

ABSTRACT
The multiplicity of infection (MOI), i.e., the number of viral genomes that infect a cell, is an important parameter in virus evolution, which for each virus and environment may have an optimum value that maximizes virus fitness. Thus, the MOI might be controlled by virus functions, an underexplored hypothesis in eukaryote-infecting viruses. To analyze if the MOI is controlled by virus functions, we estimated the MOI in plants coinfected by two genetic variants of Tomato bushy stunt virus (TBSV); by TBSV and a TBSV-derived defective interfering RNA (DI-RNA); or by TBSV and a second tombusvirus, Cymbidium ringspot virus (CymRSV). The MOI was significantly larger in TBSV-CymRSV coinfections (∼4.0) than in TBSV-TBSV or TBSV–DI-RNA coinfections (∼1.7 to 2.2). Coinfections by CymRSV or TBSV with chimeras in which an open reading frame (ORF) of one virus species was replaced by that of the other identified a role of viral proteins in determining the MOI, which ranged from 1.6 to 3.9 depending on the coinfecting genotypes. However, no virus-encoded protein or genomic region was the sole MOI determinant. Coinfections by CymRSV and TBSV mutants in which the expression of the gene-silencing suppressor protein p19 was abolished also showed a possible role of gene silencing in MOI determination. Taken together, these results demonstrate that the MOI is a quantitative trait showing continuous variation and that as such it has a complex determination involving different virus-encoded functions.
IMPORTANCE The number of viral genomes infecting a cell, or the multiplicity of infection (MOI), is an important parameter in virus evolution affecting recombination rates, selection intensity on viral genes, evolution of multipartite genomes, or hyperparasitism by satellites or defective interfering particles. For each virus and environment, the MOI may have an optimum value that maximizes virus fitness, but little is known about MOI control in eukaryote-infecting viruses. We show here that in plants coinfected by two genotypes of Tomato bushy stunt virus (TBSV), the MOI was lower than in plants coinfected by TBSV and Cymbidium ringspot virus (CymRSV). Coinfections by CymRSV or TBSV with TBSV-CymRSV chimeras showed a role of viral proteins in MOI determination. Coinfections by CymRSV and TBSV mutants not expressing the gene-silencing suppressor protein also showed a role of gene silencing in MOI determination. The results demonstrate that the MOI is a quantitative trait with a complex determination involving different viral functions.
INTRODUCTION
Strong evidence supports the fact that, in nature, mixed rather than single viral infections may predominate in both plant and animal hosts (1, 2). Mixed infections result in a variety of virus-virus interactions, from cooperation to competition for host resources, which affect each virus's fitness. Thus, mixed infections are a factor in virus evolution, and there is evidence supporting the prediction that the within-host dynamics of microparasites in mixed infections affect virulence evolution (3–5). Mixed infections may also affect the genetic diversity and structure of virus populations, as coinfection is required for genetic exchange to occur, either by recombination or by genome segment reassortment between different virus species or genotypes (6, 7). Accordingly, interactions between viruses are relevant to understand virus evolution and epidemiology, as well as for the development of efficient control strategies against viral infections (7). At the cellular level, the extent of these interactions is mostly influenced by the number of viral genomes that infect the same cell, a parameter called the multiplicity of infection (MOI) (8–11).
The MOI is a key parameter in virus evolution, because the extent of cell coinfection determines the extent of competition among virus variants and, hence, their fitness compared with single infections. Also, as the MOI is equivalent to the ploidy level, it determines the rates of genetic exchange, and thus genetic diversity, and of trans-complementation of defective mutants, slowing down selection against them (9, 12, 13). Despite its relevance, there are very few experimental estimates of the MOI. For plant-infecting viruses, six estimates have been reported. The first was presented by González-Jara et al. (14) for the colonization of Nicotiana benthamiana Domin. by Tobacco mosaic virus (TMV) (genus Tobamovirus). The numbers of singly and doubly infected cells were monitored in inoculated and systemically infected leaves using two TMV genotypes, each labeled with a different fluorescent protein. The results confirmed the existence of a strong spatial structure of both viral variants, as was reported during mixed infections by viruses with different genome organizations and gene expression strategies (15, 16). Hence, the fraction of doubly infected cells was low, and the MOI level was about 2 during the progress of infection (14, 17, 18). Miyashita and Kishino (19) estimated the MOI as the sizes of genetic bottlenecks during cell-to-cell movement of Soilborne wheat mosaic virus (SBWMV) (genus Furovirus) in inoculated leaves of Chenopodium quinoa Willd. The study also detected cells singly and doubly infected with two variants of the RNA2 genomic segment, labeled with different fluorescent proteins. The numbers of viral genomes that established the infection in adjacent cells after the first and second rounds of cell-to-cell movements from an initially infected cell were above 6 and 5, respectively. These numbers decreased rapidly in subsequent cell colonization rounds, leading to the spatial segregation of the two SBWMV RNA2 variants (19). Gutiérrez et al. (20) estimated the MOI during colonization of Brassica rapa L. by Cauliflower mosaic virus (CaMV) (genus Caulimovirus) using two CaMV variants tagged with two different oligonucleotide markers, which allowed the specific detection of each variant in mixed infections by single-cell nested PCR (20, 21). It was found that the MOI varied between about 2 and 13 during the course of infection, with values starting close to 2, increasing until flowering, and decreasing toward the end of the plant life cycle (20). Tromas et al. (22) estimated the MOI of Tobacco etch virus (TEV) (genus Potyvirus) in leaves of Nicotiana tabacum L. cv. Xanthi using flow cytometry to count cells singly and doubly infected with TEV and labeled with two fluorescent proteins. The frequency of coinfected cells was similar to that obtained for TMV, and the predicted MOI was also low, ranging from 1 to 1.5 in different leaf levels (22). Bergua et al. (23) estimated the MOI in phloem-associated cells of young citrus trees that were infected with a variant of Citrus tristeza virus (CTV) (genus Closterovirus) and later superinfected with a second variant, with each CTV variant being tagged with two fluorescent proteins and both lacking the p33 protein. The MOI value was also low, ranging from 1.06 to 1.07 in different plant sections (23). Finally, Gutiérrez et al. (24) obtained different estimates of the MOI at two phases of the systemic infection of a single systemically infected leaf of turnip plants by Turnip mosaic virus (TuMV). In the cells with a primary infection from the vasculature, the estimated MOI was very low (0.07); however, the estimation of the MOI during the subsequent cell-to-cell movement increased substantially, to between 21.7 and 41.5 (24). There are also few estimates of the MOI in viruses infecting hosts other than plants, and they show values in the same range as for plant viruses: 2 to 3 for bacteriophages (8, 10, 11), about 3 for HIV (25), and 4 to 5 for a polyhedrosis virus infecting moths (26). If MOI estimates are scarce, even less is known about the mechanisms controlling the MOI. the MOI could be determined by (i) competitive exclusion, where species or genotypes that exploit the same resource cannot coexist (27), and (ii) superinfection exclusion, defined as the ability of an established virus to interfere with a secondary infection by the same or a closely related virus at the host or cell level, which could be the basis of cross-protection in plants (1). Several mechanisms have been postulated to explain exclusion of plant viruses, including competition for host factors or intracellular replication sites (reviewed in reference 27), prevention of disassembly of the challenge virus due to the expression of the coat protein (CP) of the primary virus (1, 28), and RNA silencing that leads to the sequence-specific degradation of the challenge virus (29, 30). There is some evidence pointing to a requirement for the expression of viral proteins to prevent superinfection in viruses infecting bacteria, mammals, and plants. The exclusion by bacteriophage λ (genus λ-like viruses) of superinfection by other bacteriophages requires the products of two λ genes, rexA and rexB (31). The RexA/RexB ratio affects the effectiveness of exclusion (32). The Sim protein of bacteriophage P1 (genus P1-like viruses) appears to block injection of nucleic acid by a superinfecting phage at the cell membrane (33). The lpt gene of phage TP-J34 encodes a lipoprotein whose expression in Streptococcus thermophilus J34 could be involved in superinfection exclusion (34). The exclusion of Vesicular stomatitis virus (VSV) (genus Vesiculovirus) defective interfering particles by VSV in cell culture is at the level of virus penetration and requires the expression of viral genes and a VSV transmembrane glycoprotein (35). Superinfection exclusion of CTV at the whole-organism level, but not at the cellular level, requires the CTV protein p33, which is not essential for CTV infection in most host plants (23, 28). Thus, the information available on virus-encoded functions regulating cell infection by different viruses or genotypes is quite limited.
