore, the recruitment of IFI16 probably represents assembly of IFI16 oligomers on the viral DNA at this stage of infection, soon after it is released into the nucleus in an unchromatinized form. It was not possible to test whether deletion of both HIN domains also inhibited recruitment, as further deletion upstream of HIN2 caused loss of expression in this system.
Depletion of PML does not compromise recruitment of IFI16 to HSV-1 genomes.
It was of interest to determine whether or not PML is involved in the recruitment of IFI16 to HSV-1 genomes. HFs depleted of PML using a lentiviral shRNA vector (7) were infected with ICP0-null mutant HSV-1, and the distributions of hDaxx and IFI16 in cells at the edges of developing plaques were investigated. Infected cells could be identified by the characteristic redistribution of hDaxx (see Fig. 1), and, as expected, IFI16 was similarly redistributed in such cells (Fig. 2F, left). The same distributions of hDaxx and IFI16 were observed in infected PML-depleted cells, while hDaxx was dispersed throughout the nucleoplasm in uninfected cells (Fig. 2F, right). These data demonstrate that while PML is required for hDaxx to be present in PML NB foci in uninfected cells (as reported previously [16, 17]), PML depletion does not compromises recruitment of either hDaxx (in agreement with previous data [8]) or IFI16. Thus, recruitment of IFI16 to HSV-1 genomes is independent of the events that lead to the transient colocalization of IFI16 with PML during the early stages of wt HSV-1 infection (22, 23).
Rapid response of IFI16 to HSV-1 genomes as they enter the nucleus.
With a cell line expressing EYFP-linked wt IFI16 having been developed, it was now possible to examine the behavior of the protein in live infected cells. An unexpected observation was that high-multiplicity ICP0-null mutant HSV-1 infection induced the formation of small, discrete novel IFI16 foci, often just inside the nuclear periphery. These were highly transient, in that they could appear and then disappear within 5 min, with others subsequently appearing similarly in a different part of the cell (Fig. 3A; see also Video S1 in the supplemental material). Such events also occurred during wt HSV-1 infection (Fig. 3B; see also Video S2). The appearance of these foci was completely dependent on infection and generally occurred within the first hour or two after addition of the virus to the cell monolayer. Given their size and location, it seemed possible that they represented transient recruitment of IFI16 onto HSV-1 genomes at the time and point that they were released into the nucleus. Support for this explanation came from study of cells at the edge of a developing ICP0-null mutant plaque. Such cells are infected at much higher multiplicity than can be achieved by adding a virus inoculum to the cell monolayer and would thus be expected to display more of such foci. This was indeed the case, with many examples of such transient foci being plainly visible in Fig. 3C and in Video S3.
FIG 3.

Detection of the rapid response of IFI16 to HSV-1 infection. (A) HFs expressing EYFP-IFI16 were infected with ICP0-null mutant HSV-1 (MOI of 50), and then images were captured at the indicated times after addition of the virus. (B) A similar experiment was conducted using wt HSV-1 infection (MOI of 20). (C) Images from a sequence of a cell close to the edge of an ICP0-null mutant HSV-1 plaque. (D) HFs expressing EYFP-IFI16 were infected with virus dl0C4, and images from a sequence of a cell at the edge of a developing plaque, showing the IFI16, ICP4, and merged signals as indicated, are presented.
Further support for this hypothesis came from infection of HFs expressing EYFP-IFI16 with dl0C4, an ICP0-null mutant HSV-1 expressing ECFP-linked ICP4 (4). The compilation in Fig. 3D and in Video S4 in the supplemental material shows a cell at the edge of a developing plaque from an image sequence that began before any IFI16 foci were detected. The displayed images illustrate the appearance of transient IFI16 foci at various locations within the first 42 min, mostly in the lower left quadrant of the cell but also elsewhere. As time continued, the foci in this region became more stable and prominent and developed into an arc-like mass. The ECFP signal indicates that these apparently more stable IFI16 accumulations were very close to where ICP4 foci began to accumulate, marking the positions of viral genomes. These data are consistent with the interpretation that this cell was being infected by genomes arriving mostly in the lower left quadrant of the nucleus which initially induced transient foci of IFI16. As infection developed, and ICP4 expression became detectable (and DNA replication likely began, as judged from the expansion of the ICP4 compartments), the accumulations of IFI16 became more marked and longer lasting. From previous results, it can also be deduced that this second phase was inhibited by ICP0 but that the first phase took place too rapidly for it to be inhibited, as it probably occurred before ICP0 was expressed in sufficient quantities.
