

ABSTRACT
In establishing a respiratory infection, vaccinia virus (VACV) initially replicates in airway epithelial cells before spreading to secondary sites of infection, mainly the draining lymph nodes, spleen, gastrointestinal tract, and reproductive organs. We recently reported that interferon gamma (IFN-γ) produced by CD8 T cells ultimately controls this disseminated infection, but the relative contribution of IFN-γ early in infection is unknown. Investigating the role of innate immune cells, we found that the frequency of natural killer (NK) cells in the lung increased dramatically between days 1 and 4 postinfection with VACV. Lung NK cells displayed an activated cell surface phenotype and were the primary source of IFN-γ prior to the arrival of CD8 T cells. In the presence of an intact CD8 T cell compartment, depletion of NK cells resulted in increased lung viral load at the time of peak disease severity but had no effect on eventual viral clearance, disease symptoms, or survival. In sharp contrast, RAG−/− mice devoid of T cells failed to control VACV and succumbed to infection despite a marked increase in NK cells in the lung. Supporting an innate immune role for NK cell-derived IFN-γ, we found that NK cell-depleted or IFN-γ-depleted RAG−/− mice displayed increased lung VACV titers and dissemination to ovaries and a significantly shorter mean time to death compared to untreated NK cell-competent RAG−/− controls. Together, these findings demonstrate a role for IFN-γ in aspects of both the innate and adaptive immune response to VACV and highlight the importance of NK cells in T cell-independent control of VACV in the respiratory tract.
IMPORTANCE Herein, we provide the first systematic evaluation of natural killer (NK) cell function in the lung after infection with vaccinia virus, a member of the Poxviridae family. The respiratory tract is an important mucosal site for entry of many human pathogens, including poxviruses, but precisely how our immune system defends the lung against these invaders remains unclear. Natural killer cells are a type of cytotoxic lymphocyte and part of our innate immune system. In recent years, NK cells have received increasing levels of attention following the discovery that different tissues contain specific subsets of NK cells with distinctive phenotypes and function. They are abundant in the lung, but their role in defense against respiratory viruses is poorly understood. What this study demonstrates is that NK cells are recruited, activated, and contribute to protection of the lung during a severe respiratory infection with vaccinia virus.
INTRODUCTION
Poxviruses are large, brick-shaped, enveloped viruses, each containing a linear double-stranded DNA genome (1). Unlike most other DNA viruses, poxviruses encode transcription and replication machinery that facilitates their life cycle entirely in the cytoplasm of the infected cell (1). There are several public health and biological defense reasons to improve our ability to prevent or treat poxvirus infections. Poxviruses that can cause disease in humans are variola virus (VARV), the causative agent of smallpox, monkeypox, cowpox, and vaccinia virus (VACV) (1–3). Notably, variola and monkeypox viruses are transmitted to humans by the respiratory route and cause profound local and systemic pathological conditions with high fatality rates (1, 4). Progressive bronchiolitis/bronchopneumonia is considered the most frequent and serious complication of respiratory infection and is often the cause of death (4). Despite progress in understanding viral determinants of pathogenicity, we still lack crucial information on the cellular and molecular mechanisms of host defense against respiratory poxvirus infections.
Historically, human smallpox and monkeypox have been modeled in mice using the highly virulent, mouse-adapted Western Reserve strain of VACV (VACV-WR) (5–9). After inhalation, VACV-WR infects multiple cell types in the lung, including alveolar macrophages (10), dendritic cells (DCs) (11), and bronchiolar and alveolar epithelial cells (12). Over the next few days, the virus replicates exponentially in the lung, resulting in perivascular and peribronchial inflammation, hypertrophy and hyperplasia of epithelial cells, and diffuse alveolar damage (9). Notably, VACV has the capacity to enter the bloodstream and disseminate to many organs, including lymph nodes, brain, liver, kidneys, spleen, gastrointestinal tract, and reproductive organs (9). After replicating in these organs, high levels of VACV are again shed into the blood, resulting in a secondary viremia that closely mimics human disease (9). In general, it is believed that recovery from a respiratory VACV infection requires a tightly coordinated response by both the innate and adaptive immune systems. In this regard, a limited number of studies have shown that pattern recognition receptors (13–16), alveolar macrophages (10), and dendritic cells direct the early response to VACV (11, 17, 18). As the adaptive immune response develops, virus-specific CD8 T cells play a vital role in restricting lung pathology and virus dissemination to visceral tissues and are necessary for complete clearance of virus and protection against death (19). CD8 T cells contribute to this protective immunity via the release of interferon gamma (IFN-γ) (20), and there is evidence that IFN-γ is sufficient to protect mice in the absence of CD4 T cells and B cells (20). In the context of early infection, IFN-γ mRNA and total protein are elevated in the lungs of immunocompetent animals in the first 72 to 96 h after VACV infection (20), which is before any virus-specific T cells can be detected in the lung (19, 20). Although the critical cellular source of IFN-γ in late infection is now recognized as VACV-specific CD8 T cells (20), the cellular source and relative importance of early IFN-γ in the lung remains unclear.
Natural killer (NK) cells are large, granular innate lymphocytes that, in contrast to naive T cells, transcribe and modify the IFN-γ gene during early development in the bone marrow (BM) (21). Consequently, mature tissue-resident NK cells are poised to release IFN-γ within minutes to hours of infection (21). Among peripheral tissues, the lung contains the largest percentage of NK cells, indicating potential crucial involvement of NK cells in respiratory infections (22). Indeed, NK cells have been implicated in early defense against several viral (respiratory syncytial virus [RSV] and influenza) and bacterial (Mycobacterium tuberculosis and Bordetella pertussis) pathogens that cause pneumonia (23–25). NK cells may participate in defense against respiratory pathogens via various mechanisms, including direct lysis of infected cells, production of chemokines that amplify the host's inflammatory response, production of antiviral cytokines, such as IFN-γ and tumor necrosis factor alpha (TNF), and activation of DCs that mediate T cell differentiation and trafficking to the lung (23, 26). In some instances, particularly during more severe respiratory infections, NK cells can be responsible for acute lung immune injury and mortality (27–29). While these studies suggest that NK activities can lead to both beneficial and deleterious outcomes, the role of NK cells and innate IFN-γ and their relationship in early host defense against respiratory poxvirus infection have not been directly investigated. In the present study, we performed a thorough characterization of the NK response induced in the lung microenvironment following respiratory VACV-WR infection. We report that NK cells proliferate and display a highly activated phenotype in the lung after VACV infection. Early in infection, NK cells are the main population of innate immune cells in the lung capable of producing IFN-γ. We provide evidence that NK cells can limit both VACV replication and its spread from the lungs and that IFN-γ is an important mediator in this process.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of animals in biomedical research. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Florida, Gainesville, FL (OLAW assurance number A3377-01).
Mice.
Eight- to 12-week-old female wild-type (WT) C57BL/6J, interferon gamma reporter with endogenous poly(A) transcript (GREAT), RAG−/−, and RAG−/− common γ chain−/− mice were purchased from Jackson Laboratory, USA.
Vaccinia virus and stock preparation.
Vaccinia virus Western Reserve (VACV-WR) was purchased from the American Type Culture Collection (ATCC) and grown in HeLa cells, and then the titer was determined on VeroE6 cells as described previously (30).
Respiratory VACV-WR infection model.
Naive mice were anesthetized by isoflurane inhalation and infected intranasally (i.n.) with 1.25 × 104 PFU, with daily measurements of body weight, lung pathology, and survival as described previously (19, 31). No animals were allowed to die of natural causes; therefore, the time of death indicated on the survival curves is the time at which an animal was euthanized due to severe disease (weight loss of >25%). Body weight was calculated as the percentage of the weight for each mouse on the day of challenge.
VACV-WR titer assay.
After VACV infection, specified tissues from individual mice were homogenized and sonicated for 1 min, with a pause every 10 s, using an ultrasonic cleaner (1210 Branson). Serial dilutions were made, and virus titers were determined by plaque assay on confluent VeroE6 cells (32).
Peptides and tetramers.
We utilized the VACV-WR peptide B8R (positions 20 to 27; TSYKFESV) to stimulate CD8 T cells (33, 34). Major histocompatibility complex (MHC)/peptide tetramers containing the B8R peptide (positions 20 to 27; TSYKFESV)/H-2Kb, conjugated to allophycocyanin (APC), were obtained from the National Institutes of Health Tetramer Core Facility (Emory University, Atlanta, GA).
Flow cytometric analysis.
All tissues were aseptically removed from euthanized mice, and single-cell suspensions were prepared by mechanically dispersing the tissues through 70-μm cell strainers (Falcon BD Labware) into Hanks balanced salt solution (HBSS). Lung tissue was treated for 45 min at 37°C with 200 μg Liberase TL and 100 μg DNase I grade II (Roche), followed by treatment for 5 min at 4°C with 5 mM EDTA-supplemented medium. Following red blood cell lysis (Sigma-Aldrich), cells were resuspended in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal calf serum (FCS; Omega Scientific), 1% l-glutamine (Invitrogen), 100 μg/ml streptomycin, 100 U/ml penicillin, and 50 μM 2-mercaptoethanol (2-ME; Sigma-Aldrich) and enumerated using a BD automated Vicell counter.
NK cell and T cell staining.