Until now, there has been no empirical evidence to assess the role of competitive or superinfection exclusion and the underlying mechanisms in the regulation of the MOI during virus infection in eukaryotic hosts. In the present study, we used two species of tombusviruses, Tomato bushy stunt virus (TBSV) and Cymbidium ringspot virus (CymRSV), and one TBSV-derived defective interfering RNA (DI-RNA) to analyze the mechanisms controlling the MOI in N. benthamiana. Tombusviruses have messenger sense, single-stranded RNA genomes of approximately 4.7 kb with five open reading frames (ORFs). The 5′-proximal ORF, p33, and its readthrough product, p92, are translated directly from the RNA genome and are required for viral replication (36). ORF3 encodes the 41-kDa CP, which is synthesized from a subgenomic RNA1 (sg1) (37). The two terminal ORFs are translated from another subgenomic RNA2 (sg2) and encode the viral movement protein of 22 kDa and the p19 protein that acts as an RNA-silencing suppressor (38). Upon serial passages under conditions that avoid severe bottlenecks, tombusvirus infections are associated with the appearance of DI-RNAs (39, 40), which are incomplete viral genomes requiring coinfection with their parental helper virus (HV) for replication and encapsidation. The presence of DI-RNAs often results in a decrease in both the titer and the virulence of the HV (41, 42). The high titer of DI-RNAs in mixed infected tissues may suggest that mechanisms excluding coinfection with the helper virus would not operate, but this possibility has not been explored to date.
We have found that the number of coinfecting genomes (NCG) during leaf colonization by two variants of one tombusvirus species, TBSV, or a derived DI-RNA was much lower than that during coinfections with two tombusvirus species. To determine the possible role of viral proteins in the determination of MOI levels, the NCG was estimated in coinfections by either CymRSV or TBSV and a series of CymRSV-TBSV chimeras exchanging entire ORFs. Also, to determine the possible role of gene silencing in the determination of MOI levels, the MOI was estimated in coinfections by CymRSV and TBSV in which the expression of the p19 silencing suppressor was abolished. The results show that MOI determination is a complex phenomenon in which both specific virus-encoded proteins/ORFs and gene silencing participate.
MATERIALS AND METHODS
Viral vectors, virus purification, and plant inoculation.
Infectious clones of the cherry strain of TBSV (TBSV-C) and its derived DI-RNA, DI-72, were kindly provided by K. Andrew White and have been described previously (41, 43). Infectious clones of the pepper strain of TBSV (TBSV-P), CymRSV, and chimeras L1, L2, L4, L5, L9, L10, L11, and L12 between TBSV-P and CymRSV have also been described (36, 44). Infectious clones of TBSV-TAG1, TBSV-TAG2, and DI-72–TAG3 were constructed by introducing the oligonucleotide markers TAG1 (5′-CCGTCCAGAGTCGACC-3′), TAG2 (5′-GTGGTTCATCCCGTAA-3′), and TAG3 (5′-CAAGGACCACAGTCGA-3′) into TBSV-C (between the end of the 5′ untranslated region [UTR] and the start codon of ORF1) or wild-type DI-72 (between regions I and II) by assembly PCR using specific primers and subsequent ligation in the pCR-XL-TOPO vector (Life Technologies). TBSV-Cstop19, TBSV-Pstop19, and CymRSVstop19 were constructed by introducing two stop codons in the third and sixth codons of the p19 ORF by site-directed mutagenesis, as described previously (45), without disturbing the amino acid sequence of the p22 protein, using the commercial Quikchange II XL site-directed mutagenesis kit (Agilent Technologies), following the manufacturer's instructions. All plasmids were linearized with SmaI and transcribed to RNA using T7 RNA polymerase (New England BioLabs). In vitro RNA transcripts were used to inoculate N. benthamiana plants as described previously (36), and these plants were used for virus multiplication and for purification of virus particles and virion RNA (46).
For MOI estimates, the first three leaves of 3-week-old N. benthamiana plants were mechanically inoculated with a 1:1 mixture of virion RNAs (a total of 1 μg of viral RNA per inoculated leaf) of two different virus species or variants, depending on the treatment. We checked that, at this 1:1 ratio in the inoculum, the accumulations of both variants were similar in the coinfected plants (data not shown). We also checked that the inoculum concentration was such as to saturate the number of initially infected cells. In treatments involving DI-RNA, TBSV-TAG1 and DI-72–TAG3 were at a 1:2 ratio to ensure optimal DI-RNA accumulation in infected plants. The inoculated N. benthamiana plants were grown in chambers at 24 to 25°C with a 16-h photoperiod.
Protoplast preparations and single-cell quantitative PCR (qPCR).
In all experiments, protoplasts were prepared from the sixth leaf, i.e., the third one over the inoculated leaves, which was systemically infected. This leaf was harvested 7 days postinoculation (dpi), and the accumulations of both viral variants were quantified in the sampled leaf using reverse transcriptase (RT)-qPCR with specific primers (data not shown). For each treatment, three independent replicated experiments were performed, involving four coinfected plants. For each replica, protoplasts were prepared from 8 2-cm-diameter leaf discs from the pool of four plants, with 2 discs excised from each plant. The leaf discs were sterilized with a 20-fold-diluted bleach solution for 5 min and washed 3 times with sterile water. Tissue strips were digested with 1% Cellulase Onozuka R10 and 0.25% Macerozyme R10 (Duchefa) overnight at 25°C under dark conditions. To eliminate viral RNA from disturbed cells in the supernatant, the protoplast preparations were incubated at room temperature for 1 h to allow RNA degradation by endogenous RNases. Afterward, the protoplasts were sedimented through a 21% sucrose cushion with 5 min centrifugation at 100 × g and, to prevent further cell disruption, were fixed and washed with 1× phosphate-buffered saline (PBS), as described previously (21). Finally, the protoplast preparations were stained for 5 min using EZBlue gel-staining reagent (Sigma-Aldrich) and diluted in RNase-free water to 1 to 2 cells per 10 μl, using a microscope to count the cells. The diluted protoplast suspensions were distributed onto plastic strips, and pieces of plastic containing only one protoplast were excised using a binocular stereomicroscope (Leica MZ10F) and transferred into 0.2-ml PCR tubes as described previously (21). Forty-eight protoplasts were analyzed per replica (i.e., 144 protoplasts per treatment). Empty plastic pieces in regions next to protoplasts were used as negative controls (32 per treatment), and protoplasts prepared from N. benthamiana plants singly infected with all the virus variants used in this work were used as positive controls (data not shown).
