

ABSTRACT
The ability of Epstein-Barr virus (EBV) to spread and persist in human populations relies on a balance between host immune responses and EBV immune evasion. CD8+ cells specific for EBV late lytic cycle antigens show poor recognition of target cells compared to immediate early and early antigen-specific CD8+ cells. This phenomenon is due in part to the early EBV protein BILF1, whose immunosuppressive activity increases with lytic cycle progression. However, published data suggest the existence of a hitherto unidentified immune evasion protein further enhancing protection against late EBV antigen-specific CD8+ cells. We have now identified the late lytic BDLF3 gene as the missing link accounting for efficient evasion during the late lytic cycle. Interestingly, BDLF3 also contributes to evasion of CD4+ cell responses to EBV. We report that BDLF3 downregulates expression of surface major histocompatibility complex (MHC) class I and class II molecules in the absence of any effect upon other surface molecules screened, including CD54 (ICAM-1) and CD71 (transferrin receptor). BDLF3 both enhanced internalization of surface MHC molecules and reduced the rate of their appearance at the cell surface. The reduced expression of surface MHC molecules correlated with functional protection against CD8+ and CD4+ T cell recognition. The molecular mechanism was identified as BDLF3-induced ubiquitination of MHC molecules and their subsequent downregulation in a proteasome-dependent manner.
IMPORTANCE Immune evasion is a necessary feature of viruses that establish lifelong persistent infections in the face of strong immune responses. EBV is an important human pathogen whose immune evasion mechanisms are only partly understood. Of the EBV immune evasion mechanisms identified to date, none could explain why CD8+ T cell responses to late lytic cycle genes are so infrequent and, when present, recognize lytically infected target cells so poorly relative to CD8+ T cells specific for early lytic cycle antigens. The present work identifies an additional immune evasion protein, BDLF3, that is expressed late in the lytic cycle and impairs CD8+ T cell recognition by targeting cell surface MHC class I molecules for ubiquitination and proteasome-dependent downregulation. Interestingly, BDLF3 also targets MHC class II molecules to impair CD4+ T cell recognition. BDLF3 is therefore a rare example of a viral protein that impairs both the MHC class I and class II antigen-presenting pathways.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus found in more than 90% of the human population. Primary infection with EBV is usually followed by establishment of a lifelong latent infection, with occasional reactivation (1). The balance between host immune responses, including CD4+ and CD8+ T cells, and viral immune evasion of these responses is key to the spread and survival of EBV in human populations. Passive evasion through the ability to establish antigenically silent latent infections is an important characteristic of all herpesviruses, including EBV. In addition, active evasion mechanisms are an important feature of herpesviruses. Because these active evasion mechanisms are observed predominantly during the lytic phase of the herpesvirus life cycle, they are presumed to be particularly important for enabling virus spread. There have been a number of EBV immune evasion genes identified that are expressed in the lytic cycle and target the major histocompatibility complex (MHC) class I or class II antigen presentation pathway (2, 3). The genes responsible for interfering with MHC class I antigen presentation encode BGLF5, BNLF2a, and BILF1, which act upon different elements of the MHC class I antigen presentation pathway (3–7). The EBV proteins BGLF5, BZLF1, and gp42 have been shown to interfere with MHC class II antigen presentation (5, 8–10).
The above-mentioned MHC class I evasion proteins encoded by EBV have been well studied and shown to act via different mechanisms upon different elements of the MHC class I antigen presentation pathway. Briefly, BGLF5 is a host shutoff protein that has been shown to induce the degradation of MHC class I mRNA, thereby reducing cell surface MHC class I peptide presentation (5, 11). BILF1 is known to target both cell surface MHC class I molecules and those en route to the surface for degradation, thus reducing the presentation of peptides to CD8+ T cells (7, 12, 13). Finally, BNLF2a inhibits the function of the transporter associated with antigen processing (TAP), which reduces the supply of peptides for loading onto MHC class I molecules, thus reducing the level of MHC class I molecule-peptide presentation to CD8+ T cells (4, 14, 15).