Cells were washed with fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] and 2% FCS) and incubated with anti-Fc II/III receptor monoclonal antibody 2.4G2 for 15 min at 4°C. After an additional FACS buffer wash, the following antibodies were incubated with the cells for 30 min at 4°C: antibodies to CD4 (RM4-5; BD Pharmingen), CD8 (53-6.7; BD Pharmingen), CD3ε (145-2C11; eBioscience), CD44 (IM7; eBioscience), CD62L (MEL-14; eBioscience), and B8R tetramer (NIH tetramer facility) were used to determine T cell subsets. Anti-mouse antibodies to NK1-1 (PK136; Southern Biotech), CD49b (DX5; eBioscience), NKp46 (29A1.4, fluorescein isothiocyanate [FITC]; eBioscience), CD11b (M1/70; eBioscience), CD27 (LG.7F9; eBioscience), and CD69 (H1.2F3; eBioscience) delineated NK cell populations. All samples were acquired on a FACSCanto II or Fortessa (BD Bioscience) and analyzed using FlowJo (Tree Star).
NK cell IFN-γ cytokine production.
Briefly, after lysing red blood cells (RBCs), splenocytes and lung cells from infected mice were plated in round-bottom 96-well microtiter plates in 200 μl of RPMI 1640 medium (Invitrogen) supplemented with 10% FCS (Omega Scientific), 1% l-glutamine (Invitrogen), 100 μg/ml streptomycin, 100 U/ml penicillin, and 50 μM 2-ME (Sigma-Aldrich). Cells were then either left unstimulated (medium), or received phorbol 12-myristate 13-acetate (PMA) (100 ng/ml)–ionomycin (300 ng/ml) or recombinant interleukin-12 (IL-12) (5 ng/ml) and IL-18 (2.5 ng/ml) and incubated for 1 h at 37°C. GolgiPlug (BD Biosciences) was then added to the cultures according to the manufacturer's instructions, and the incubation was continued for 4 h. Cells were then stained with anti-NK1-1 antibody (PK136; eBioscience) or anti-CD3ε antibody (145-2C11; eBioscience), followed by fixation with Cytofix-Cytoperm (BD Biosciences) for 20 min at 4°C. Fixed cells were subjected to intracellular cytokine staining in Perm/Wash buffer (BD Biosciences) for 30 min at 4°C. Cells were stained with anti-IFN-γ antibody (XMG1.2; eBioscience) for 30 min at 4°C. Samples were analyzed for their proportion of cytoplasmic cytokines after gating on CD3ε− NK1-1+ cells by using a FACSCalibur flow cytometer and FlowJo software.
In vivo depletion studies.
Groups of VACV-WR-infected mice were depleted of NK1-1+ cells with anti-NK1-1 monoclonal antibody (PK136; BioXcell) (200 μg/mouse) given in one intravenous (i.v.) injection 3 days before and intraperitoneal (i.p.) injections on day −1 and every 2 days thereafter until the termination of the experiment. NK cell depletion was confirmed by flow cytometry of spleen and lung tissues for the presence of NKp46+ CD49b+ NK cells.
In vivo IFN-γ neutralization.
IFN-γ was neutralized in VACV-WR-infected RAG−/− mice by using an anti-IFN-γ antibody (clone XMG1.2) (200 μg/mouse) given in one i.v. injection 3 days before and i.p. injections on day −1 and every 2 days thereafter until the termination of the experiment.
Immunofluorescence studies.
At the times indicated below, VACV-infected lungs were inflated with 500 μl 50/50 OCT-PBS embedding solution and immediately snap-frozen on dry ice. Six-micrometer-thick cryosections of OCT-embedded lung and spleen tissues were cut using a Microtome HM 505E cryostat and prepared on SuperFrost glass slides for immunofluorescence microscopy. Each cryosection slide was washed with 1 ml PBS, fixed with 4% paraformaldehyde for 15 min at 4°C, and subsequently permeabilized with 0.1% Triton X-100 for 5 min at 4°C. The sections were washed with cold PBS and incubated overnight in the dark at 4°C with rat anti-mouse NK1.1 phycoerythrin (PE)-conjugated antibody and 500 ng/ml DAPI (4′,6-diamidino-2-phenylindole; Calbiochem). Sections were washed three times with PBS, mounted with Cytoseal, and covered with glass coverslips. The stained lung and spleen sections were observed and analyzed at wavelengths of 488 nm for yellow fluorescent protein (YFP) (green), 543 nm for PE (red), and 405 nm for DAPI (blue) staining, as described previously (35). Using an EVOS fl (Advanced Microscopy Group inverted immunofluorescence microscope), images were captured by 20× and 40× objectives, keeping all the conditions of microscopy and software settings identical for all treatments and controls.
Statistical analysis.
Tests were performed using Prism 5.0 (GraphPad, San Diego, CA). Statistics were done using the two-tailed, unpaired Student's t test with 95% confidence intervals unless otherwise indicated. Two-way analysis of variance (ANOVA) was used to determine differences in weight loss profiles, and the Mantel-Cox test was utilized for survival analysis. Unless otherwise indicated, data represent the mean results ± standard errors of the means (SEM); a P value of <0.05 is considered statistically significant.
RESULTS
VACV-WR infection of the lung elicits a robust NK cell response.
It is known that vaccinia virus (VACV) is a potent inducer of IL-12 and IL-18 (36), which can augment NK activity (37). We were interested in determining whether control of primary respiratory VACV infection in immunocompetent mice correlated with the presence and/or recruitment of NK cells to the site of infection. To do this, cohorts of naive, wild-type (WT) C57BL/6 mice were infected intranasally (i.n.) with the highly virulent, mouse-adapted VACV Western Reserve strain (VACV-WR) (1.25 × 104 PFU), and the kinetics of NK cell recruitment to the lung and peripheral tissues were determined.
In mice, NK cell-committed progenitors have a CD122+ CD3− phenotype and differentiate into mature NK cells through the sequential acquisition of cell surface receptors such as NKp46 (also known as CD335), NK1.1 (a C-type lectin, also called NKR-P1C), CD11b (Mac-1 α; integrin αM chain), and CD49b (α2 integrin; recognized by DX5 monoclonal antibody) (38). Our gating strategy for identifying NK cells in the lung and spleen is shown in Fig. 1A. Dead and doublet cells were excluded through the acquisition of Live/Dead violet dye and forward and side scatter characteristics. Initially, we identified NK cells as CD3− NKp46+ cells, because NKp46 has been found to be the most useful inclusive marker for NK cells for mice (39). In uninfected mice, a sizeable fraction of CD3− cells recovered from the lung (Fig. 1B, top) and spleen (Fig. 1C, top) expressed NKp46. The majority (>90%) of CD3− NKp46+ cells coexpressed markers, including CD11bhigh/bright, associated with developmentally mature cells (40) (Fig. 1B and C, top), NK1.1, and DX5 (Fig. 1B and C, bottom) (41, 42). As the VACV-WR infection proceeded, the relative frequency of NKp46+ cells recovered from the lung increased between days 1 and 4, peaked between days 4 and 6, and contracted by day 8 (Fig. 1B), a time point that correlated well with virus clearance (19). In contrast, the frequency of splenic NK cells did not change significantly over the course of VACV infection (Fig. 1C). Translating these frequencies into total numbers, we observed that the absolute number of NK cells recovered from the lung increased by approximately 6- to 7-fold, compared to 2- to 3-fold in the spleen (Fig. 1D and E).
FIG 1.

Lung and splenic NK cell responses to a respiratory VACV-WR infection. Wild-type C57BL/6J mice were infected intranasally with 1 × 104 PFU of VACV-WR. (A) Representative gating strategy to identify NK cells among live mononuclear cells. NK cell subsets were identified by sequentially gating on live cells, the lymphocytic singlet population, and then CD3− cells. FSC, forward scatter; SSC, side scatter; A, signal area; W, signal width. Kinetic changes in lung (B and D) and spleen (C and E) NK cells were examined over 8 days postinfection (n = 3 per time point). (B and C) Top, representative FACS plots showing NK cells (CD11b+ NKp46+) as percentages of CD3− lymphocytes. Bottom, representative FACS plots showing NK1.1+ CD49b+ cells as percentages of NKp46+ NK cells. Total lung (D) and spleen (E) NK cell numbers are presented as the mean results ± SEM from three separate experiments containing 3 mice per group. Student's t test with Bonferroni correction was used to determine statistical significance. *, P < 0.05, control versus infected mice.
To extend our flow cytometry data, we also evaluated how VACV infection alters the frequency and localization of NK cells within the lung tissue. In naive WT mice, few NK cells were sporadically dispersed in the lung parenchyma (Fig. 2A, day 0). In striking contrast, on day 4 p.i., high numbers of NK1.1+ cells could be readily visualized in the lamina propria of bronchioles, in the surrounding connective tissue, and in the interalveolar septa (Fig. 2B). These cells were seen as both scattered and in clusters. Thus, a nonlethal respiratory VACV-WR infection triggered a vigorous NK response within the parenchymal and peribronchiolar regions of the lung.
FIG 2.