Synthesis of viral cDNA was done using a PrimeScript RT reagent kit (Perfect Real Time; TaKaRa). Briefly, 7.3 μl of RNase-free water was added to each tube containing a single protoplast or an empty plastic piece, and to ensure accessibility to the RT, viral RNA was denatured at 65°C for 5 min. After a 2-min incubation on ice, 2 μl of 5× PrimeScript buffer, 0.2 μl of reverse primer at 5 mM, and 0.5 μl of PrimeScript RT Enzyme Mix I were added to each tube. The reaction mixture was incubated for 15 min at 50°C, followed by 2 s at 85°C to inactivate the enzyme. The primers used for RT reactions were designed to specifically anneal to sequences conserved between the two virus variants in each experiment and are reported in Table S1 in the supplemental material. The RT reaction mixtures were 1/5 diluted with RNase-free water and used as the template for two qPCRs using primers specific for each virus variant (see Table S1 in the supplemental material). The efficiency of each pair of primers used was estimated by calculating the slope of a standard curve of serial dilutions of infected RNA samples, and the curves were similar for all the virus species or genotypes analyzed. The qPCRs were done using a LightCycler 480 instrument (Roche) in plates with 384 wells using Brilliant III Ultra-Fast SYBR green QPCR master mix (Agilent Technologies). Two microliters of diluted cDNA, 5 μl of 2× master mix, 0.4 μl of each primer at 5 mM, and 2.2 μl of RNase-free water were mixed and brought to a final volume of 10 μl. The qPCR conditions were as recommended by the manufacturer. Analyses of singly and doubly infected protoplasts were done using the LightCycler 480 software under the module “Tm calling.”
Estimation of the MOI.
To estimate the number of coinfecting genomes in a cell, i.e., the MOI when referring to a single virus species, the frequencies of singly and doubly infected and noninfected cells were calculated from the qPCR data obtained from single protoplasts (see Table S2 in the supplemental material). The MOI was calculated from these frequencies according to five mathematical models (models 2, 3, 4, 5, and 6) originally proposed by Zwart et al. (18) and Tromas et al. (22). Model 2 is based on the assumption that the distribution of infecting viral genomes per cell follows a Poisson distribution, from which the number of coinfecting genomes, i.e., the MOI, is derived. The other models derive from model 2 by introducing new parameters that may explain different aspects of the dynamics of viral infection, that is, spatial segregation of both coinfecting virus variants (model 3), spatial aggregation of infected cells (model 4), superinfection exclusion at the cellular level (model 5), or a mix of spatial segregation of virus variants and spatial aggregation of infected cells (model 6) (18, 22). The introduction of these parameters makes the models more realistic, since, as plant cells are infected by cell-to-cell movement through the plasmodesmata, spatial considerations of virus distribution should increase the model's explanatory power. To choose the best model, the MOI was estimated using the mean frequency over the three independent replicas of uninfected and coinfected cells (see Tables S2 and S3 in the supplemental material). Model 4 was best supported in 10 of 16 experiments analyzed, according to a maximum-likelihood approach and to the Akaike information criterion (18, 22). Hence, model 4 was used to estimate the MOI in each of the three independent replicas per treatment, which allowed the estimation of the mean and standard error (SE) for each treatment. The calculated MOI corresponds to mI of Zwart et al. (18), i.e., the MOI considering only infected cells, with a range of 1 to infinity, first described by González-Jara et al. (14), as opposed to mT, i.e., the MOI also considering uninfected cells, with a range of 0 to infinity, described by Gutiérrez et al. (20), as discussed previously (10). MOI estimations were performed in R version 3.0.2 (47) using equations for the different models as described in reference 18, with our own R scripts. Data on the frequency of infected cells (total and singly and doubly infected) and on the MOI were normally distributed and homoscedastic, according to Kolmogoroff-Smirnoff and to Levene's test of equality of error variances, respectively. Thus, differences in the frequencies of infected cells and in the MOI depending on treatments were analyzed by means of Student's t test (48). The percentage of the total variance in the MOI accounted for by differences in the viral genotype was derived from variance component analyses, after analyses of variance (ANOVA). Correlations between variables were tested using Pearson coefficients. Comparison and correlation analyses were done using SPSS v.17.0.
Since the MOI is a property of a given virus in a given host cell, to refer to this parameter in mixed infections with two virus species, we use the term NCG.
RESULTS
Estimation of the MOI in coinfections of tombusviruses and derived DI-RNAs.
The MOI level during TBSV infection was estimated in plants infected by two TBSV variants, TBSV-TAG1 and TBSV-TAG2, which carry two different 16-nucleotide (nt) markers in the TBSV-C background (see Materials and Methods) (Fig. 1). Both markers were stable in the TBSV genome for at least three passages in N. benthamiana, and their insertion did not significantly affect the kinetics of the tagged TBSV variants compared to the wild-type virus TBSV-C (data not shown). The frequency of infected cells over the total analyzed cells was 0.672 ± 0.054, and the frequency of doubly infected cells over the total analyzed cells was 0.294 ± 0.049 (Table 1). The MOI calculated from these data using model 4 (18, 22) was 1.797 ± 0.158 (Table 1).
FIG 1.
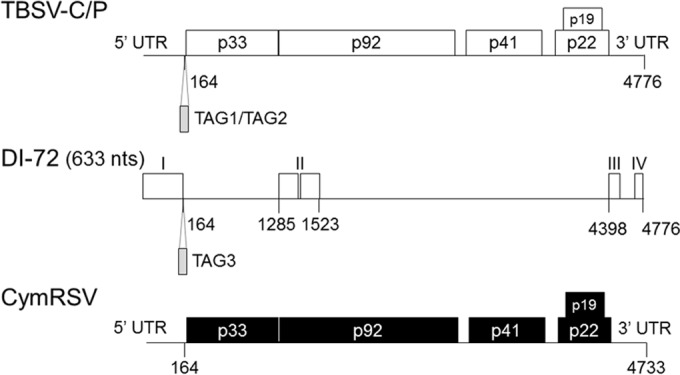
Schematic representation of the genomes of TBSV-C/P, DI-72, and CymRSV. The organization of the coding regions and UTRs of TBSV-C/P (white boxes) and CymRSV (black boxes) is shown. The four regions of DI-72 (I to IV) derived from TBSV are outlined with boxes.
TABLE 1.