Our group recently investigated the relevance of the BGLF5, BNLF2a, and BILF1 immune evasion genes in the context of lytic virus infection (16). It was concluded that BGLF5 in fact plays a minimal role in protecting EBV-infected cells against T cell recognition and that BNLF2a plays an important role in protecting cells during the immediate early (IE) and early (E) stages of the lytic cycle but contributes little protection in the late (L) stage of the lytic cycle (IE > E ≫ L) (14, 16). BILF1 was shown to contribute minimal protection during the immediate early stage of the lytic cycle, a reasonable level of protection during the early stage of the lytic cycle, and a more dramatic level of protection during the late stage of the lytic cycle (IE < E ≪ L) (16). This investigation revealed a level of cooperation between EBV-carried MHC class I immune evasion genes in order to protect cells from CD8+ T cell recognition. However, CD8+ T cell responses to late lytic cycle antigens still recognize lytically infected target cells relatively poorly, even in the absence of BILF1 expression (16, 17). This implies that another, as yet unidentified immune evasion gene or genes may function late in the lytic cycle.
In comparison to what is known about the immune evasion of MHC class I antigen presentation, the evasion of MHC class II antigen presentation by EBV is less well understood. Overexpression of the host shutoff protein BGLF5 has been shown to result in a reduced level of surface MHC class II molecules (5). In addition, the immediate early protein BZLF1 has been shown to interfere with MHC class II antigen presentation by modulating the expression of cell surface invariant chains (8). A third EBV-encoded protein, BZLF2 (gp42), has been shown to interfere with MHC class II antigen presentation to CD4+ T cells by sterically hindering MHC class II interaction with the T cell receptor, thus blocking CD4+ T cell recognition (9, 10). To date, no other EBV proteins have been identified as potential CD4+ T cell immune evasion proteins.
The present study sought to identify novel candidate EBV genes responsible for interfering with MHC class I antigen presentation during the late phase of the lytic cycle, thus providing an explanation for the pronounced immune evasion observed at that stage of the lytic cycle. Screening experiments revealed that the late lytic protein BDLF3, whose functions are unknown (18–20), was able to impair MHC class I antigen presentation. Unexpectedly, BDLF3 also impaired CD4+ T cell recognition of MHC class II-presented peptides. The molecular mechanism for the effect of BDLF3 on antigen presentation involved ubiquitination and proteasome-dependent downregulation of surface MHC class I and class II molecules.
MATERIALS AND METHODS
Plasmids.
The previously described (7) expression plasmid pCDNA3-IRES-GFP, a kind gift from Emmanuel Wiertz (Utrecht Medical Center, Netherlands), was used to subclone and express a selection of EBV genes. The p509 expression plasmid for BZLF1, a kind gift from Paul Farrell (Imperial College London, United Kingdom), has also been described previously (11), as has the cytoplasmic EBNA1 expression vector (21). The retroviral plasmid PLZRS-NGFR, also a kind gift from Emmanuel Wiertz, was used to subclone PLZRS-BDLF3-NGFR, and both were used in transient-transfection assays to allow for in-house sorting of transfected cells based on the expression of surface truncated nerve growth factor receptor (NGFR).
Cells, transfection, and electroporation.
The MJS (Mel JuSo) melanoma-derived cell line (22) and the EBV-negative Burkitt's lymphoma cell line DG75 (23) were maintained in RPMI 1640 supplemented with 10% fetal calf serum (FCS). CIITA-293 cells are HEK-293 cells stably expressing CIITA (24) and were a kind gift from Andrew Hislop, University of Birmingham. These were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS. The myelogenous leukemia cell line K562, transduced to express either HLA-A2, -B35, -Cw1, -DR, or -DQ (a kind gift from Emmanuel Wiertz), was maintained in RPMI 1640 supplemented with 10% FCS plus 400 μg/ml of Geneticin (Invitrogen). EBV-specific CD4+ and CD8+ T cell clones were grown in RPMI 1640 supplemented with 10% FCS, 5% human serum, 30% supernatant from the interleukin-2-producing MLA 144 cell line (25), and 50 U/ml recombinant interleukin-2, as described previously (17).