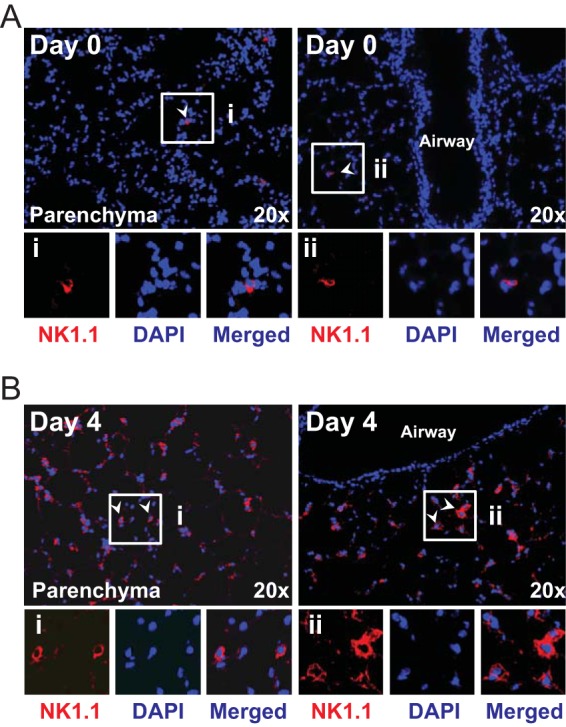
Localization of NK1.1+ cells in the lungs of VACV-WR-infected mice. Wild-type C57BL/6J mice were intranasally infected with 1.25 × 104 PFU of VACV-WR. Lung tissue sections from mice infected with VACV-WR and uninfected control (day 0) mice were stained with DAPI (blue) and for NK1.1 (red) and analyzed by confocal microscopy (20× objective; scale bar = 100 mm). NK1.1+ cells were visualized in the lung parenchyma and lamina propria of bronchioles and airway epithelial cells. Representative pictures taken from animals on day 0 (A) and day 4 (B) postinfection are shown. Arrows point at NK1.1+ cells. Insets shows NK1.1+ cells in the lung tissue of infected animals on the indicated days. Results are representative of three experiments each with three mice per group.
NK cells proliferate and become activated during respiratory VACV-WR infection.
Next, we assessed whether the accumulation of NK cells in the lung and spleen was due to an increase in total numbers of NK cells or reflected an increase in a specific stage of maturation in response to respiratory VACV infection. To do this, CD3− NK1.1+ NK cells were examined for cell surface expression of CD27, a TNF receptor superfamily member that defines distinct maturation and functional NK cell subsets when combined with CD11b (43, 44). When these two markers are analyzed in combination, three major NK cell subsets can be identified: an early mature population characterized as CD11blow CD27high (Fig. 3A, upper left quadrants); an intermediate population defined as CD11bhigh CD27high (Fig. 3A, upper right quadrants); and finally, a terminally differentiated CD11bhigh CD27low NK cell population (Fig. 3A, lower right quadrants). At steady state, the terminally differentiated CD11bhigh CD27low population comprised more than 90% of total lung-resident NK cells, which was a substantially higher fraction than was found in spleen tissues (∼70%) or bone marrow (BM) (10 to 15%) (Fig. 3A, day 0). Interestingly, approximately 10% to 25% of tissue (lung, spleen, and BM)-resident NK cells were undergoing homeostatic proliferation, as shown by costaining for the cell cycle-specific protein Ki67 (Fig. 3B, day 0). VACV infection did not substantially alter the proportions of CD27high and CD27low NK cell subsets within the lung, spleen, or BM but did induce extensive proliferation of both CD27high and CD27low cells as early as day 4 postinfection (p.i.). Strikingly, by day 6 of infection, the majority (80 to 90%) of NK cells trafficking to or within each tissue were actively proliferating. These data imply that VACV-induced cell proliferation is in part responsible for the increase in total numbers of NK cells observed in each tissue.
FIG 3.

Respiratory VACV-WR infection induces NK cell activation and proliferation. Wild-type C57BL/6J (WT) mice were intranasally infected with 1.25 × 104 PFU of VACV-WR. On the indicated days postinfection, lung, spleen, and bone marrow (BM) NK cells (CD3− NKp46+ NK1.1+) were examined for phenotypic and functional markers by flow cytometry. (A) Expression of CD11b and CD27 by gated CD3− NKp46+ NK1.1+ cells in mock-infected (day zero [D0]) and VACV-infected mice. The quadrants show the three major NK subsets (CD11blow CD27high, CD11bhigh CD27high, and CD11bhigh CD27low). (B) Representative FACS plots showing the percentages of NK cell proliferation by Ki67 staining among CD3− NKp46+ NK1.1+ cells in mock-infected (D0) and VACV-infected mice. The numbers in the plots indicate the percentages of NK1.1+ cells that stained for Ki67. (C) Representative FACS plots showing cell surface expression levels of CD69, CD44, CD43, and KLRG by lung NKp46+ NK1.1+ cells on day 5 postinfection. The results shown are representative of three experiments each with three mice per group.
To further assess the activation status of NK cells in VACV-infected tissues, we monitored the expression of the early activation marker CD69. At the peak of the NK response to VACV, a large proportion of NK cells present in the lung had upregulated CD69 (Fig. 3C). Furthermore, NK cells from VACV-infected mice displayed high expression levels of the key activation molecules CD44, CD43, and KLRG-1. Thus, NK cells responding to an acute respiratory VACV infection display signs of phenotypic activation.
Functional activity of NK cells during primary VACV infection.
To assess the relationship between maturation and cytolytic function, we assessed whether the expression of cytolytic effector molecules by NK cells was affected by VACV. As expected, low percentages of NK cells obtained directly from uninfected lungs and spleen were positive for granzyme B (Fig. 4A, day 0), CD107a (Fig. 4B, day 0), and perforin (not shown). In sharp contrast, without in vitro restimulation, NK cells from infected mice expressed high levels of intracellular granzyme B (Fig. 4A, day 4), CD107a (Fig. 4B, day 4), and perforin (not shown). Most significantly, freshly isolated lung NK cells but not spleen NK cells showed signs of extensive degranulation (as measured by cell surface expression of CD107a) on day 4 (∼25% of total NK cells) (Fig. 4B, day 4) and day 7 of infection (∼75% of total NK cells) (not shown).
FIG 4.
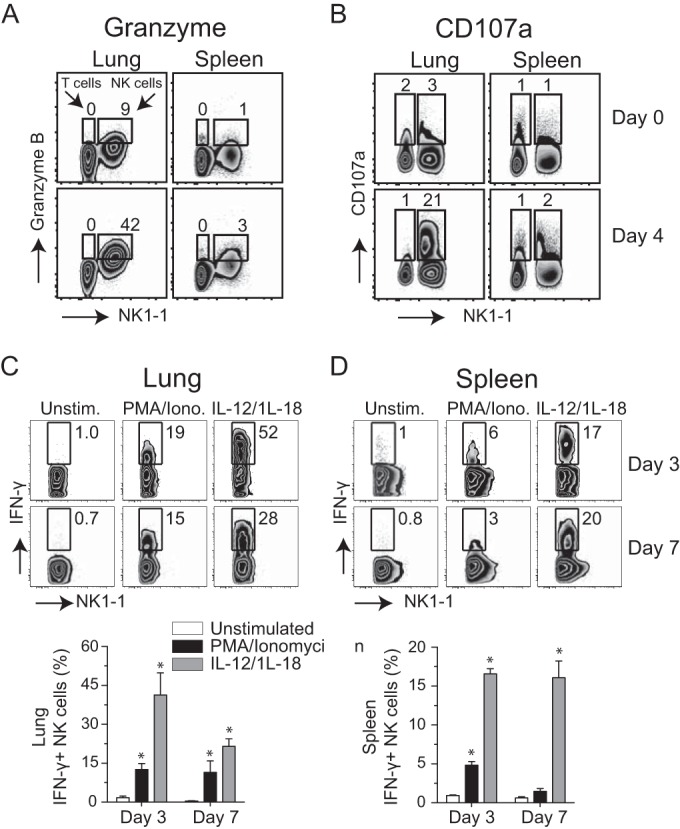
NK cells express cytolytic effector molecules and release IFN-γ after respiratory VACV-WR infection. Wild-type C57BL/6J (WT) mice were intranasally infected with 1.25 × 104 PFU of VACV-WR. On day 4 postinfection, total lung (left) and spleen (right) cells were stained for cell surface expression of NK1.1+, NKp46+, and intracellular granzyme (A) and CD107a (B). Uninfected (naive) mice (day 0) were used as controls. The numbers displayed in quadrants within the zebra plots indicate the percentages of granzyme B- or CD107a-positive cells. On days 3 and 7 postinfection, total lung (C) and spleen (D) cells were stimulated ex vivo with either PMA-ionomycin or IL-12 plus IL-18 and subsequently stained for intracellular IFN-γ. Representative zebra plots for IFN-γ staining by CD3− NK1.1+ NKp46+-gated NK cells are shown. The numbers in the plots indicate the percentages of NK1.1+ NKp46+ cells that stained for IFN-γ+. Quadrant settings were based on the results for infected cells that were not stimulated with PMA-ionomycin or IL-12 plus IL-18 (medium alone). Similar results were obtained in three separate experiments.