MOI or NCG levels of tombusviruses and derived DI-RNAs in mixed infections
| Virus Aa | Virus Ba | Frequencyb over total analyzed: |
MOI or NCGc | |||
|---|---|---|---|---|---|---|
| Total infected cells | Virus A singly infected cells | Virus B singly infected cells | Doubly infected cells | |||
| TBSV-TAG1 | TBSV-TAG2 | 0.672 ± 0.054 | 0.107 ± 0.054 | 0.272 ± 0.076 | 0.294 ± 0.049 | 1.797 ± 0.158 |
| TBSV-C | TBSV-P | 0.669 ± 0.089 | 0.267 ± 0.060 | 0.136 ± 0.054 | 0.267 ± 0.094 | 1.759 ± 0.221 |
| TBSV-TAG1 | DI-72-TAG3 | 0.676 ± 0.151 | 0.287 ± 0.149 | 0.078 ± 0.050 | 0.311 ± 0.233 | 2.392 ± 0.930 |
| CymRSV | TBSV-C | 0.903 ± 0.045 | 0.073 ± 0.048 | 0.054 ± 0.036 | 0.776 ± 0.022 | 4.230 ± 0.204 |
| CymRSV | TBSV-P | 0.951 ± 0.011 | 0.048 ± 0.026 | 0.187 ± 0.098 | 0.717 ± 0.071 | 3.978 ± 0.188 |
Virus A and virus B represent coinfecting viruses in each treatment.
The data are means ± standard errors from three replicated experiments of the frequencies of total infected cells, singly infected cells, and doubly infected cells over total analyzed cells.
MOI and NCG were computed over the fraction of infected cells using model 4 (18).
The MOI during TBSV infection was also estimated in plants coinfected by two wild-type strains of TBSV, TBSV-C and TBSV-P, whose genomes are 93% identical. The frequencies of infected cells (0.669 ± 0.089) and doubly infected cells (0.267 ± 0.094) were the same as in plants coinfected by the tagged variants derived from TBSV-C (Table 1), underscoring that the introduction of a marker in the TBSV genome did not affect the dynamics of infection. From these data, an MOI value of 1.759 ± 0.221 was estimated (Table 1), which did not differ from that in the TBSV-TAG1–TBSV-TAG2-coinfected plants (t4 = 0.662; P = 0.570).
The coinfection of TBSV-TAG1 with a tagged TBSV-derived DI-RNA (DI-72–TAG3) (Fig. 1) was analyzed next. The frequency of infected cells was 0.676 ± 0.151, and the frequency of doubly infected cells was 0.311 ± 0.233. The estimated MOI was 2.392 ± 0.930 (Table 1), which was not significantly different from the MOI values in coinfections by two TBSV variants (t7 = 0.417; P = 0.689).
To estimate the MOI in plants coinfected by two different tombusvirus species, N. benthamiana plants were inoculated with a mixture of CymRSV (Fig. 1) and either TBSV-C or TBSV-P. The results were similar in both coinfections (Table 1). The frequencies of infected cells were 0.903 ± 0.043 and 0.951 ± 0.011 for CymRSV–TBSV-C and CymRSV–TBSV-P, respectively (Table 1). The frequencies of doubly infected cells were 0.776 ± 0.022 and 0.717 ± 0.071 for CymRSV–TBSV-C and CymRSV–TBSV-P, respectively (Table 1). Thus, the frequencies of both infected cells and doubly infected cells were higher than in plants coinfected with two TBSV variants (t10 < −3.101; P < 0.011). Importantly, the fraction of doubly infected cells over total infected cells in CymRSV-TBSV coinfections (0.812) was about twice that in coinfection by two TBSV variants (0.418). The estimated NCG were 4.230 ± 0.204 and 3.978 ± 0.188 for CymRSV–TBSV-C- and CymRSV–TBSV-P-coinfected plants, respectively (Table 1). While these two values were not different (t4 = 0.488; P = 0.654), they were about 2.5-fold higher than in plants coinfected by two TBSV variants or by TBSV and a DI-RNA (t10 = −3.984, P = 0.003, and t7 = −6.366, P < 10−3) (Table 1). In all experiments involving coinfections by CymRSV and TBSV, the frequencies of cells singly infected by either CymRSV or TBSV were similar (t4 > −1.775; P > 0.150) (Table 1), which strongly suggests that no synergism occurs between the two viruses that could bias the estimation of the NCG.
Role of tombusviral genes in MOI level determination.
The hypothesis that the MOI level may be determined by the activity of viral proteins, as reported for other viruses infecting bacteria and mammalian cells, was tested. For this purpose, and since MOI or NCG levels varied greatly in plants coinfected by two TBSV variants and by two tombusvirus species, each CymRSV ORF was replaced with its orthologue ORF in the TBSV genome (Fig. 2A), and NCG levels were estimated in plants coinfected by the recombinant viruses and CymRSV. Recombinant viruses accumulated to levels similar to those of the wild-type parental viruses and caused similar symptoms in N. benthamiana plants (reference 44 and data not shown). In most treatments (L1-CymRSV, L5-CymRSV, L9-CymRSV, and L12-CymRSV), the frequency of infected cells was lower than that in CymRSV-TBSV coinfections, ranging between 0.825 ± 0.022 (for L1-CymRSV) and 0.609 ± 0.047 (for L9-CymRSV) (t7 < −2.737; P < 0.029) (Fig. 2B). The frequency of doubly infected cells over total analyzed cells was also lower than in CymRSV-TBSV coinfections (t7 < −4.042; P < 0.005), ranging from 0.449 ± 0.031 (L5-CymRSV) to 0.127 ± 0.065 (L9-CymRSV) (Fig. 2B). In all the combinations, including a virus chimera, the estimated NCG was lower than in the CymRSV-TBSV-coinfected plants (t7 < −5.043; P < 0.003), with values ranging between 2.483 ± 0.084 (L1-CymRSV) and 1.568 ± 0.089 (L9-CymRSV) (Fig. 2B). This lower value was the only one similar to that found in plants coinfected by two TBSV variants (t7 = −0.767; P = 0.468). These results suggest a major role of ORF2 in determining the low MOI values characteristic of coinfections with two variants of TBSV, although decreasing NCG values in the coinfections by CymRSV and the various chimeras also suggest the involvement of other viral genes.
FIG 2.

Schematic representation of chimeras between TBSV-P and CymRSV and the NCG in various coinfections. (A) Chimeras coinoculated with CymRSV or with TBSV-P are shown, with ORFs derived from TBSV-P indicated as white boxes and ORFs derived from CymRSV indicated as black boxes. (B) Frequencies of total infected cells, doubly infected cells, and the NCG for each coinfection. The values are means ± standard errors of the results of three replicate experiments.
To corroborate these data, experiments were performed using coinfections of TBSV-P and the corresponding recombinants in the CymRSV genome background (Fig. 2A), although chimera availability did not allow us to analyze as many recombinants as in the TBSV background. In all the coinfections, the frequency of infected cells over the total analyzed cells was similar to that in CymRSV-TBSV coinfection (t7 > −1.691; P > 0.135) (Fig. 2B). In L2–TBSV-P- and L10–TBSV-P-coinfected plants, the frequencies of doubly infected cells were similar to that in CymRSV-TBSV coinfection, 0.710 ± 0.085 (t7 = −0.479; P = 0.647) and 0.598 ± 0.079 (t7 = −2.011; P = 0.084), respectively. The estimated NCG values were also similar, 3.869 ± 0.570 (t7 = −0.462; P = 0.658) and 3.073 ± 0.460 (t7 = −2.242; P = 0.060) for L2–TBSV-P and L10–TBSV-P, respectively. However, in the L11–TBSV-P coinfection, the frequency of doubly infected cells, 0.258 ± 0.142, was lower than in the CymRSV-TBSV coinfection (t7 = −4.589; P = 0.003), and the estimated NCG, 2.571 ± 0.204, was also lower (t7 = −4.498; P = 0.003) and similar to that in the TBSV-C–TBSV-P coinfection (t7 = 0.331; P = 0.751). These results are in contrast to those for TBSV-background chimeras and could indicate a possible role of both ORF1 and ORF2 in determining MOI levels (Fig. 2B).