Transient transfection of MJS and 293-CIITA cells with plasmid DNA was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Transient expression of plasmid DNA in DG75 cells and K562 cells was performed by electroporating cells at 290 V and 950 μF in cuvettes with a 4-mm gap. In some experiments, cells transiently transfected with NGFR-expressing plasmids were positively selected for the surface expression of NGFR by use of MACSelect NGFR-transfected cell selection kits per the manufacturer's protocol (Miltenyi Biotec).
Antibodies.
For immunoprecipitation experiments and for internalization/appearance assays, unconjugated W6/32 and L243 murine monoclonal antibodies (MAbs) to human MHC molecules were obtained from Biolegend: W6/32 (26) recognizes native β2 microglobulin-associated MHC class I complexes (HLA-A, -B, and -C alleles), and L243 recognizes HLA-DR. For flow cytometry experiments, allophycocyanin (APC)- and phycoerythrin (PE)-conjugated antibodies to HLA class I (W6/32), HLA-DR (L243), ICAM1/CD54 (HCD54), and transferrin receptor (TfR)/CD71 (CY1G4) were purchased from Biolegend. For Western blotting, a mouse anti-ubiquitin MAb (P4D1) was purchased from Biolegend. Goat antibodies to calregulin were purchased from Santa Cruz Biotechnology. The BZ.1 murine MAb, specific for the EBV BZLF1-encoded protein, was generated by our laboratory (27). A rabbit anti-BDLF3 (V8) serum was a kind gift from L. Hutt-Fletcher (19).
Flow cytometry analysis of cell surface MHC class I and class II molecules.
Cell surface expression of MHC class I and class II molecules was determined by staining cells with APC- or PE-conjugated anti-HLA class I or class II antibodies and was detected on a BD Biosciences Accuri C6 flow cytometer. Data were analyzed using FlowJo software (TreeStar).
The kinetics of internalization and appearance of cell surface MHC molecules were determined essentially as described previously (12). To assay the kinetics of surface MHC class I and class II internalization, MJS cells were incubated on ice with saturating amounts of anti-MHC class I (W6/32) or anti-MHC class II (L243) MAb. Cells were then washed three times in phosphate-buffered normal saline (PBS) and placed in culture medium at 37°C for 60 min. Aliquots of cells were taken at the times shown in Results and were rapidly cooled to 0°C to inhibit further membrane trafficking. The level of W6/32 or L243 MAb remaining at the cell surface was then analyzed by staining cells with APC-conjugated goat anti-mouse IgG2a antibody (Biolegend). Cells were analyzed using flow cytometry.
To assay the kinetics of MHC class I and II appearance, MJS cells were again incubated with saturating amounts of W6/32 or L243 for 60 min on ice. Cells were washed three times with PBS, placed in warm culture medium at 37°C for 60 min, and then analyzed at the times indicated in Results. After cooling to 0°C to prevent the further appearance of molecules at the surface through membrane trafficking, cells were stained with APC-conjugated W6/32 or APC-conjugated L243 and analyzed using flow cytometry. These directly conjugated anti-MHC detection antibodies would bind only to MHC molecules that had appeared since the excess unconjugated blocking antibody was washed away immediately prior to beginning the incubations in warmed medium. Note that MHC molecules newly arrived at the cell surface are likely to be a mixture of de novo-synthesized molecules arriving at the surface for the first time and recycled molecules that had previously been endocytosed.
Flow cytometric analysis of whole-cell (intracellular) proteins.
Intracellular staining for HLA class I and class II was performed to quantify the total cellular levels of these proteins. Washed pellets of 0.5 × 106 cells were first fixed using 100 μl Ebiosciences intracellular fixative for 1 h on ice, followed by permeabilization using 100 μl (0.2%) Triton X-100 and a further 30-min incubation on ice. After washing in PBS, cells were incubated with the appropriate conjugated antibody for 1 h at 37°C. Cells were then washed in PBS and analyzed using flow cytometry.
T cell function assays.