In addition to their direct cytotoxic effector function, NK cells have been considered to be an important early source of IFN-γ in antiviral immune responses. The production of IFN-γ by NK cells in the lung may be highly relevant, as IFN-γ is known to limit VACV replication in the lung and prevent virus dissemination to visceral tissues (20). Therefore, we analyzed NK cells for their capacity to produce IFN-γ in response to a respiratory VACV infection. On day 3 or 7 of infection, total lung and spleen cells were harvested and stimulated directly ex vivo for 5 h with phorbol 12-myristate 13-acetate (PMA) and ionomycin or left unstimulated. As IL-12 and IL-18 can directly induce NK cells to produce IFN-γ (45, 46), we also stimulated lung and spleen cells with both cytokines. Without ex vivo stimulation, few IFN-γ-expressing NK cells (<1%) could be detected in either the lung (Fig. 4C) or spleen (Fig. 4D). In contrast, PMA-ionomycin or IL-12 plus IL-18 readily induced IFN-γ production in lung and splenic NK cells; however, the combination of IL-12 plus IL-18 was far superior to treatment with PMA-ionomycin (Fig. 4C and D). This was reflected in both the percentages (Fig. 4C and D) and absolute numbers (not shown) of IFN-γ-positive NK cells present in the lung and spleen of VACV-infected mice. Compared with spleen cells, a greater percentage of lung NK cells were capable of producing IFN-γ, which is consistent with the respiratory nature of the infection. Taken together, these results demonstrate that the large number of NK cells that accumulate in the lung prior to viral clearance display many of the phenotypic and functional markers that would be consistent with an ability to mediate direct cytolytic activity and to produce IFN-γ when the appropriate signals were presented.
IFN-γ expression in natural killer cells precedes lung CD4 and CD8 T cell recruitment in response to VACV infection.
To determine directly whether NK cells were producing IFN-γ within the lung microenvironment, we utilized the interferon gamma reporter with endogenous poly(A) transcript (GREAT) mouse. These mice possess an internal ribosome entry site-enhanced yellow fluorescent protein (IRES-eYFP) reporter cassette inserted between the translational stop codon and 3′ untranslated region (UTR)/poly(A) tail of the interferon gamma gene (Ifng) (47). This allows precise identification of cells that have transcribed Ifng in vivo in response to VACV-WR infection without the need for in vitro restimulation. As expected, very few eYFP-positive (eYFP+) (i.e., IFN-γ+) CD4 or CD8 T cells were detectable in the lungs of naive mice. In striking contrast, in GREAT (B6.129S4-Ifngtm3.1Lky/J) mice, approximately 50% of lung- and spleen-resident NK cells were eYFP+ (Fig. 5A, day 0). In response to VACV infection, the numbers of eYFP+ NK cells in the lung and spleen increased, peaking between day 3 and day 6 and then declining nearly to background levels over the next 7 days (Fig. 5A and B). eYFP+ NK cells were readily visualized in lung parenchyma (Fig. 5E) and in splenic red pulp, white pulp, and bridging channels (Fig. 5F). The numbers of eYFP+ CD8 T cells increased ∼56-fold between day 3 and day 6 and peaked during the resolution of infection (days 6 to 9) (Fig. 5C). Recruitment of eYFP+CD4 T cells to the lung followed kinetics similar to those of eYFP+ CD8 T cells (Fig. 5D). Of note, despite similar proportions of eYFP+ cells between CD4 and CD8 T cell populations at the acute stage of infection, the total number of eYFP+ CD8 T cells was approximately 10-fold higher than the number of eYFP+ CD4 T cells (Fig. 5C and D). We did not detect any eYFP+ B cells, inflammatory monocytes, macrophages, eosinophils, neutrophils, or dendritic cells in the lung at any time points analyzed (Fig. 6). Thus, these data provide compelling evidence that NK1.1+ cells in the lung expressed IFN-γ before CD4 or CD8 T cells did, consistent with the idea that NK cells are among the early effectors activated in the lung after respiratory VACV infection.
FIG 5.

In vivo expression of IFN-γ by NK cells following a respiratory VACV-WR infection. Interferon-gamma reporter with endogenous poly(A) transcript (GREAT) (B6.129s4-Ifngtm3.1Lky/J) mice were intranasally (i.n.) infected with a sublethal inoculum of VACV-WR (1.25 × 104 PFU/mouse). On the specified days postinfection, the percentages and absolute numbers of lung and spleen eYFP–IFN-γ+ NK1.1+ cells (A and B), CD8 T cells (C), and CD4 T cells (D) were determined. C57BL/6 mice were used as negative controls (B6). Representative FACS plots and total cell counts from one experiment are presented. Similar results were obtained in three separate experiments. The numbers in the plots indicate the percentages of NK1.1+ cells that stained for eYFP–IFN-γ+. Student's t test with Bonferroni correction was used to determine statistical significance. *, P < 0.05, control versus infected mice. On day 6 postinfection, frozen lung (E) and spleen (F) sections were stained with rat anti-mouse NK1.1 antibodies and DAPI as indicated. The images were captured by a 20× objective using the EVOS fl inverted microscope. The micrographs show localization of NK1.1+ cells (red), DAPI (purple), and eYFP–IFN-γ+ (green) in lung parenchyma (C) and the splenic red pulp, white pulp, and bridging channels (D).
FIG 6.

In vivo expression of IFN-γ by lung innate immune and B cells following a respiratory VACV-WR infection. Interferon gamma reporter with endogenous poly(A) transcript (GREAT) (B6.129s4-Ifngtm3.1Lky/J) mice were intranasally (i.n.) infected with a sublethal inoculum of VACV-WR (1.25 × 104 PFU/mouse). (A) Gating strategy to identify cell subsets in VACV-infected lungs. DC, dendritic cells; MHC-II+, major histocompatibility complex II-positive cells; Macro, macrophages; Eo, eosinophils; Mono, monocytes; Neutro, neutrophils. (B) On the specified days postinfection, the percentages of lung eYFP–IFN-γ+ B cells, inflammatory monocytes (Inflam. Mono.), DC, neutrophils, eosinophils, and macrophages were determined. VACV-infected C57BL/6 mice were used as the negative control. The numbers in the plots indicate the percentages of cells that stained for eYFP–IFN-γ+.
Depletion of NK cells leads to an increase in VACV replication in the lung but does not alter eventual viral clearance.
To determine whether the emergence of highly activated NK cells in lung tissue and their secretion of IFN-γ in these sites contributed to virus control and recovery, groups of WT mice were depleted of NK cells using a previously well characterized monoclonal antibody (MAb) to NK1.1 (PK136) (48). We injected anti-NK1.1 MAb 1 day before and every 48 h after intranasal infection with VACV-WR, which resulted in robust elimination of NK cells in the lung and spleen (Fig. 7A). Contrary to our expectations, NK depletion did not modify the kinetics of the initial weight loss (Fig. 7B, days 0 to 7), recovery (Fig. 7B, days 7 to 14), or survival profiles of VACV-infected mice (Fig. 7C).
FIG 7.
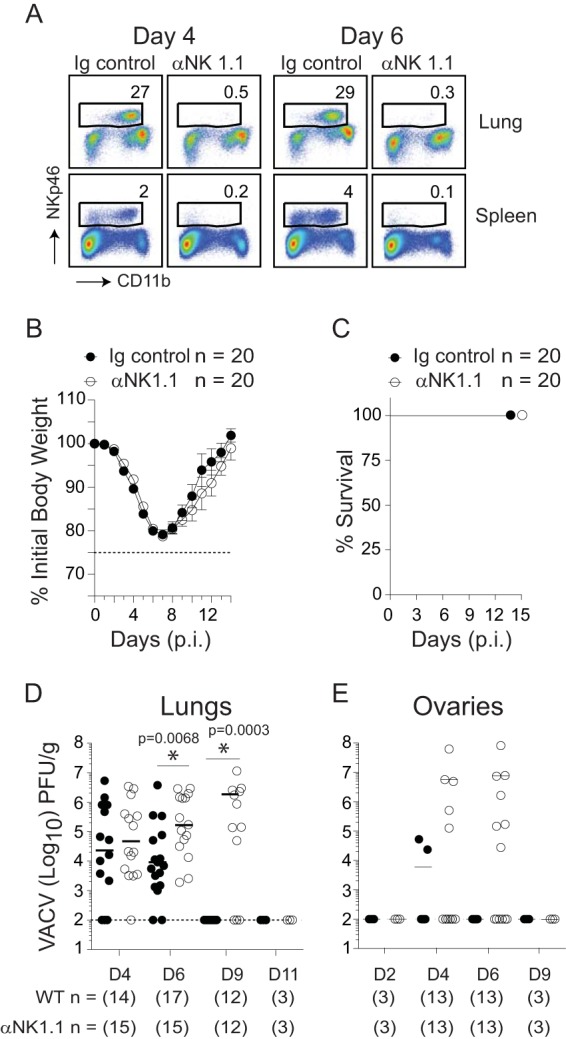
Depletion of NK cells leads to an increase in VACV replication in the lung but does not alter disease symptoms, viral clearance, or survival. Wild-type (WT) C57BL/6J mice were intranasally infected with VACV-WR (1.25 × 104 PFU/mouse). Mice were treated with a depleting anti-NK1.1 MAb (clone PK136) or PBS starting 1 day before infection (day −1) and then every 2 days over the course of the study. (A) Four and 6 days after infection, mice were sacrificed, and lung tissues (top) and splenocytes (bottom) were stained with anti-CD3, anti-CD11b, and anti-NKp46 antibodies. The events were gated on CD3− cells, and the percentages of NKp46+ cells among the gated population are shown. Body mass (B) and survival (C) were monitored daily in both PBS control and NK1.1-depleted mice. At the indicated times after infection, lung (D) and ovary (E) tissue homogenates were prepared and assayed for infectious VACV by plaque assay on VERO cells. The dashed lines indicate the minimum detection limit of the plaque assay. Symbols indicate the results for individual mice, and lines indicate the mean results. Student's t test with Bonferroni correction was used to determine statistical significance. *, P < 0.05, NK cell-depleted mice versus nondepleted control mice.