Rapid recruitment of hDaxx from the nucleoplasm to HSV-1 genomes.
Previous work using fluorescence recovery after photobleaching (FRAP) had demonstrated that hDaxx is highly mobile in the nucleus, with an exchange rate between PML NBs and the general nucleoplasm measured in terms of seconds, while that for PML was slower but still in the low numbers of minutes (11, 35–37). It had previously been suggested that this dynamic behavior would enable the formation of novel PML NB-like foci in association with HSV-1 genomes without the need for movement of preexisting PML NBs (11). To address this point further, lentiviral vectors were constructed that expressed either hDaxx or PML linked to a fusion protein that included mCherry and a photoactivatable version of EGFP (30). Thus, the expressed proteins constitutively expressed red fluorescence but also expressed green fluorescence after photoactivation by pulses of a 405-nm laser. In this way, the movement of molecules from activated to unactivated parts of the nucleus could be monitored. This technique was therefore adopted to examine the dynamics of recruitment of both hDaxx and PML from one region of the nucleus to another in which viral genome foci were present. Experiments in uninfected cells illustrated that photoactivation of both the PML and hDaxx fusion proteins occurred as expected and that the subsequent migration of activated molecules to the unactivated part of the nucleus could be observed. In the case of PML, this took place over a period of minutes, while with hDaxx the time scale was in seconds, to the extent that some transfer into the unactivated part of the nucleus was seen as soon as an image could be captured after photoactivation (Fig. 4A). These results are consistent with the previous FRAP experiments (11, 35–37). By applying this technique to cells at the edge of developing ICP0-null mutant HSV-1 plaques, characteristic recruited patterns of hDaxx and PML could be seen via the mCherry fluorescence. After photoactivation of molecules in the opposite part of the nucleus, the presence of activated hDaxx in the viral genome-associated foci could be detected at the earliest possible time point, becoming more pronounced within 15 s (Fig. 4B, upper row). The same occurred with PML over a longer time course (Fig. 4B, lower row). Therefore, recruitment of hDaxx in particular to viral-genome-associated foci is extremely rapid.
FIG 4.

Dynamics of PML.I and hDaxx in uninfected cells and their rapid recruitment to sites associated with HSV-1 genomes. (A). Uninfected HepaRG cells transduced to express either G(PA)C-PML.I or G(PA)C-hDaxx were imaged before and after photoactivation of the PA-EGFP moiety within the red boxed area. Detection of PA-EGFP at intervals after photoactivation reveals migration of activated molecules from the activated region to the rest of the nucleoplasm. (B). Experiments were performed as described for panel A, but cells at the edge of developing ICP0 mutant HSV-1 plaques with characteristic asymmetrically distributed foci were examined under the same activation and time course conditions. Activated hDaxx was detectable in the asymmetric foci at the first time point after bleaching (about 2 s), becoming more prominent as time progressed. Activated PML accumulated in the foci at the nuclear periphery more slowly.
IFI16 and hDaxx respond to HSV-1 genomes more rapidly than PML.
Given that both IFI16 and hDaxx can be recruited very rapidly to sites associated with HSV-1 genomes, it was of interest to determine whether a temporal order of IFI16 and PML NB components could be determined. Therefore, cells were constructed that express EYFP-IFI16 and either ECFP-linked hDaxx or PML. The EYFP-IFI16/ECFP-hDaxx cells were infected at high multiplicity with ICP0-null mutant HSV-1, and then an image sequence was captured during the very early stages of infection (Fig. 5). As seen before, in a proportion of cells, transient virus-induced small foci of IFI16 appeared to which hDaxx was also recruited de novo either at the same time point or very shortly thereafter. Therefore, these proteins respond with similar kinetics to HSV-1 infection. That hDaxx is also recruited to these novel, virus-induced IFI16 foci is further evidence that they are associated with viral genomes because it has been established that hDaxx responds in this manner. As time proceeds, the IFI16 signal is lost from these foci but they remain hDaxx positive.
FIG 5.