RAK CD8+ T cell clones, specific for the RAKFKQLL peptide originating from the BZLF1 protein, and SNP CD4+ clones, specific for the SNPKFENIAEGLRVLLARSH epitope from the EBNA1 protein, were generated as previously described (16). Targets for RAK-specific CD8+ T cells were generated by cotransfection of MJS cells with BZLF1 and the control-GFP or BDLF3-GFP expression plasmid. At 24 h posttransfection, cells were used as targets for RAK-specific CD8+ T cell clones. T cell recognition was determined by a gamma interferon-specific enzyme-linked immunosorbent assay (IFN-γ ELISA), using a previously described protocol (16). Targets for SNP-specific CD4+ T cell clones were generated by transfection of MJS cells with the cytoplasmic EBNA1 expression plasmid EBNA1ΔNLS, which generates a target protein that is efficiently processed via the MHC antigen presentation pathway (21). Cells were reseeded at 24 h posttransfection, and 24 h later these cells were transfected with the control-NGFR or BDLF3-NGFR expression plasmid. After another 24 h, cells were harvested and sorted as described above, and the recognition of target cells by SNP-specific CD4+ T cell clones was determined by IFN-γ ELISA.
Immunoprecipitation.
Positively selected control-NGFR- and BDLF3-NGFR-expressing MJS cells (2 × 106) were used for surface MHC class I and class II immunoprecipitation. Cells were incubated for 2 h on ice with anti-HLA class I MAb (W6/32) or anti-HLA class II MAb (L243) and then washed and lysed using 400 μl of NP-40 buffer (0.5% Nonidet P-40, 5 mM MgCl2, and 50 mM Tris-HCl, pH 7.5) with protease inhibitor cocktail (Sigma) at 4°C for 45 min. Nuclei and insoluble debris were removed by centrifugation, and the supernatants were incubated with 20 μl of Dynabeads protein A and 20 μl of Dynabeads protein G (Invitrogen) at 4°C overnight. Beads were then washed four times with NET buffer (0.5% NP-40, 150 mM NaCl, 5 mM EDTA, and 50 mM Tris-HCl, pH 7.5), and the precipitated proteins were eluted by boiling in reducing sample buffer for 5 min. Finally, samples were separated by SDS-PAGE on 4 to 12% Bis-Tris NuPage minigels with morpholinepropanesulfonic acid (MOPS) electrolysis buffer (Invitrogen).
Western blotting.
Total cell lysates were denatured in reducing sample buffer and then sonicated and heated to 100°C for 5 min. Solubilized proteins equivalent to 2 × 105 cells/20-μl sample were separated by SDS-PAGE on 4 to 12% acrylamide gradient bis-Tris NuPage minigels with MOPS running buffer (Invitrogen).
RESULTS
The late lytic protein BDLF3 is an immune evasion protein.
As previously demonstrated, EBV encodes a number of immune evasion proteins that cooperate to afford the protection of EBV-infected cells against recognition by CD8+ T cells during the lytic cycle. However, it has been hypothesized that an as yet unidentified EBV lytic gene may be responsible for the ultimate protection of EBV-infected cells during the late stage of the lytic cycle (16). In order to identify other potential EBV immune evasion proteins involved in protecting infected cells against CD8+ T cell recognition, more than 25 EBV genes expressed during the lytic cycle were transiently expressed in MJS cells by using bicistronic plasmid vectors that coexpressed green fluorescent protein (GFP) with the test gene (see Fig. S1A in the supplemental material). The expression of GFP allowed for the identification of transfected cells by flow cytometry. At 24 h posttransfection, flow cytometric analysis was used to analyze surface expression levels of MHC class I on GFP-positive cells. Of all the EBV lytic genes included in this screen, the only one encoding a protein that reproducibly affected surface levels of MHC class I was the BDLF3 gene (Fig. 1A). Figure 1A shows a representative selection of these screens. As a control, the level of surface MHC class II was also analyzed in this screen (see Fig. S1B). Interestingly, BDLF3 also affected the surface expression of MHC class II (Fig. 1B).
FIG 1.

Screening of EBV lytic genes to identify potential MHC class I immune evasion genes. MJS cells were transiently transfected with pCDNA3.1-IRES-GFP plasmids carrying a selection of EBV lytic genes. At 24 h posttransfection, surface levels of MHC class I (A) and MHC class II (B) on GFP-positive cells were analyzed using two-color flow cytometry. (C) AKBM cells were induced into the lytic cycle by cross-linking of B cell receptors for 1 h at 37°C and were analyzed by Western blotting at the indicated time points postinduction. Levels of BZLF1 (upper blot), BDLF3 (middle blot; stars indicate monomeric and trimeric forms of BDLF3), and the loading control calregulin (lower blot) are shown.