Although we did not see any role for NK cells in the recovery phase of infection, we reasoned that they might be crucial for early control of VACV in the lung and that weight loss measurements may not accurately reflect subtle changes in viral titers during this phase of the response. In immunocompetent hosts, viral titers in the lung increase 1 to 2 days postinfection, reach a maximum at 3 to 5 days, start to decline at day 7, and are cleared between day 9 and day 14 (19). Early (day 2) p.i., the viral titers in the lungs of WT and NK cell-depleted mice were identical (not shown). As viral replication reached more significant levels at day 4 p.i., there was a trend toward increased viral loads in NK cell-depleted animals, but these did not reach statistical significance (Fig. 7D). At day 6 p.i., a time point at which peak numbers of IFN-γ+ NK cells occur in the lung, a significant increase in VACV titer was observed in anti-NK1.1 antibody-treated mice, and this high titer continued in some animals through to day 9. Despite this, both NK cell-depleted and untreated, NK cell-sufficient WT controls cleared VACV between day 9 and day 11, directly correlating with the kinetics of weight loss recovery. To further assess the antiviral activities of NK cells, we investigated the dissemination of VACV from the lung to peripheral tissues. At day 4 p.i., there was an increase in VACV titers in ovaries isolated from 5 of 13 NK cell-depleted mice (Fig. 7E). Again, VACV titers continued to increase over the next 2 days but were minimally detectable by day 9 (Fig. 7E). Notably, NK depletion did not affect all organs, because an increase in infectious virus was not found in the liver, spleen, heart, or kidneys (not shown). These data suggest that NK cells provide a temporary barrier limiting VACV replication and dissemination during the acute phase of infection.
Delayed clearance of VACV from the lungs of NK cell-depleted mice is not due to impaired CD8 T cell responses.
In addition to their direct cytolytic activity, NK cells also provide helper signals for CD8 T cells by inducing DC maturation and cytokine production (26). Because both NK cells and CD8 T cells are required during the acute phase of VACV infection (19), it was possible that the increased viral titers observed in NK cell-depleted mice were related to impaired activation and/or recruitment of CD8 T cells to the lungs. Thus, we performed a number of phenotypic and functional analyses to quantitate the extent of CD8 T cell priming in the absence of NK cells. Analysis of virus-induced total CD8 and CD4 T cells in NK cell-depleted mice showed no significant differences from untreated control mice (Fig. 8A and B). To assess the induction of virus-specific CD8 T cells, the immunodominant VACV-reactive population was tracked with a tetramer containing the B8R (positions 20 to 27; TSYKFESV) peptide (33, 34) (Fig. 8C and D) and by IFN-γ production following stimulation with the B8R peptide epitope (not shown). Again, we found that the percentages and total numbers of B8R-reactive CD8 T cells in the lung (Fig. 8C) and spleen (Fig. 8D) were comparable between NK cell-depleted and control mice. This showed that NK cell help was not required for the generation and migration of VACV-reactive CD8 effector T cells to the lungs.
FIG 8.

NK cell depletion does not influence the CD8 T cell response elicited in the lungs of mice infected with VACV. Wild-type (WT) C57BL/6J and NK cell-depleted mice were intranasally infected with 1.25 × 104 PFU of VACV-WR. On days 4 and 6 postinfection, total lung (A and C) and spleen (B and D) cells were stained for CD3ε, CD4 CD8α, CD44, and B8R tetramer. Representative plots for CD4 and CD8 staining gated on CD3ε+ cells (A and B) and B8R tetramer staining (C and D) gated on CD8α+ cells are shown. The percentages and total numbers of CD3ε+ CD4+, CD3ε+ CD8α, CD3ε+ CD8α+ CD44high, and B8R tetramer-positive T cells are also quantified. The results shown are the means ± SEM (n = 3 mice/group) from one experiment. Similar results were obtained in three separate experiments. Student's t test with Bonferroni correction was used to determine statistical significance.
NK cells and innate IFN-γ are required for virus control and prolong survival of VACV-infected T cell-deficient mice.
We next asked whether NK cells and IFN-γ contribute to the host immune response to respiratory VACV infection in the absence of T lymphocytes. To do so, we examined RAG−/− mice that contain NK cells but lack T and B cells due to the deletion of the recombination activation gene. We first determined the magnitude and quality of the NK cell response in RAG−/− mice. Utilizing the same gating strategy as described in the legend to Fig. 1, we observed that following high- (1.25 × 104 PFU) or low-dose (1.25 × 103 PFU) VACV infection, greater frequencies and absolute numbers of lung NK cells were found in RAG−/− mice than in WT controls (Fig. 9A). Importantly, lung NK cells proliferated (Fig. 9B), displayed an activated phenotype (Fig. 9B), and rapidly expressed intracellular IFN-γ following ex vivo stimulation with either PMA-ionomycin or IL-12 plus IL-18 (Fig. 9C). The effect of T and B cell deficiency on the induction of total lung IFN-γ mRNA levels was also examined by real-time reverse transcription (RT)-PCR. At day 9 p.i., a time point at which peak IFN-γ mRNA expression and protein levels are seen in WT mice (20), comparable IFN-γ mRNA levels were observed in the lungs of RAG−/− mice (Fig. 9D). Enhanced IFN-γ mRNA expression in RAG−/− mice correlated with a marked influx or proliferation of NK cells within lung tissue seen at day 9 p.i. (Fig. 9A), suggesting that NK cells are the major cellular source of IFN-γ in RAG−/− mice. As expected, no IFN-γ mRNA expression was detected in lungs isolated from VACV-infected RAG-2−/− IL-2 receptor γ common chain−/− (γc−/−) mice, which are deficient in NK cells, as well as T and B cells (Fig. 9D).
FIG 9.
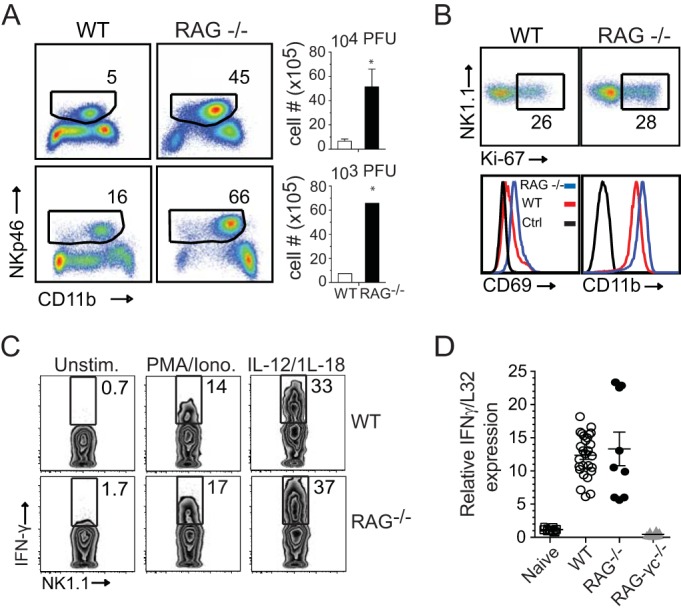
Enhanced NK cell responses in T cell-deficient RAG−/− mice infected with VACV-WR. Wild-type (WT), RAG−/−, and RAG−/− IL-2Rγ−/− C57BL/6J mice were infected intranasally with 1.25 × 104 or 1.25 × 103 PFU of VACV-WR, as indicated. (A) Representative FACS plots from day 9 postinfection showing NK cells (CD11b+ NKp46+) as percentages of CD3− lymphocytes. Right, histogram showing NK cell numbers presented as the mean results ± SEM from two separate experiments; n = 3 mice/group. Student's t test with the Bonferroni correction was used to determine statistical significance. *, P < 0.05, WT versus RAG−/−-infected mice. (B) On day 9 postinfection, lung cells were stained with anti-CD3, anti-NK1.1, and anti-Nkp46 antibodies and for Ki67 and assayed for NK cell proliferation and CD11b and CD69 expression on NK cells. (C) On day 9 postinfection with VACV-WR (1.25 × 104 PFU), total lung cells were stimulated ex vivo with either PMA-ionomycin or IL-12 plus IL-18 for 5 h and then stained for intracellular IFN-γ. Representative zebra plots are shown for IFN-γ staining of CD3− NK1.1+-gated NK cells. The numbers in the plots indicate the percentages of NK1.1+ cells that stained for IFN-γ. Quadrant settings were based on the results for controls, using infected cells that were not stimulated (medium alone) with PMA-ionomycin or IL-12 and IL-18. Similar results were obtained in two separate experiments. (D) On day 9 postinfection with VACV-WR (1.25 × 104 PFU), lungs were isolated and total RNA was collected. IFN-γ transcript levels were measured by real-time RT-PCR. Levels of mRNA were standardized to the level of the L32 housekeeping gene. Symbols indicate the results for individual mice, and bars- and whiskers indicate the mean results ± SEM. Student's t test with Bonferroni correction was used to determine statistical significance. *, P < 0.05 for RAG−/− versus RAG−/−/IL-2Rγ−/− mice.