The NCG values in the set of treatments presented in Fig. 2 and in the CymRSV–TBSV-P coinfection (Table 1) show a continuous unimodal distribution, indicating that the NCG is not determined by a single tombusvirus gene or encoded protein. Rather, tombusvirus genes or proteins are quantitative determinants of the MOI, and the genotypes of the coinfecting viruses explained a large fraction of the variance in the MOI across treatments (62.98%). When the set of data in which CymRSV coinfected with TBSV or CymRSV-TBSV chimeras was analyzed separately from the data in which TBSV coinfected with CymRSV or TBSV-CymRSV chimeras, the fractions of the NCG variance explained by the interacting genotypes were 78.03% and 38.23%, respectively, underscoring the asymmetry of gene interactions in MOI determination depending on the genomic background of the virus.
Next, we wondered if the effect of the genotype on the MOI or NCG could be explained by differences in the RNA sequences of the coinfecting viral genomes. To test this hypothesis, we analyzed if the MOI or NCG was correlated with the sequence identity of the coinfecting viruses. A negative linear correlation between the estimated MOI or NCG and the sequence identity of the coinfecting viruses was found (R = −0.642; P = 0.024, in a Pearson correlation analysis) (Fig. 3). Still, NCG values could vary greatly among plants coinfected with viruses with very similar sequence identities of ∼80% (i.e., L1-CymRSV, L4-CymRSV, L9-CymRSV, L2-TBSV-P, and L10–TBSV-P) (Fig. 3), indicating that the effect of the virus genotype in MOI/NCG determination could not be explained solely on the basis of genome similarity and strongly suggesting the involvement of viral proteins. This result further indicated that more than one mechanism could be involved in MOI/NCG determination.
FIG 3.
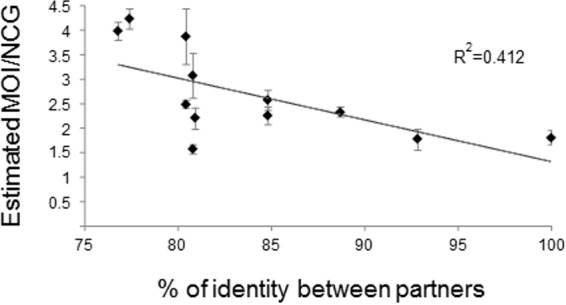
Correlation between sequence identity of the genomes of coinfecting viruses and the estimated MOI. Correlation analysis between the percentage of sequence identity and the estimated MOI or NCG is shown as an R2 statistic (df = 10). Tendency is represented by a dashed line. Standard error bars are represented.
Role of RNA silencing in the determination of MOI levels.
The negative correlation between the sequence identity of the coinfecting genomes and MOI or NCG levels could be explained by the degradation activity of the plant RNA-silencing machinery on the genomes of the same or related viruses. To test this hypothesis, we estimated MOI or NCG values in plants coinfected with different combinations of three mutant viruses that did not express the silencing suppressor protein p19, CymRSVstop19, TBSV-Cstop19, and TBSV-Pstop19 (45). The rationale for this approach was that if RNA silencing has a role in determining the MOI or NCG, increasing the RNA-silencing activity by inactivation of the silencing suppressor proteins would result in lower MOI or NCG levels than in coinfections with wild-type viruses, due to more efficient degradation of the genomic RNAs of both viruses. N. benthamiana plants infected with these mutants showed symptoms of infection at 6 to 7 dpi, but they developed a recovery-like phenotype in the new leaves in contrast to the lethal necrosis caused by the wild-type viruses, as described previously (45). Table 2 presents the results of these coinfections in comparison with the previous results for the wild-type viruses. Abolishing the expression of p19 did not have a gross effect on the frequencies of cells singly infected by each virus, which were similar (t4 < 4.831; P > 0.080), except in the case of CymRSVstop19–TBSV-Cstop19 (t4 = 4.416; P = 0.021) (Table 2). In the CymRSVstop19–TBSV-Cstop19 and CymRSVstop19–TBSV-Pstop19 coinfections, the frequencies of infected cells were the same as in the CymRSV-TBSV coinfections (Table 2), showing that the kinetics of infection was not affected by the lack of expression of the p19 protein. However, the frequencies of doubly infected cells were dramatically reduced (0.367 ± 0.051 for TBSV-Cstop19 and 0.375 ± 0.082 for TBSV-Pstop19) relative to the CymRSV-TBSV coinfection (t4 > 3.157; P < 0.034), and so was the estimated NCG: 2.234 ± 0.046 for CymRSVstop19–TBSV-Cstop19 and 2.239 ± 0.238 for CymRSVstop19–TBSV-Pstop19 (t4 > 3.285; P < 0.030) (Table 2). On the other hand, in the TBSV-Cstop19–TBSV-Pstop19 coinfection, the frequency of infected cells (0.687 ± 0.063), the frequency of doubly infected cells (0.263 ± 0.075), and the MOI (1.757 ± 0.145) were not significantly different from those in the TBSV-C–TBSV-P coinfections (t4 > −0.168; P > 0.875) (Table 2). Thus, these data show a role of RNA silencing in MOI determination.
TABLE 2.
Comparison of the MOI or NCG levels of wild-type tombusvirus and p19 mutants
| Virus Aa | Virus Ba | Frequencyb over total analyzed: |
MOI or NCGc | |||
|---|---|---|---|---|---|---|
| Total infected cells | Virus A singly infected cells | Virus B singly infected cells | Doubly infected cells | |||
| CymRSV | TBSV-C | 0.903 ± 0.045 | 0.073 ± 0.048 | 0.054 ± 0.036 | 0.776 ± 0.022 | 4.230 ± 0.204 |
| CymRSVp19 | TBSV-Cp19 | 0.837 ± 0.006 | 0.404 ± 0.069 | 0.066 ± 0.033 | 0.367 ± 0.051 | 2.234 ± 0.046 |
| CymRSV | TBSV-P | 0.951 ± 0.011 | 0.048 ± 0.026 | 0.187 ± 0.098 | 0.717 ± 0.071 | 3.978 ± 0.188 |
| CymRSVp19 | TBSV-Pp19 | 0.821 ± 0.048 | 0.378 ± 0.062 | 0.067 ± 0.018 | 0.375 ± 0.082 | 2.239 ± 0.238 |
| TBSV-C | TBSV-P | 0.669 ± 0.089 | 0.267 ± 0.060 | 0.136 ± 0.054 | 0.267 ± 0.094 | 1.759 ± 0.221 |
| TBSV-Cp19 | TBSV-Pp19 | 0.687 ± 0.063 | 0.174 ± 0.042 | 0.250 ± 0.059 | 0.263 ± 0.075 | 1.757 ± 0.145 |
Virus A and virus B represent coinfecting viruses in each treatment. TBSV-Cp19, TBSV-Pp19, and CymRSVp19 correspond to TBSV-Cstop19, TBSV-Pstop19, and CymRSVstop19.