BDLF3 was previously classified as a late expressed lytic protein (19). We confirmed the late expression kinetics by using the EBV-positive cell line AKBM (9), which can be induced into the lytic cycle by cross-linking of the B cell receptor. Following induction, aliquots of induced AKBM cells were taken at 0 h, 1 h, 6 h, 12 h, 24 h, and 48 h, and immunoblotting was performed in order to detect the expression of BDLF3 and the immediate early protein BZLF1. As shown in Fig. 1C, BDLF3 protein expression was detected weakly at 12 h postinduction, but stronger expression was seen at 24 h. These expression kinetics are consistent with previous findings (19) and suggest that BDLF3 could be the missing link immune evasion protein responsible for interfering with MHC class I antigen presentation during the late stage of the lytic cycle to protect these cells against CD8+ T cell recognition. In addition, the effect on expression of MHC class II molecules raised the possibility that BDLF3 also plays a role in CD4+ T cell immune evasion. To confirm that the levels of BDLF3 protein expression in our transfected cells were physiologically relevant, the BDLF3 protein expression level in transfected MJS cells was compared with the BDLF3 expression level in induced AKBM cells after adjusting for the percentage of GFP+ cells in MJS cells and the percentage of VCA+ cells in induced AKBM cells. Quantification of Western blots (see Fig. S2 in the supplemental material) showed that the expression of BDLF3 in MJS cells was around 70% of that in induced AKBM cells.
In order to confirm that BDLF3 acts specifically on MHC class I and MHC class II, MJS cells transiently expressing BDLF3 were harvested at 24 h posttransfection and were analyzed in more detail by using flow cytometry to detect surface levels of MHC class I, MHC class II, and other cell surface proteins. The results in Fig. 2A indicate that BDLF3-expressing cells (dashed line) exhibited a 60% decrease in surface MHC class I mean fluorescence intensity (MFI) compared to control cells (solid black line). A similar result was observed for surface levels of MHC class II on BDLF3-positive cells (Fig. 2B), where there was a 50% reduction in MHC class II MFI. Importantly, the levels of two other surface proteins tested, transferrin receptor (TfR) (Fig. 2C) and ICAM1 (Fig. 2D), were not affected by the expression of BDLF3.
FIG 2.
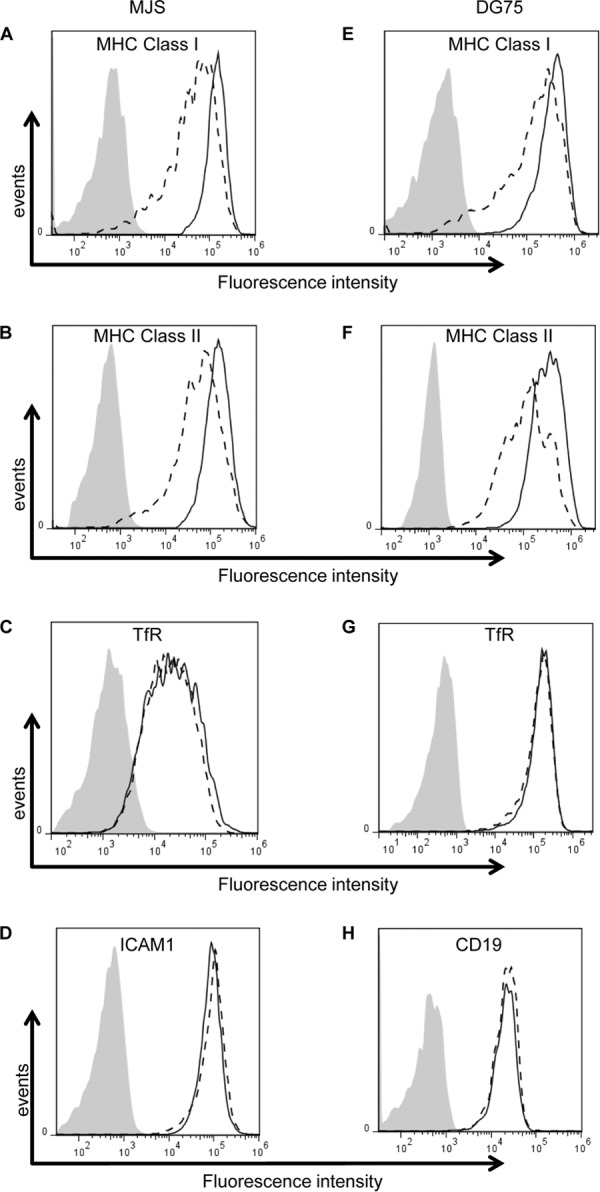
BDLF3 expression induces the downregulation of surface MHC class I and MHC class II. MJS cells (A to D) and DG75 cells (E to H) were transiently transfected with the control-GFP or BDLF3-GFP plasmid. At 24 h posttransfection, two-color flow cytometry was used to measure surface levels of MHC class I (A and E), MHC class II (B and F), TfR (C and G), ICAM1 (D), and CD19 (H) in the GFP+ populations of control-GFP-transfected cells (solid lines) and BDLF3-GFP-transfected cells (dashed lines). The gray histograms denote background staining obtained with an isotype control antibody.