WT mice infected with 1.5 × 103 PFU of VACV-WR experienced moderate weight loss (10 to 15%) but quickly recovered and went on to maintain normal weight after day 11 p.i. (Fig. 10A). In sharp contrast, RAG−/− mice were found to be highly susceptible to infection (Fig. 10A). By day 9 p.i., RAG−/− mice exhibited severe cachexia and immobility, which resulted in 100% mortality by day 17 postinfection (Fig. 10B), despite the presence of substantially higher NK cell numbers than in WT mice (Fig. 9A). Correlating with increased susceptibility, RAG−/− mice exhibited elevated titers of infectious virus in their lungs (Fig. 10C) and ovaries (Fig. 10D) compared with the levels in WT controls. These results clearly demonstrate that lymphocytes are required to clear VACV from the lung and complement our recent studies showing that CD8 T cells are the lymphocyte subset required for protection against respiratory VACV infection.
FIG 10.
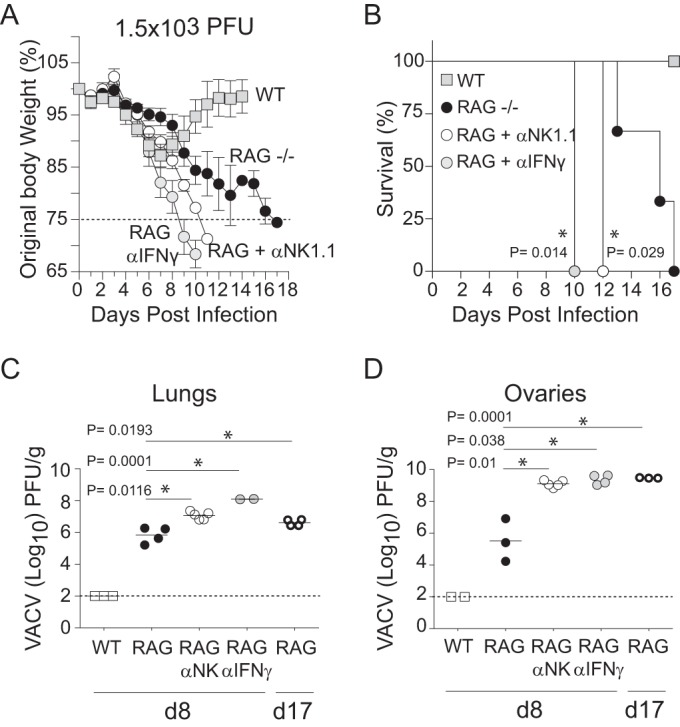
NK cells and IFN-γ participate in innate defense against respiratory VACV infection. Wild-type C57BL/6J, and RAG−/− mice were infected intranasally with 1.25 × 103 of VACV-WR. Weight loss (A) and survival (B) were monitored in RAG−/− mice that were continuously treated with a depleting anti-NK1.1 MAb (clone PK136), neutralizing anti-IFN-γ MAb (clone XMG.1), or PBS control starting 1 day before infection and then every 2 days over the course of the study. Weight loss data are presented as the mean results ± SEM from two separate experiments with three to five mice per group and analyzed using two-way ANOVA to determine statistical significance. Survival data comprise combined survival data across combined experiments analyzed using the Mantel-Cox test. *, P < 0.05. At the indicated times after infection, lung (C) and ovary (D) tissue homogenates were prepared and assayed for infectious VACV by plaque assay on VERO cells. The dashed line indicates the minimum detection limit of the plaque assay. Data are presented as the mean results ± 1 SEM from two independent experiments with three to five mice per group. Student's t test with Bonferroni correction was used to determine statistical significance of viral titer results. *, P < 0.05, NK cell-depleted or IFN-γ-depleted versus nondepleted RAG−/− mice.
To formally test whether NK cells are required for T cell-independent resistance to VACV infection, we compared the degrees of susceptibility of anti-NK1.1 MAb-treated and untreated RAG−/− mice to VACV infection. We found that RAG−/− mice depleted of NK cells developed accelerated clinical manifestations and succumbed more rapidly to infection than did untreated NK cell-sufficient RAG−/− controls (Fig. 10A and B). No NK cell-depleted RAG−/− mice survived past day 12 after infection (Fig. 10B). Again correlating with our weight loss and survival data, the VACV titers in the lungs (Fig. 10C) and ovaries (Fig. 10D) were significantly higher in NK cell-depleted RAG−/− mice than in NK-competent WT and RAG−/− controls. Likewise, IFN-γ-depleted RAG−/− mice failed to control VACV replication in the lungs and ovaries, developed clinical symptoms similar to those observed in NK cell-depleted RAG−/− mice, and succumbed to infection by day 10 (Fig. 10A to D). Together, these results provide compelling evidence that NK cells in the absence of T and B cells can participate in the innate immune response to respiratory VACV infection and indicate that IFN-γ is an important mediator in this process.
DISCUSSION
In the present study, we have identified an NK cell- and innate-IFN-γ-mediated mechanism of host resistance to respiratory VACV infection. Previous studies of the host adaptive immune response to respiratory VACV infection have indicated that trafficking of virus-specific CD8 T cells into the lung tissue and their secretion of IFN-γ in these sites is necessary for complete clearance of virus from the lungs of infected mice and for protection against death (19, 20). VACV-specific CD8 T cells begin entering the lung at day 4.5, increase significantly by day 6 postinfection, and reach peak numbers by day 10 (19, 20). In contrast, we found that the frequency of NK cells in the infected lung tissue increased over the first 2 days postinfection, peaked between days 4 and 6, and decreased thereafter. Proliferation of NK cells trafficking to or within the lung was associated with increased expression of activation and maturation markers. Moreover, lung-resident NK cells expressed high levels of cytolytic effector molecules (perforin and granzyme B) and were the main source of innate IFN-γ before the arrival of CD8 T cells. We show that the prominent role of local IFN-γ release by NK cells is to limit VACV replication during the time needed for optimal expansion and migration of virus-specific CD8 T cells to the lung, where there are few resident CD8 T cells in naive mice. Together, our observations indicate that recovery from a highly virulent respiratory poxvirus infection requires a coordinated response by both NK cells and cytotoxic CD8 T lymphocytes.
The majority of what we know about the role of NK cells in recovery from primary infection with poxviruses has been drawn from footpad or i.v. inoculation of mice with ectromelia virus (ECTV; mousepox) (49–54). In the footpad model, ECTV rapidly spreads from the initial site of infection through the draining popliteal lymph nodes and the lymphatics to reach the spleen and liver (9, 55). Depletion of NK cells before or soon after infection leads to an increase in viral titers in both liver and spleen as early as day 4 and eventual death of the animals by day 7 to 10 (52, 53, 56). Notably, the antiviral activity of NK cells appears to be essential only in the first 5 days of infection (56), as depletion of NK cells after this time has little impact on disease outcome (56). These results and others (reviewed by Burshtyn [54]) obtained after systemic (i.v. or i.p) infection with relatively high infectious doses of VACV (106 to 107 PFU) support the view that NK cells are generally required in the early phase of poxvirus infection to limit viral spread from the initial site of infection and to prevent death.
The data presented in the current study indicate different outcomes when VACV enters the host via the respiratory tract. We found that VACV infection in the lung of NK cell-depleted mice exhibited different kinetics of viral clearance than ECTV or systemic VACV infection. Unlike ECTV and systemic VACV infection, nearly equivalent titers of VACV were observed in the lungs of NK cell-depleted and control mice during the early phase (days 2 to 4) of infection. It was not until day 6 or day 9 postinfection that NK cell-depleted mice showed much higher titers than NK cell-competent control animals. Despite this, NK cell deficiency appeared to have no impact upon viral clearance or survival. A similar conclusion was recently made following footpad infection with cowpox, where systemic depletion of NK cells resulted in substantially higher viral titers in the draining popliteal lymph nodes but had no effect on morbidity and mortality (57). The overall influence of NK cells on host survival as a consequence of poxvirus infections is not known for certain, but there are data to suggest it might be related to the impact of NK cells on adaptive immunity. For example, following ECTV infection, virus-specific CD4 and CD8 T cell proliferation and cytokine production are dramatically reduced in the absence of NK cells (56). Similarly, it has been well documented in influenza virus infection that depletion of NK cells results in defective CD8 T cell responses, with a concomitant increase in morbidity and mortality (58). In contrast, we found no evidence for this being the case in the context of a respiratory VACV infection. Indeed, many features of VACV-specific T cell responses, including activation, expansion, effector function, and migratory capacity, were completely intact in the absence of NK cells. Whether NK cells influence the development of cowpox-specific CD8 T cells is not known; however, following a respiratory murine gammaherpesvirus-68 (MHV-68) (59) infection—another model where NK cells are not required for antiviral T cell responses—depletion of NK cells did not have any effect on recovery and survival. These studies suggest that there are markedly different roles for NK cells following infection with distinct viruses.