The data are means ± standard errors from three replicated experiments of the frequencies of total infected cells, singly infected cells, and doubly infected cells over total analyzed cells.
MOI and NCG were computed over the fraction of infected cells using model 4 (18).
DISCUSSION
The MOI is an important parameter in virus evolution, as it affects evolutionary processes, such as genetic exchange, selection intensity on viral genes, hybrid incompatibility, the evolution of segmented and multipartite genomes, and hyperparasitism by molecular parasites, such as satellites and defective interfering nucleic acids or particles (10, 12, 13, 49, 50). A high MOI results in high genetic diversity, which would be advantageous to viruses in a new environment, but limiting the MOI would be advantageous for the fittest genomes and reduces the pressure of molecular hyperparasites on the virus. Hence, it could be speculated that the MOI would evolve to an optimum value and/or vary during host colonization (14, 20, 22, 24). The corollary is that the MOI would be controlled at least in part by virus functions, and evidence derived mostly from the study of bacteriophages (31, 33–35) indicates that this is the case. Although it has been reported that the CTV-encoded protein p33 was required for superinfection exclusion at the plant and tissue level, it did not seem to have a role in MOI determination (23, 28), and for plant viruses, there is no evidence of a role of viral functions in the control of the MOI.
In this work, we present the first attempt to decipher the molecular mechanisms involved in the control of the MOI in a plant virus. We used coinfections by two species or variants of tombusviruses, thus comparing systems where exclusion mechanisms are highly efficient, resulting in low MOIs and, consequently, in a small fraction of coinfected cells (e.g., in coinfections by variants of the same virus species, TBSV), with systems where the efficiency of exclusion mechanisms is lower, resulting in high NCG and in a large fraction of coinfected cells (e.g., in coinfections by two virus species, CymRSV and TBSV). We estimated the MOI considering only infected cells, i.e., MOI estimates corresponding to mI (18), because only infected cells are relevant in the dynamics and evolution of viral populations (18). The MOI for TBSV infecting N. benthamina plants, estimated from coinfections with two TBSV variants, was very similar to those reported for other plant viruses (14, 20, 22), which strongly suggests that the MOI for CymRSV, which was not estimated, is similar. This low MOI results in the spatial segregation within the infected leaf of genetic variants of the same tombusvirus species, as indicated by a fraction of coinfected cells over total infected cells of about 40%. The spatial segregation of genetic variants within the leaf agrees with reports for other plant viruses (14–16, 22). On the other hand, the model for MOI estimation that best fit the experimental results was model 4 of Zwart et al. (18), which considers the aggregation of infected cells through the value of parameter β, a parameter varying between 1 (no aggregation) and 0. The good fit to the data of model 4 emphasizes the relevance of the aggregation of infected cells during the progress of infection in a leaf, as shown for TEV (22). This is not an unexpected result, as leaf colonization occurs by cell-to-cell movement of infection (51). Still, the estimated values of β were much higher than those reported for TEV infecting N. tabacum (0.94 to 0.99 versus 0.125 to 0.544), indicating less aggregation, in agreement with the greater fraction of infected cells in leaves infected by TBSV (Table 1) than in those infected by TEV (22).
The higher NCG estimated from coinfection of CymRSV and TBSV indicates less efficient exclusion mechanisms between genetically distant genomes, resulting in less spatial segregation (about 80% of infected cells were coinfected by both viruses), again as reported for other systems (15). The higher NCG in CymRSV-TBSV coinfections allowed the analysis of viral functions involved in the control of the MOI. Coinfections by CymRSV with chimeras in which each CymRSV ORF was replaced by that of TBSV identified the product of ORF2 as a major determinant, because the NCG in the L9-CymRSV coinfection was not significantly different from that in coinfections by two TBSV variants (Fig. 2). It is noteworthy that the effect of the product of ORF1 seems to counter that of ORF2, since the NCG in L12-CymRSV coinfection was higher than in L9-CymRSV coinfection. This result points to epistatic interactions between the products of ORF1 and -2, i.e., between proteins p33 and p92, in the determination of the MOI. Both the p33 and p92 proteins are essential for viral replication; p33 is an RNA-binding protein involved in functions other than viral replication, such as subgenomic-RNA synthesis or RNA recombination, while the readthrough p92, which includes the whole sequence of the p33 protein, is the viral RNA-dependent RNA polymerase (RdRp). The interaction of p33 and p92 has been demonstrated to be necessary for some aspects of tombusvirus replication (52). Interestingly, the L10-TBSV coinfection does not indicate a role of the product of ORF2 in MOI determination. The asymmetry in the role of the ORF2 product shown by results obtained in L9-CymRSV and L10-TBSV coinfections can also be explained by epistasis, which would change sign according to the origin of the ORF and the genetic background, CymRSV or TBSV. This result agrees with the coadaptation of the different genetic modules within the genomes of viruses (53–58), which could explain the different roles of these ORFs/proteins depending on the genetic background. Note also that proteins with a major role in MOI determination, encoded by ORFs 1 and 2, are involved in genome replication, which might have different rates for TBSV and CymRSV. Another interesting finding is that the proteins encoded by other tombusviral ORFs also have a role in the determination of the MOI, which varies quantitatively, and the genotypes involved in the coinfections explain a high fraction of MOI variance. Again, the separate analyses of coinfection with chimeras in the CymRSV or TBSV genetic background underscore the asymmetry of the interactions.
The experimental approach followed here to detect singly and doubly infected cells allowed us to estimate the MOI in TBSV and DI-RNA coinfections. The MOI and the frequency of coinfected cells were the same as in coinfections by two TBSV variants. This efficient exclusion of DI-RNA from cells infected by TBSV agrees with observations on the restriction of DI-RNA superinfection in cells with primary infections with VSV (35). This result shows that the high accumulation of DI-RNAs relative to TBSV, or to other HVs in infected plants (references 41 and 42 and results not shown), is not due to a large fraction of coinfected cells. Rather, a low MOI efficiently prevents molecular hyperparasitism in TBSV-infected cells, and high DI-RNA accumulation must be explained by faster replication of the DI-RNA in subpopulations of the plant cells infected by the HV.
The low MOI in DI-RNA–TBSV coinfections and the correlation between the MOI and sequence identity between the genomes of coinfecting tombusviral species and genotypes (Fig. 3) strongly suggest a role of RNA silencing in the exclusion mechanism leading to a low MOI. This hypothesis is supported by the analyses of coinfections with mutant CymRSV and TBSV in which the expression of the silencing suppressor p19 was abolished. Although other possible roles of p19 cannot be discarded as a cause of these results, it seems unlikely that the different MOI/NCG observed when p19 expression was abolished were not related to differences in silencing activities: p19 has been reported to be a determinant of pathogenicity and systemic movement (37), well-known roles of viral silencing suppressors that are associated with their suppressor activities. It should be pointed out that the MOI in coinfections by two TBSV mutants not expressing p19 was the same as in coinfection by the wild-type viruses. However, this observation does not deny a role of RNA silencing in MOI control, and it could be explained by saturation of the RNA-silencing machinery. That is, during the exclusion process of two TBSV genomes, RNA silencing would have the same high efficiency against both viral genomes, regardless of the expression of the viral suppressor protein, which would not be the case with two different viruses, which would share a smaller fraction of viral small interfering RNAs (vsiRNAs). Also, it could be speculated that the MOI in coinfections by variants of the same virus has evolved to a lower threshold optimum and that enhancement of RNA silencing would not have a further effect in reducing the MOI. In any case, our results clearly show a role of a virus-encoded suppressor of silencing in MOI determination.