Since B cells are the natural reservoir for EBV, we next investigated the phenotype of BDLF3 in B cells. To this end, the effect of transient BDLF3 expression on surface MHC molecules on the EBV-negative B cell line DG75 was investigated using flow cytometry. In this instance, the ICAM1 control could not be included, as its expression on DG75 cells is negligible; therefore, the effect of BDLF3 on expression of CD19 was analyzed along with TfR expression. As shown in Fig. 2E to H, results similar to those seen in MJS cells were obtained. BDLF3-expressing DG75 cells showed a 43% reduction in the MFI of MHC class I (Fig. 2E) and a 30% reduction in that of MHC class II (Fig. 2F) compared to the MFI of control cells not expressing BDLF3. There was no observed effect of BDLF3 on the expression of surface TfR (Fig. 2G) or CD19 (Fig. 2H).
BDLF3 induces downregulation of all screened MHC class I and MHC class II alleles.
Since some viral immune evasion proteins, including BILF1, have been shown to preferentially target specific HLA class I alleles (13), we next sought to investigate the HLA specificity of BDLF3. For this purpose, MHC class I-negative K562 cells engineered to stably express HLA-A2, -B35, or -Cw1 were transiently transfected to express either the BDLF3 or control vector. At 24 h posttransfection, the surface levels of HLA-A2, -B35, and -Cw1 on positively transfected cells were detected using flow cytometry. As shown in Fig. 3A, cells expressing BDLF3 showed decreases in the cell surface levels of HLA-A2 (upper histograms) (27% reduction in MFI), HLA-B35 (middle histograms) (34% reduction in MFI), and HLA-Cw1 (lower histograms) (26% reduction in MFI) compared to those in control cells. A similar approach was then used to test the specificity of BDLF3 for HLA class II alleles. In this case, HLA class II-negative HEK-293 cells engineered to stably express CIITA, thus driving the surface expression of HLA-DR and -DQ, were transiently transfected to express the BDLF3 or control vector. Similar to the results seen for HLA class I alleles, BDLF3 induced reductions in both HLA-DR (47% reduction in MFI) (Fig. 3B, upper histograms) and HLA-DQ (32% reduction in MFI) (Fig. 3B, lower histograms) compared to the levels in control transfected cells. In all examples, the levels of surface TfR remained similar between BDLF3- and control-transfected cells (data not shown). These results indicate that BDLF3 is not selective in downregulating HLA molecules but instead acts more broadly to downregulate all HLA class I and HLA class II molecules.
FIG 3.