IFI16 and hDaxx respond to HSV-1 infection with similar kinetics. HFs expressing EYFP-IFI16 and ECFP-hDaxx were infected with ICP0-null mutant HSV-1 (MOI of 100). After a 15-min absorption period, the cells were examined by live-cell microscopy, with images captured every 90 s. A selection of images is shown, with the times after adding the virus indicated in the top row. The upper three rows show the IFI16, hDaxx, and merged channels, with the numbered arrows pointing to IFI16 foci that transiently appeared during the sequence. The corresponding foci are also indicated on the hDaxx and merged panels. The sets of smaller images show details from each time point of the boxed areas marked for the 48-min sample. The left and right columns of these images show the images corresponding to the lower and upper boxes, respectively, for each time point, using the same numbering system. In this sequence covering just 9 min, four IFI16 foci appeared simultaneously with an hDaxx signal, although, as shown at dot 3, the latter was weak at the 55.5-min time point. The presence of IFI16 was transient as shown at dots 1 and 2 in this sequence, while hDaxx was more stable. A longer sequence from the same time course is shown in Video S5 in the supplemental material, in which several other examples of the same phenomena can be seen.
A similar experiment was performed using EYFP-IFI16/ECFP-PML cells infected with ICP0-null mutant HSV-1. Figure 6 shows a cell infected at a high MOI in which an example of an event that commonly occurs in such cells was observed. First, a small accumulation of IFI16 appeared near the nuclear periphery, which contained no detectable PML at this point (Fig. 6; 3-min time point). As before, IFI16 appeared only transiently, but at the 7-min time point, this dot also contained PML. At later time points, IFI16 was no longer detectable in this dot, whereas PML remained in this novel focus. Over the following minutes, the new PML dot became closer to a preexisting PML NB and then appeared to merge with it. The top three rows of Fig. 6 show the IFI16, PML, and merged signals of this cell over an 11-min period, while the lower three panels show enlargements of these images covering the event in question. Examination of similar IFI16 foci over a longer period indicated that these events were quite common, although the novel foci did not always merge with preexisting PML NBs (see Video S6 in the supplemental material). A possible interpretation of these observations is presented in the Discussion, but for now it can be concluded that these events are most easily explained by a local and transient accumulation of IFI16 at viral genomes after their entry into the nucleus, followed by a more stable recruitment of PML.
FIG 6.

IFI16 responds to HSV-1 infection more rapidly than PML. HFs expressing EYFP-IFI16 and ECFP-PML were infected with ICP0-null mutant HSV-1 (MOI of 25). After a 20-min absorption period, the cells were examined by live-cell microscopy, with images captured every minute, starting 35 min after addition of the virus. A selection of images from the time course is presented, showing a cell in which a novel focus of IFI16 appeared, which later became PML positive before merging with a preexisting PML NB structure. The lower set of three rows presents expansions for each time point of the boxes in the merged image corresponding to the 82-min time point. Further details are provided in the text.
Recruitment of hDaxx is detectable before that of PML.
The results represented in Fig. 5 and 6 suggest that hDaxx responds more quickly than PML to virus infection, because there was a delay before the latter was detectable at the novel IFI16 foci, while the former appeared commonly within the same time frame. To test this directly, the relative dynamics of recruitment of hDaxx and PML to HSV-1-induced foci were compared in HFs constructed to express ECFP-hDaxx and EYFP-PML. A common phenomenon observed in such cells near the periphery of developing ICP0-null mutant HSV-1 plaques was the appearance of novel foci that first included hDaxx, following which PML was also seen to be present at a slightly later time. Many such examples are visible in the images of a time course presented in Fig. 7. A longer period of the time course of this cell is shown in Video S7 in the supplemental material. A detail of one such example (boxed in the merged image from the first time point) is shown in the lowest row of Fig. 7. At the first time point, a dot which was green only was present; at 15 min, the green signal in the dot was less intense (probably because the dot had moved slightly out of the focal plane), but by this time the dot also included PML. At the 35-min time point, another new green dot had appeared at the lower left of the detail (also clearly seen in the image of the whole-cell nucleus; this also later accumulated PML), while one of the preexisting PML NBs had moved out of this segment, leaving the dot that was green only at the start in the center of the detail. By the 45-min time point, this appears to have merged with the nearest neighboring PML NB. Detailed examination of the complete time course (see Video S7) illustrates that these events were very common in such cells; novel foci that included only hDaxx appeared that later included both hDaxx and PML and sometimes, but not always, merged with neighboring PML NBs.