Convincing evidence of the importance of NK cells in innate immunity to respiratory VACV infection was provided by the observation that NK depletion in lymphopenic RAG−/− mice resulted in substantially higher lung viral titers, as well as increased dissemination of VACV to the ovaries, compared with the lung viral titers and viral dissemination in NK cell-competent RAG−/− mice. Of significance, we found that NK cell-depleted RAG−/− mice succumbed more rapidly to infection than NK cell-competent RAG−/− controls. These results establish the existence of an NK cell-mediated, T and B cell-independent mechanism of host defense against respiratory VACV infection. NK cells mediate their antiviral effects through the release of cytolytic granules, the induction of target cell death through cell surface receptors, or the production of antiviral cytokines (60). Following respiratory VACV infection, we found considerable increases in intracellular granzyme and perforin expression and cell surface CD107a. The expression of CD107a by NK cells indicates a recent release of cytolytic granules (61), suggesting that they may have used perforin/granzyme B-mediated killing to limit VACV replication in the lungs. However, in our earlier study, we found no difference in the course of respiratory VACV infection in mice deficient in perforin (20). Complete recovery was also seen in the absence of FAS-, FASL-, or TRAIL-mediated lysis (20). In striking contrast, mice deficient in or depleted of IFN-γ at the time of VACV infection were highly susceptible to a respiratory infection by VACV-WR (20). Consistent with this, we found that neutralization of IFN-γ in VACV-infected RAG−/− mice abolished the resistance of control mice to this virus, closely mirroring the response observed in NK cell-depleted RAG−/− mice. Using IFN-γ eYFP reporter and NK cell-deficient RAG−/− (RAG−/− γc−/−) mice, we found that NK cells were the main source of innate IFN-γ in lungs of infected animals. These results point toward the release of IFN-γ as the critical nonredundant component of NK cell-mediated host resistance to respiratory VACV infection. Interestingly, NK cells limit ECTV spread from the popliteal lymph nodes to the liver using both perforin-mediated killing of infected cells and release of IFN-γ (53, 56). Thus, the relative importance of these alternative antiviral pathways appears to depend on the poxvirus infection model being studied, which is likely a reflection of key differences in virus-specific (virulence mechanisms) and host-specific (genetic background, cytokine microenvironment, and inhibitory/stimulatory receptors) factors in distinct target tissues.
Of importance in the current study is the observation that intranasal inoculation with as few as 103 PFU of VACV in RAG−/− mice resulted in widespread and persistent infection and eventual death by day 17. Remarkably, this occurred despite the presence of substantially higher (∼10-fold) numbers of IFN-γ-producing NK cells in the lung than in WT controls. These results demonstrate that the NK response alone was not sufficient to resolve a highly virulent respiratory VACV infection without the help of T cell-mediated host defense mechanisms. Consistent with this, selective engraftment of RAG−/− mice with naive polyclonal CD8 T cells afforded protection against death, closely mirroring the recovery observed in WT mice (19). In marked contrast, RAG−/− mice that received CD8 T cells from IFN-γ−/− mice failed to clear the virus, and all succumbed to infection (20). Considered collectively, these results provide compelling evidence that CD8 T cell-derived IFN-γ cannot be functionally compensated for by IFN-γ released by NK cells. Paradoxically, when CD8 T cells from WT mice were transferred into naive IFN-γ−/− hosts (20) or, as shown here, when WT mice were depleted of NK cells, all mice were fully protected from death, indicating that CD8 T cells are capable of clearing VACV from the lungs of infected mice in the absence of either host- or NK cell-derived IFN-γ.
We do not know why IFN-γ produced by NK cells is not sufficient for recovery from a highly virulent respiratory VACV infection. Of potential interest here are some older studies by Ramshaw et al. that suggest that this phenomenon might be related to the respiratory nature of VACV infection. Accordingly, insertion of the IFN-γ gene into the VACV genome allowed both T cell-deficient athymic nude and sublethally irradiated euthymic mice to survive a systemic (i.v.) infection (62, 63). Moreover, NK cell-derived IFN-γ, triggered by recombinant VACV expressing IL-2, contributed to rapid clearance of infection in nude mice (64–66). In considering these studies, the simplest explanation for our results may be that in the absence of T cells, IFN-γ levels in the infected lungs never reach the threshold necessary to completely eliminate VACV. Although we cannot rule out this possibility, our observation that IFN-γ mRNA is equally induced in the presence or absence of T cells argues (at least in the context of a respiratory VACV infection) that IFN-γ levels alone do not account for the failure of NK cells to clear virus. The finding that RAG−/− mice had greatly increased numbers of IFN-γ-producing NK cells compared to the numbers in WT controls also argues against the absolute numbers of lung-resident NK cells as being the major limiting factor of NK function. A more likely explanation for our results is that restricted accessibility of infectious foci in the lung to NK cells is responsible for their failure to completely clear virus. This hypothesis is consistent with a previous report demonstrating that IFN-γ must act in a highly localized manner to prevent further virus replication in infected cells (64). It will be important to address the means by which NK cells and T cells reach and infiltrate lung cells that could function as a reservoir for VACV replication and to determine why some types of virus infections are more accessible for NK cells than others.
In conclusion, our studies help to define the precise immune mechanisms that mediate viral clearance and recovery from primary poxvirus respiratory tract infection. In addition to the established role of CD8 T cell-derived IFN-γ in protection against respiratory VACV infection (20), our findings suggest that IFN-γ production by NK cells limits VACV replication and prolongs survival but does not eliminate the virus without the help of CD8 T cells. Future studies to define the precise mechanism of this response will be important for the design of therapeutic interventions against existing and emerging respiratory poxvirus infections.
ACKNOWLEDGMENTS
We thank the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of the major histocompatibility complex (MHC)/peptide tetramer containing the B8R peptide (positions 20 to 27; TSYKFESV)/H-2Kb conjugated to allophycocyanin (APC). We thank John Goulding for performing real-time RT-PCR and intracellular IFN-γ assays.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Buller RM, Palumbo GJ. 1991. Poxvirus pathogenesis. Microbiol Rev 55:80–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Giulio DB, Eckburg PB. 2004. Human monkeypox: an emerging zoonosis. Lancet Infect Dis 4:15–25. doi: 10.1016/S1473-3099(03)00856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Souza Trindade G, Drumond BP, Guedes MI, Leite JA, Mota BE, Campos MA, da Fonseca FG, Nogueira ML, Lobato ZI, Bonjardim CA, Ferreira PC, Kroon EG. 2007. Zoonotic vaccinia virus infection in Brazil: clinical description and implications for health professionals. J Clin Microbiol 45:1370–1372. doi: 10.1128/JCM.00920-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cann JA, Jahrling PB, Hensley LE, Wahl-Jensen V. 2013. Comparative pathology of smallpox and monkeypox in man and macaques. J Comp Pathol 148:6–21. doi: 10.1016/j.jcpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson JB. 1938. The behavior of pox viruses in the respiratory tract. I. The response of mice to the nasal instillation of vaccinia virus. J Exp Med 68:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner GS. 1967. Respiratory infection of mice with vaccinia virus. J Gen Virol 1:399–402. doi: 10.1099/0022-1317-1-3-399. [DOI] [PubMed] [Google Scholar]
- 7.Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. 1990. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol 71(Pt 11):2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- 8.Schriewer J, Buller RM, Owens G. 2004. Mouse models for studying orthopoxvirus respiratory infections. Methods Mol Biol 269:289–308. doi: 10.1385/1-59259-789-0:289. [DOI] [PubMed] [Google Scholar]
- 9.Chapman JL, Nichols DK, Martinez MJ, Raymond JW. 2010. Animal models of orthopoxvirus infection. Vet Pathol 47:852–870. doi: 10.1177/0300985810378649. [DOI] [PubMed] [Google Scholar]
- 10.Rivera R, Hutchens M, Luker KE, Sonstein J, Curtis JL, Luker GD. 2007. Murine alveolar macrophages limit replication of vaccinia virus. Virology 363:48–58. doi: 10.1016/j.virol.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonduelle O, Duffy D, Verrier B, Combadiere C, Combadiere B. 2012. Cutting edge: protective effect of CX3CR1+ dendritic cells in a vaccinia virus pulmonary infection model. J Immunol 188:952–956. doi: 10.4049/jimmunol.1004164. [DOI] [PubMed] [Google Scholar]