Thus, the results presented here show that the MOI in tombusvirus infections is controlled by virus-encoded functions. RNA silencing also seems to have a role in MOI determination. As the MOI has an important role in virus fitness and evolution, this result can be interpreted as yet another instance of virus manipulation of the host plant RNA-silencing machinery to optimize its fitness (59). Our results also show that RNA-silencing manipulation is not the only way for the virus to control the MOI but that functions encoded by viral ORFs, and most probably due to the expressed proteins, also have a role in MOI determination. The complexity of the interactions between viral proteins for MOI determination and the underlying mechanisms require further investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Andrew White (York University, Toronto, Canada) for generously supplying infectious clones of TBSV-C and DI-72, Mark P. Zwart for helpful discussion of his MOI models and how to implement them, and Israel Pagán for helpful suggestions and criticisms of an earlier version of this work.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02345-15.
REFERENCES
- 1.Syller J. 2012. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol Plant Pathol 13:204–216. doi: 10.1111/j.1364-3703.2011.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waner JL. 1994. Mixed viral infections: detection and management. Clin Microbiol Rev 7:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.May RM, Nowak MA. 1995. Coinfection and the evolution of parasite virulence. Proc R Soc B Biol Sci 261:209–215. doi: 10.1098/rspb.1995.0138. [DOI] [PubMed] [Google Scholar]
- 4.Gandon S, Jansen VAA, van Baalen M. 2001. Host life history and the evolution of parasite virulence. Evolution 55:1056–1062. doi: 10.1554/0014-3820(2001)055[1056:HLHATE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 5.Escriu F, Fraile A, García-Arenal F. 2003. The evolution of virulence in a plant virus. Evolution 57:755–765. [DOI] [PubMed] [Google Scholar]
- 6.DaPalma T, Doonan B, Trager N, Kasman L. 2010. A systematic approach to virus-virus interactions. Virus Res 149:1–9. doi: 10.1016/j.virusres.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García-Arenal F, Fraile A, Malpica JM. 2001. Variability and genetic structure of plant virus populations. Annu Rev Phytopathol 39:157–186. doi: 10.1146/annurev.phyto.39.1.157. [DOI] [PubMed] [Google Scholar]
- 8.Olkkonen VM, Bamford DH. 1989. Quantitation of the adsorption and penetration stages of bacteriophage Phi6 infection. Virology 171:229–238. doi: 10.1016/0042-6822(89)90530-8. [DOI] [PubMed] [Google Scholar]
- 9.Turner PE, Chao L. 1998. Sex and the evolution of intrahost competition in RNA virus Phi6. Genetics 150:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner PE, Burch CL, Hanley KA, Chao L. 1999. Hybrid frequencies confirm limit to coinfection in the RNA bacteriophage Phi6. J Virol 73:2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Susskind MM, Botstein D, Wright A. 1974. Superinfection exclusion by P22 prophage in lysogens of Salmonella typhimurium. III. Failure of superinfecting phage DNA to enter sieA+ lysogens. Virology 62:350–366. [DOI] [PubMed] [Google Scholar]
- 12.Chao L. 1991. Levels of selection, evolution of sex in RNA viruses, and the origin of life. J Theor Biol 153:229–246. doi: 10.1016/S0022-5193(05)80424-2. [DOI] [PubMed] [Google Scholar]
- 13.Frank SA. 2001. Multiplicity of infection and the evolution of hybrid incompatibility in segmented viruses. Heredity 87:522–529. doi: 10.1046/j.1365-2540.2001.00911.x. [DOI] [PubMed] [Google Scholar]
- 14.González-Jara P, Fraile A, Canto T, García-Arenal F. 2009. The multiplicity of infection of a plant virus varies during colonization of its eukaryotic host. J Virol 83:7487–7494. doi: 10.1128/JVI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich C, Maiss E. 2003. Fluorescent labelling reveals spatial separation of potyvirus populations in mixed infected Nicotiana benthamiana plants. J Gen Virol 84:2871–2876. doi: 10.1099/vir.0.19245-0. [DOI] [PubMed] [Google Scholar]
- 16.Takeshita M, Shigemune N, Kikuhara K, Furuya N, Takanami Y. 2004. Spatial analysis for exclusive interactions between subgroups I and II of Cucumber mosaic virus in cowpea. Virology 328:45–51. doi: 10.1016/j.virol.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 17.González-Jara P, Fraile A, Canto T, García-Arenal F. 2013. The multiplicity of infection of a plant virus varies during colonization of its eukaryotic host. J Virol 87:2374. doi: 10.1128/JVI.03255-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zwart MP, Tromas N, Elena SF. 2013. Model-selection-based approach for calculating cellular multiplicity of infection during virus colonization of multi-cellular hosts. PLoS One 8:e64657. doi: 10.1371/journal.pone.0064657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyashita S, Kishino H. 2010. Estimation of the size of genetic bottlenecks in cell-to-cell movement of Soil-borne wheat mosaic virus and the possible role of the bottlenecks in speeding up selection of variations in trans-acting genes or elements. J Virol 84:1828–1837. doi: 10.1128/JVI.01890-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutiérrez S, Yvon M, Thébaud G, Monsion B, Michalakis Y, Blanc S. 2010. Dynamics of the multiplicity of cellular infection in a plant virus. PLoS Pathog 6:e1001113. doi: 10.1371/journal.ppat.1001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yvon M, Monsion B, Martin J-F, Gutiérrez S, Blanc S. 2009. PCR-based amplification and analysis of specific viral sequences from individual plant cells. J Virol Methods 159:303–307. doi: 10.1016/j.jviromet.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 22.Tromas N, Zwart MP, Lafforgue G, Elena SF. 2014. Within-host spatiotemporal dynamics of plant virus infection at the cellular level. PLoS Genet 10:e1004186. doi: 10.1371/journal.pgen.1004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergua M, Zwart MP, El-Mohtar C, Shilts T, Elena SF, Folimonova SY. 2014. A viral protein mediates superinfection exclusion at the whole-organism level but is not required for exclusion at the cellular level. J Virol 88:11327–11338. doi: 10.1128/JVI.01612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutiérrez S, Pirolles E, Yvon M, Baecker V, Michalakis Y, Blanc S. 2015. The multiplicity of cellular infection changes depending on the route of cell infection in a plant virus. J Virol 89:9665–9675. doi: 10.1128/JVI.00537-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung A, Maier R, Vartanian J-P, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. 2002. Multiply infected spleen cells in HIV patients. Nature 418:144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- 26.Bull JC, Godfray HCJ, O'Reilly DR. 2001. Persistence of an occlusion-negative recombinant nucleopolyhedrovirus in Trichoplusia ni indicates high multiplicity of cellular infection. Appl Environ Microbiol 67:5204–5209. doi: 10.1128/AEM.67.11.5204-5209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardin G. 1960. The competitive exclusion principle. Science 131:1292–1297. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- 28.Folimonova SY. 2012. Superinfection exclusion is an active virus-controlled function that requires a specific viral protein. J Virol 86:5554–5561. doi: 10.1128/JVI.00310-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ratcliff F, Harrison BD, Baulcombe DC. 1997. A similarity between viral defense and gene silencing in plants. Science 276:1558–1560. doi: 10.1126/science.276.5318.1558. [DOI] [PubMed] [Google Scholar]