FIG 7.

hDaxx responds more rapidly than PML to HSV-1 infection. HFs expressing both ECFP-hDaxx and EYFP-PML were infected with ICP0-null mutant HSV-1, and cells at the edge of a developing plaque were examined. A selection of images from an image sequence are presented, indicating the time (arbitrary) after the first image shown (which is image 54 of the original sequence [frame 24 as presented in Video S7 in the supplemental material]). The lowermost row shows a magnified view of the region that is boxed in the merged image of the leftmost column. Further details are provided in the text.
Such events can be detected only by live-cell analysis, which reveals the transient and subtle events that occur in cells close to developing plaques. Previous examination of such cells using fixed-cell methods had concentrated on those in which marked viral-genome-associated foci in arcs near the edge of the nucleus had been formed, because in these cases it was clear that these novel foci were virus induced. The live-cell analysis extends these findings to reveal virus-induced events in many cells that do not form or have not yet formed the characteristic perinuclear arcs of foci and which could not be detected at such time points by costaining for ICP4.
DISCUSSION
The results presented in this paper allow an extension of previous models of the interactions between HSV-1 genomes and PML NB proteins during the earliest stages of infection (summarized in Fig. 8). At a very early stage of infection, probably within seconds and certainly within minutes of the release of the viral genome through the nuclear pore into the nucleoplasm, IFI16 can be seen to accumulate in distinct foci that are very likely to be closely associated with the viral genomes. This event is PML independent, but it requires the pyrin domain of IFI16 which is involved in its oligomerization on naked DNA (29). Equally rapidly, hDaxx also accumulates in these foci, colocalizing with IFI16 transiently, while the IFI16 signal is dispersed after a few minutes. As the IFI16 signal weakens, PML then also accumulates in the foci, colocalizing with hDaxx. All these events can be observed in both wt and ICP0-null mutant infections, but the recruitment of hDaxx and PML is much more difficult to detect in the latter case because of the activity of ICP0 (9, 11, 12). In the absence of ICP0, at a high multiplicity of infection and in cells which enter the lytic cycle, hDaxx and PML remain associated at sites close to the viral genomes as replication compartments develop, and a second and more stable phase of IFI16 recruitment occurs. However, it is known that the majority of ICP0-null mutant HSV-1 genomes are repressed at a low multiplicity of infection. The evidence here suggests that these genomes may remain stably associated with hDaxx and PML, perhaps sequestered within these modified PML NB-like structures, which in some cases appear to fuse with preexisting PML NBs. This hypothesis is consistent with the observation that quiescent HSV-1 genomes can be detected within enlarged PML NBs in fibroblasts (38) (and in similar structures in latently infected mouse neurones [39]). In contrast to what is seen in the case of active replication, in which IFI16 more stably accumulates close to the viral genomes (Fig. 2), IFI16 was not detected within PML NBs in quiescently infected cells (data not shown). Therefore, recruitment of IFI16 appears to be a transient event.
FIG 8.
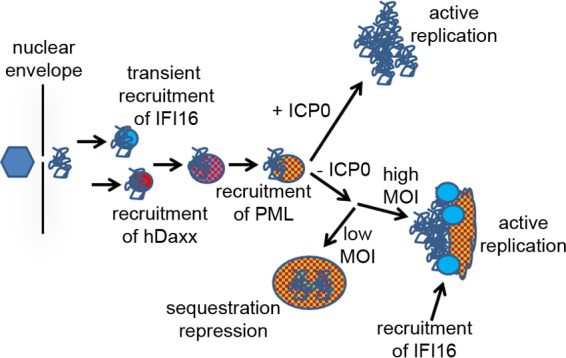
A model for the dynamics of IFI16 and PML NB components with respect to HSV-1 genomes. A full explanation is provided in the text. The hexagon on the left represents a full capsid bound to the outer side of a nuclear pore through which the naked viral genome (tangled line) is transferred into the nucleoplasm.