- 12.Rice AD, Turner PC, Embury JE, Moldawer LL, Baker HV, Moyer RW. 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol 85:550–567. doi: 10.1128/JVI.00254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutchens MA, Luker KE, Sonstein J, Nunez G, Curtis JL, Luker GD. 2008. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathog 4:e1000153. doi: 10.1371/journal.ppat.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez J, Huang X, Yang Y. 2010. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog 6:e1000811. doi: 10.1371/journal.ppat.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Martinez J, Huang X, Yang Y. 2007. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Nunez G, Curtis JL, Luker GD. 2008. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol 180:483–491. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yammani RD, Pejawar-Gaddy S, Gurley TC, Weimer ET, Hiltbold EM, Alexander-Miller MA. 2008. Regulation of maturation and activating potential in CD8+ versus CD8- dendritic cells following in vivo infection with vaccinia virus. Virology 378:142–150. doi: 10.1016/j.virol.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beauchamp NM, Busick RY, Alexander-Miller MA. 2010. Functional divergence among CD103+ dendritic cell subpopulations following pulmonary poxvirus infection. J Virol 84:10191–10199. doi: 10.1128/JVI.00892-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. 2012. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol 189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goulding J, Abboud G, Tahiliani V, Desai P, Hutchinson TE, Salek-Ardakani S. 2014. CD8 T cells use IFN-gamma to protect against the lethal effects of a respiratory poxvirus infection. J Immunol 192:5415–5425. doi: 10.4049/jimmunol.1400256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM. 2003. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 198:1069–1076. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Li F, Zheng M, Sun R, Wei H, Tian Z. 2012. Lung natural killer cells in mice: phenotype and response to respiratory infection. Immunology 137:37–47. doi: 10.1111/j.1365-2567.2012.03607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hesker PR, Krupnick AS. 2013. The role of natural killer cells in pulmonary immunosurveillance. Front Biosci (Schol Ed) 5:575–587. doi: 10.2741/S391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs A, Colonna M. 2011. Natural killer (NK) and NK-like cells at mucosal epithelia: mediators of anti-microbial defense and maintenance of tissue integrity. Eur J Microbiol Immunol (Bp) 1:257–266. doi: 10.1556/EuJMI.1.2011.4.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Culley FJ. 2009. Natural killer cells in infection and inflammation of the lung. Immunology 128:151–163. doi: 10.1111/j.1365-2567.2009.03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferlazzo G, Morandi B. 2014. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol 5:159. doi: 10.3389/fimmu.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, Chew MV, Chan T, Al-Garawi AA, Jordana M, Ashkar AA. 2012. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis 206:167–177. doi: 10.1093/infdis/jis340. [DOI] [PubMed] [Google Scholar]
- 28.Zhou G, Juang SW, Kane KP. 2013. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol 43:929–938. doi: 10.1002/eji.201242620. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Zhu H, Sun R, Wei H, Tian Z. 2012. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J Virol 86:2251–2258. doi: 10.1128/JVI.06209-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salek-Ardakani S, Moutaftsi M, Crotty S, Sette A, Croft M. 2008. OX40 drives protective vaccinia virus-specific CD8 T cells. J Immunol 181:7969–7976. doi: 10.4049/jimmunol.181.11.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salek-Ardakani S, Moutaftsi M, Sette A, Croft M. 2011. Targeting OX40 promotes lung-resident memory CD8 T cell populations that protect against respiratory poxvirus infection. J Virol 85:9051–9059. doi: 10.1128/JVI.00619-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salek-Ardakani S, Arens R, Flynn R, Sette A, Schoenberger SP, Croft M. 2009. Preferential use of B7.2 and not B7.1 in priming of vaccinia virus-specific CD8 T cells. J Immunol 182:2909–2918. doi: 10.4049/jimmunol.0803545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med 201:95–104. doi: 10.1084/jem.20041912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui HH, Grey H, Sette A. 2006. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol 24:817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 35.Flynn R, Hutchinson T, Murphy KM, Ware CF, Croft M, Salek-Ardakani S. 2013. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS One 8:e77991. doi: 10.1371/journal.pone.0077991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Chaudhri G, Jackson RJ, Karupiah G. 2009. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J Immunol 183:3324–3331. doi: 10.4049/jimmunol.0803985. [DOI] [PubMed] [Google Scholar]
- 37.Gherardi MM, Ramirez JC, Esteban M. 2003. IL-12 and IL-18 act in synergy to clear vaccinia virus infection: involvement of innate and adaptive components of the immune system. J Gen Virol 84:1961–1972. doi: 10.1099/vir.0.19120-0. [DOI] [PubMed] [Google Scholar]
- 38.Di Santo JP. 2006. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol 24:257–286. doi: 10.1146/annurev.immunol.24.021605.090700. [DOI] [PubMed] [Google Scholar]
- 39.Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, Moretta L, Moretta A. 1997. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med 186:1129–1136. doi: 10.1084/jem.186.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, Yokoyama WM. 2002. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol 3:523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- 41.Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. 2001. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol 31:1900–1909. doi: 10.1002/1521-4141(200106)31:6<1900::AID-IMMU1900>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 42.Arase H, Saito T, Phillips JH, Lanier LL. 2001. Cutting edge: the mouse NK cell-associated antigen recognized by DX5 monoclonal antibody is CD49b (alpha 2 integrin, very late antigen-2). J Immunol 167:1141–1144. doi: 10.4049/jimmunol.167.3.1141. [DOI] [PubMed] [Google Scholar]
- 43.Hayakawa Y, Smyth MJ. 2006. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol 176:1517–1524. doi: 10.4049/jimmunol.176.3.1517. [DOI] [PubMed] [Google Scholar]
- 44.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. 2009. Maturation of mouse NK cells is a 4-stage developmental program. Blood 113:5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- 45.Tomura M, Zhou XY, Maruo S, Ahn HJ, Hamaoka T, Okamura H, Nakanishi K, Tanimoto T, Kurimoto M, Fujiwara H. 1998. A critical role for IL-18 in the proliferation and activation of NK1.1+ CD3- cells. J Immunol 160:4738–4746. [PubMed] [Google Scholar]
- 46.Lauwerys BR, Renauld JC, Houssiau FA. 1999. Synergistic proliferation and activation of natural killer cells by interleukin 12 and interleukin 18. Cytokine 11:822–830. doi: 10.1006/cyto.1999.0501. [DOI] [PubMed] [Google Scholar]
- 47.Reinhardt RL, Liang HE, Locksley RM. 2009. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10:385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koo GC, Dumont FJ, Tutt M, Hackett J Jr, Kumar V. 1986. The NK-1.1(-) mouse: a model to study differentiation of murine NK cells. J Immunol 137:3742–3747. [PubMed] [Google Scholar]
- 49.Jacoby RO, Bhatt PN, Brownstein DG. 1989. Evidence that NK cells and interferon are required for genetic resistance to lethal infection with ectromelia virus. Arch Virol 108:49–58. doi: 10.1007/BF01313742. [DOI] [PubMed] [Google Scholar]
- 50.Karupiah G, Fredrickson TN, Holmes KL, Khairallah LH, Buller RM. 1993. Importance of interferons in recovery from mousepox. J Virol 67:4214–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niemialtowski MG, Spohr de Faundez I, Gierynska M, Malicka E, Toka FN, Schollenberger A, Popis A. 1994. The inflammatory and immune response to mousepox (infectious ectromelia) virus. Acta Virol 38:299–307. [PubMed] [Google Scholar]
- 52.Delano ML, Brownstein DG. 1995. Innate resistance to lethal mousepox is genetically linked to the NK gene complex on chromosome 6 and correlates with early restriction of virus replication by cells with an NK phenotype. J Virol 69:5875–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parker AK, Parker S, Yokoyama WM, Corbett JA, Buller RM. 2007. Induction of natural killer cell responses by ectromelia virus controls infection. J Virol 81:4070–4079. doi: 10.1128/JVI.02061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burshtyn DN. 2013. NK cells and poxvirus infection. Front Immunol 4:7. doi: 10.3389/fimmu.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panchanathan V, Chaudhri G, Karupiah G. 2008. Correlates of protective immunity in poxvirus infection: where does antibody stand? Immunol Cell Biol 86:80–86. doi: 10.1038/sj.icb.7100118. [DOI] [PubMed] [Google Scholar]
- 56.Fang M, Lanier LL, Sigal LJ. 2008. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog 4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pak-Wittel MA, Yang L, Sojka DK, Rivenbark JG, Yokoyama WM. 2013. Interferon-gamma mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proc Natl Acad Sci U S A 110:E50–E59. doi: 10.1073/pnas.1220456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGill J, Heusel JW, Legge KL. 2009. Innate immune control and regulation of influenza virus infections. J Leukoc Biol 86:803–812. doi: 10.1189/jlb.0509368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Usherwood EJ, Meadows SK, Crist SG, Bellfy SC, Sentman CL. 2005. Control of murine gammaherpesvirus infection is independent of NK cells. Eur J Immunol 35:2956–2961. doi: 10.1002/eji.200526245. [DOI] [PubMed] [Google Scholar]
- 60.Mullbacher A. 2003. Cell-mediated cytotoxicity in recovery from poxvirus infections. Rev Med Virol 13:223–232. doi: 10.1002/rmv.381. [DOI] [PubMed] [Google Scholar]
- 61.Alter G, Malenfant JM, Altfeld M. 2004. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Kohonen-Corish MR, King NJ, Woodhams CE, Ramshaw IA. 1990. Immunodeficient mice recover from infection with vaccinia virus expressing interferon-gamma. Eur J Immunol 20:157–161. doi: 10.1002/eji.1830200123. [DOI] [PubMed] [Google Scholar]
- 63.Ramshaw I, Ruby J, Ramsay A, Ada G, Karupiah G. 1992. Expression of cytokines by recombinant vaccinia viruses: a model for studying cytokines in virus infections in vivo. Immunol Rev 127:157–182. doi: 10.1111/j.1600-065X.1992.tb01413.x. [DOI] [PubMed] [Google Scholar]
- 64.Karupiah G, Blanden RV, Ramshaw IA. 1990. Interferon gamma is involved in the recovery of athymic nude mice from recombinant vaccinia virus/interleukin 2 infection. J Exp Med 172:1495–1503. doi: 10.1084/jem.172.5.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karupiah G, Coupar BE, Andrew ME, Boyle DB, Phillips SM, Mullbacher A, Blanden RV, Ramshaw IA. 1990. Elevated natural killer cell responses in mice infected with recombinant vaccinia virus encoding murine IL-2. J Immunol 144:290–298. [PubMed] [Google Scholar]
- 66.Karupiah G, Woodhams CE, Blanden RV, Ramshaw IA. 1991. Immunobiology of infection with recombinant vaccinia virus encoding murine IL-2. Mechanisms of rapid viral clearance in immunocompetent mice. J Immunol 147:4327–4332. [PubMed] [Google Scholar]