- 30.Ratcliff FG, MacFarlane SA, Baulcombe DC. 1999. Gene silencing without DNA: RNA-mediated cross-protection between viruses. Plant Cell 11:1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Susskind MM, Botstein D. 1980. Superinfecion exclusion by lambda prophage in lysogens of Salmonella typhimurium. Virology 100:212–216. doi: 10.1016/0042-6822(80)90571-1. [DOI] [PubMed] [Google Scholar]
- 32.Snyder L, McWilliams K. 1989. The rex genes of bacteriophage lambda can inhibit cell function without phage superinfection. Gene 81:17–24. doi: 10.1016/0378-1119(89)90332-6. [DOI] [PubMed] [Google Scholar]
- 33.Kliem M, Dreiseikelmann B. 1989. The superimmunity gene sim of bacteriophage P1 causes superinfection exclusion. Virology 171:350–355. doi: 10.1016/0042-6822(89)90602-8. [DOI] [PubMed] [Google Scholar]
- 34.Sun X, Göhler A, Heller KJ, Neve H. 2006. The ltp gene of temperate Streptococcus thermophilus phage TP-J34 confers superinfection exclusion to Streptococcus thermophilus and Lactococcus lactis. Virology 350:146–157. doi: 10.1016/j.virol.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Whitaker-Dowling P, Youngner JS, Widnell CC, Wilcox DK. 1983. Superinfection exclusion by Vesicular stomatitis virus. Virology 131:137–143. doi: 10.1016/0042-6822(83)90540-8. [DOI] [PubMed] [Google Scholar]
- 36.Dalmay T, Rubino L, Burgyán J, Kollár Á, Russo M. 1993. Functional analysis of Cymbidium ringspot virus genome. Virology 194:697–704. doi: 10.1006/viro.1993.1310. [DOI] [PubMed] [Google Scholar]
- 37.Russo M, Burgyán J, Martelli G. 1994. Molecular biology of tombusviridae. Adv Virus Res 44:381–428. doi: 10.1016/S0065-3527(08)60334-6. [DOI] [PubMed] [Google Scholar]
- 38.White KA, Nagy PD. 2004. Advances in the molecular biology of tombusviruses: gene expression, genome replication and recombination. Prog Nucleic Acid Res Mol Biol 78:187–226. doi: 10.1016/S0079-6603(04)78005-8. [DOI] [PubMed] [Google Scholar]
- 39.Burgyán J, Rubino L, Russo M. 1991. De novo generation of Cymbidium ringspot virus defective interfering RNA. J Gen Virol 72:505–509. doi: 10.1099/0022-1317-72-3-505. [DOI] [PubMed] [Google Scholar]
- 40.Knorr D, Mullin R, Hearne P, Morris T. 1991. De novo generation of defective interfering RNAs of Tomato bushy stunt virus by high multiplicity passage. Virology 181:193–202. doi: 10.1016/0042-6822(91)90484-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Havelda Z, Szittya G, Burgyán J. 1998. Characterization of the molecular mechanism of defective interfering RNA-mediated symptom attenuation in tombusvirus-infected plants. J Virol 72:6251–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scholthof KBG, Scholthof HB, Jackson AO. 1995. The effect of defective interfering RNAs on the accumulation of Tomato bushy stunt virus proteins and implications for disease attenuation. Virology 211:324–328. doi: 10.1006/viro.1995.1410. [DOI] [PubMed] [Google Scholar]
- 43.Hearne P, Knorr D, Hillman B, Morris T. 1990. The complete genome structure and synthesis of infectious RNA from clones of Tomato bushy stunt virus. Virology 177:141–151. doi: 10.1016/0042-6822(90)90468-7. [DOI] [PubMed] [Google Scholar]
- 44.Szittya G, Burgyán J. 2001. Cymbidium ringspot tombusvirus coat protein coding sequence acts as an avirulent RNA. J Virol 75:2411–2420. doi: 10.1128/JVI.75.5.2411-2420.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szittya G, Molnár A, Silhavy D, Hornyik C, Burgyán J. 2002. Short defective interfering RNAs of tombusviruses are not targeted but trigger post-transcriptional gene silencing against their helper virus. Plant Cell 14:359–372. doi: 10.1105/tpc.010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luis-Arteaga M, Rodríguez-Cerezo E, Fraile A, Sáez E, García-Arenal F. 1996. Different Tomato bushy stunt virus strains that cause disease outbreaks in solanaceous crops in Spain. Phytopathology 86:535–542. doi: 10.1094/Phyto-86-535. [DOI] [Google Scholar]
- 47.R Development Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 48.Sokal RR, Rohlf FJ. 1995. Biometry: the principles and practice of statistics in biological research. Freeman and Company, New York, NY. [Google Scholar]
- 49.Szathmáry E. 1992. Natural selection and dynamical coexistence of defective and complementing virus segments. J Theor Biol 157:383–406. doi: 10.1016/S0022-5193(05)80617-4. [DOI] [PubMed] [Google Scholar]
- 50.Szathmáry E. 1992. Viral sex, levels of selection, and the origin of life. J Theor Biol 159:99–109. doi: 10.1016/S0022-5193(05)80770-2. [DOI] [PubMed] [Google Scholar]
- 51.Harries P, Ding B. 2011. Cellular factors in plant virus movement: at the leading edge of macromolecular trafficking in plants. Virology 411:237–243. doi: 10.1016/j.virol.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 52.Nagy PD, Barajas D, Pogany J. 2012. Host factors with regulatory roles in tombusvirus replication. Curr Opin Virol 2:691–698. doi: 10.1016/j.coviro.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Martin DP, van der Walt E, Posada D, Rybicki EP. 2005. The evolutionary value of recombination is constrained by genome modularity. PLoS Genet 1:e51. doi: 10.1371/journal.pgen.0010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Escriu F, Fraile A, García-Arenal F. 2007. Constraints to genetic exchange support gene coadaptation in a tripartite RNA virus. PLoS Pathog 3:e8. doi: 10.1371/journal.ppat.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elena SF, Solé RV, Sardanyés J. 2010. Simple genomes, complex interactions: epistasis in RNA virus. Chaos 20:026106. doi: 10.1063/1.3449300. [DOI] [PubMed] [Google Scholar]
- 56.Sanjuán R, Moya A, Elena SF. 2004. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc Natl Acad Sci U S A 101:15376–15379. doi: 10.1073/pnas.0404125101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lalić J, Elena SF. 2012. Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 109:71–77. doi: 10.1038/hdy.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Monjane AL, Martin DP, Lakay F, Muhire BM, Pande D, Varsani A, Harkins G, Shepherd DN, Rybicki EP. 2014. Extensive recombination-induced disruption of genetic interactions is highly deleterious but can be partially reversed by small numbers of secondary recombination events. J Virol 88:7843–7851. doi: 10.1128/JVI.00709-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brigneti G, Voinnet O, Li W, Ji L, Ding S, Baulcombe DC. 1998. Viral pathogenicity determinants are suppressors of transgene silencing in Nicotiana benthamiana. EMBO J 17:6739–6746. doi: 10.1093/emboj/17.22.6739. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.