In previous studies on the recruitment of PML NB proteins to incoming HSV-1 genomes, the analysis concentrated on cells in which there are abundant numbers of ICP4 foci close to the edge of the nucleus. This was because the asymmetric pattern of PML NB protein foci could be clearly distinguished from their normal distribution in uninfected cells; therefore, clear unequivocal conclusions could be made. The observation of virus-induced IFI16 foci in this study has allowed a more detailed analysis, because IFI16 never forms small punctate foci in uninfected cells. The use of dually labeled cells, combining EYFP-IFI16 with either ECFP-hDaxx or ECFP-PML, also emphasizes the compelling nature of the observations. Because of these factors, the virus-induced foci could be studied in cells in which they are neither abundant nor routinely close to the nuclear envelope, within cells at the early stages of a normal infection as well as within cells at the edges of developing plaques, and also within a background of preexisting PML NBs. The live-cell sequences thus clearly show the formation of novel virus-induced PML NB-like structures that, in some cases, later fuse with preexisting PML NBs, events that could underlie the detection of multiple quiescent viral genomes within enlarged PML NBs (38).
The mechanism of the recruitment of IFI16 to HSV-1 genomes involves several factors. One important aspect is likely to be that the viral DNA is not chromatinized at the point of delivery into the nucleoplasm, and this naked configuration is likely to underlie its recognition by IFI16 (29). Chromatin immunoprecipitation (ChIP) assays performed by others have confirmed a direct association between IFI16 and HSV-1 DNA (24). IFI16 binds to DNA through its HIN domains, and it can form oligomers on DNA through pyrin domain interactions (29). A single HIN domain was sufficient for IFI16 to accumulate on HSV-1 genomes, and the pyrin domain was essential for this abundant accumulation (Fig. 2). It is possible that the pyrin domain mutant protein still binds to viral DNA, but lack of oligomerization may reduce its accumulation on viral genomes to below a level detectable by microscopy. The recruitment of several PML NB proteins to HSV-1 genomes occurs through SUMO-related pathways, but whether sumoylation is also involved in the recruitment of IFI16 remains to be determined. IFI16 has been identified as a substrate for sumoylation in proteomic screens (15, 40), but an abundantly sumoylated form is not evident in Western blot analyses. Whatever the mechanisms involved, they are clearly inhibited by ICP0. As abundant recruitment of IFI16 does not occur in cells infected by wt HSV-1 even when it remains easily detectable by fluorescence (23), this inhibition is not a simple consequence of the degradation of IFI16 that occurs at later times of wt HSV-1 infection (22, 23).
The transient nature of IFI16 accumulation in the virus-induced foci is intriguing. It is not a simple case of the entity in question moving out of focus (although this can occur), because the hDaxx or PML signal remains. The recruitment of IFI16 is clearly a very early event, and it is possible that the viral genome is assembled into more-complex nucleoprotein complexes, including nucleosomal structures, such that the parental viral DNA ceases to be in the naked form required for IFI16 accumulation. The more stable accumulation of IFI16 that occurs as replication compartments develop in ICP0-null mutant infections could reflect the production of newly replicated naked DNA in these structures. However, IFI16 does not colocalize with viral replication compartments in the same way as ICP8 and ICP4 even in ICP0-null mutant infections, but it instead often forms tangled filament-like structures that are associated with the replication compartments (23). These thread-like structures are likely to represent oligomers of IFI16, but how they relate to the viral DNA remains to be determined.
A fundamental issue that is beyond the scope of this current study is whether or not IFI16 plays an important role in the recruitment of other proteins to the HSV-1 genome. The hypothesis that it does indeed play an important role would provide an interesting aspect to the role of IFI16 as a sensor of pathogen DNA. It was previously shown that the recruitment of hDaxx appeared to be delayed in cells depleted of IFI16, in that it was much more difficult to detect hDaxx in the asymmetric staining pattern characteristic of infected cells prior to detectable expression of ICP4 (in other words, cells of the phenotype of the cell labeled “d” in Fig. 1B were very rare in IFI16-depleted cells) (23). However, recruitment of both PML and hDaxx was readily detectable in the majority of IFI16-depleted cells once ICP4 was expressed (23). Although depletion of IFI16 was efficient in these cells, it is possible that the remaining trace levels were sufficient for a hypothetical function in which recruitment of IFI16 is a necessary step prior to recruitment of the other proteins. Alternatively, the recruitment of IFI16 may be unlinked to that of PML NB proteins and may be more involved in regulation of chromatin assembly on the viral genome, as proposed by others (24, 25). Whatever the functions of the events described here, it is clear that detailed examination in live cells reveals striking and unexpected aspects of the dynamic nature of the interaction between cellular proteins and the invading HSV-1 genomes.
Supplementary Material
ACKNOWLEDGMENTS
I am very grateful for the excellent technical assistance of Anne Orr, and I thank Jungsan Sohn (IFI16 mutant m3) and Arkadiusz Welman and Margaret Frame [the G(PA)C vector] for the indicated materials. I also acknowledge the many colleagues who over the years have contributed to understanding of virus interactions with PML NBs, particularly the late Gerd Maul, who initiated this whole story.
Funding Statement
The Zeiss Axio Observer Z1 microscopy system was purchased via a grant from the Medical Research Foundation (grant reference C0446). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02249-15.
REFERENCES
- 1.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 2.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 3.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Everett RD, Boutell C, Orr A. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J Virol 78:1763–1774. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J Gen Virol 67:2571–2585. doi: 10.1099/0022-1317-67-12-2571. [DOI] [PubMed] [Google Scholar]
- 6.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J Virol 84:4026–4040. doi: 10.1128/JVI.02597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuchet-Lourenço D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog 7:e1002123. doi: 10.1371/journal.ppat.1002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloan E, Tatham MH, Groslambert M, Glass M, Orr A, Hay RT, Everett RD. 2015. Analysis of the SUMO2 proteome during HSV-1 infection. PLoS Pathog 11:e1005059. doi: 10.1371/journal.ppat.1005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752. [PubMed] [Google Scholar]
- 18.Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, Everett RD. 2011. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J Cell Sci 124:280–291. doi: 10.1242/jcs.075390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7:e1002084. doi: 10.1371/journal.ppat.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Unterholzner L, Bowie AG. 2011. Innate DNA sensing moves to the nucleus. Cell Host Microbe 9:351–353. doi: 10.1016/j.chom.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuchet-Lourenço D, Anderson G, Sloan E, Orr A, Everett RD. 2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87:13422–13432. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. 2014. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10:e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diner BA, Lum KK, Javitt A, Cristea IM. 18 February 2015, posting date Interactions of the antiviral factor IFI16 mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol Cell Proteomics doi: 10.1074/mcp.M114.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Diner BA, Chen J, Cristea IM. 2012. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. 2014. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci U S A 111:E62–E71. doi: 10.1073/pnas.1313577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welman A, Serrels A, Brunton VG, Ditzel M, Frame MC. 2010. Two-color photoactivatable probe for selective tracking of proteins and cells. J Biol Chem 285:11607–11616. doi: 10.1074/jbc.M110.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J Cell Sci 101:773–784. [DOI] [PubMed] [Google Scholar]
- 32.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect Immun 34:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishov AM, Maul GG. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J Cell Biol 134:815–826. doi: 10.1083/jcb.134.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 35.Brand P, Lenser T, Hemmerich P. 2010. Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys 3:3. doi: 10.1186/1757-5036-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weidtkamp-Peters S, Lenser T, Negorev D, Gerstner N, Hofmann TG, Schwanitz G, Hoischen C, Maul G, Dittrich P, Hemmerich P. 2008. Dynamics of component exchange at PML nuclear bodies. J Cell Sci 121:2731–2743. doi: 10.1242/jcs.031922. [DOI] [PubMed] [Google Scholar]
- 37.Wiesmeijer K, Molenaar C, Bekeer IM, Tanke HJ, Dirks RW. 2002. Mobile foci of Sp100 do not contain PML: PML bodies are immobile but PML and Sp100 proteins are not. J Struct Biol 140:180–188. doi: 10.1016/S1047-8477(02)00529-4. [DOI] [PubMed] [Google Scholar]
- 38.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P. 2012. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog 8:e1002852. doi: 10.1371/journal.ppat.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tammsalu T, Matic I, Jaffray EG, Ibrahim AF, Tatham MH, Hay RT. 2014. Proteome-wide identification of SUMO2 modification sites. Sci Signal 7:rs2. doi: 10.1126/scisignal.2005146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.