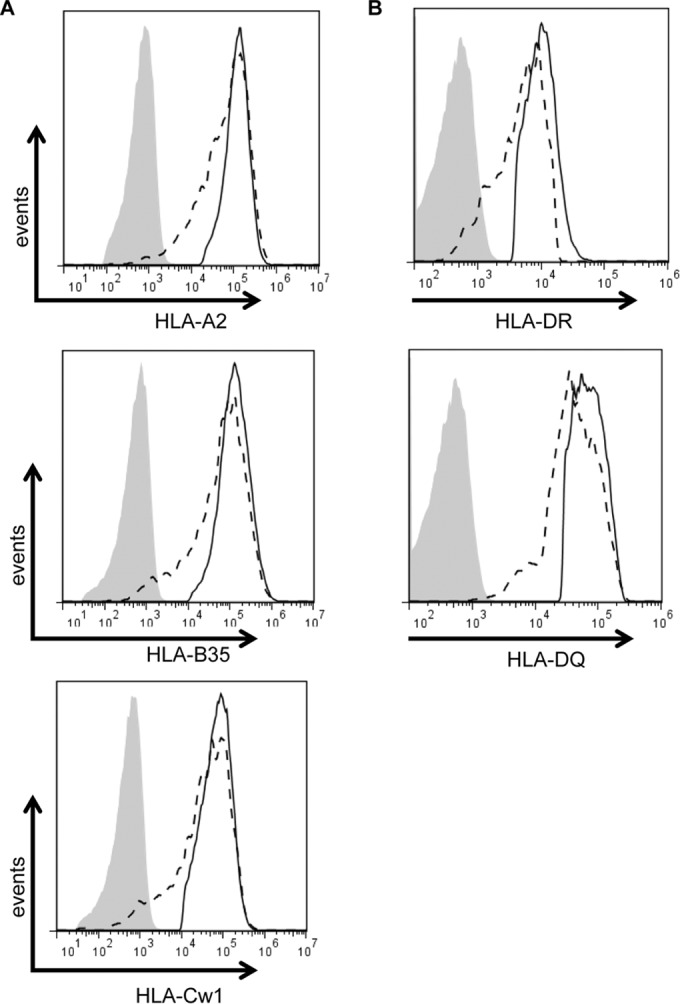
BDLF3 induces downregulation of all HLA class I and class II alleles. (A) The MHC class I-negative cell line K562, transduced to stably express either HLA-A2, -B35, or -Cw1, was electroporated with the control-GFP or BDLF3-GFP plasmid. At 24 h posttransfection, two-color flow cytometry was used to measure surface MHC class I levels in the GFP+ populations in the control-GFP-transfected cells (solid lines) and BDLF3-GFP-transfected cells (dashed lines). (B) HEK-293 cells stably expressing CIITA were transiently transfected with the control-GFP or BDLF3-GFP plasmid. At 24 h posttransfection, two-color flow cytometry was used to measure surface HLA-DR and HLA-DQ levels in GFP+ populations in the control-GFP-transfected cells (solid lines) and the BDLF3-GFP-transfected cells (dashed lines). The gray histograms denote background staining obtained with an isotype control antibody.
BDLF3-mediated reductions in surface MHC class I and class II confer protection against both CD8+ and CD4+ T cell recognition.
Since BDLF3 induces reductions in the levels of surface MHC class I and class II molecules, we next investigated whether BDLF3 expression provided protection against recognition by EBV-specific CD8+ and CD4+ T cells. In order to address this, the HLA-B8-positive cell line MJS was cotransfected with BZLF1 and either a BDLF3-GFP or control-GFP vector plasmid (Fig. 4A). At 24 h posttransfection, these cells were used as targets in a T cell assay with CD8+ T cell clones restricted through HLA-B8 and specific for the peptide RAKFKQLL, contained within the BZLF1 antigen. T cell recognition was measured by the amount of IFN-γ release in the IFN-γ ELISA. As shown in one representative experiment (n = 3) in Fig. 4A, the expression of BDLF3 resulted in a significant decrease in IFN-γ release by RAK-specific T cell clones, from ∼1,100 pg/ml to ∼500 pg/ml. The BDLF3-mediated reduction in BZLF1-specific CD8+ T cell recognition was not due to any change in the expression of BZLF1 target protein expression (Fig. 4B).
FIG 4.

BDLF3 can inhibit EBV-specific CD8+ and CD4+ T cell recognition. MJS cells were cotransfected with the p509 plasmid (BZLF1 expression vector) together with control-GFP or BDLF3-GFP. At 24 h posttransfection, the MJS cells were cocultured with effector T cells, i.e., a BZLF1 (RAK)-specific CD8+ T cell clone, for a further 18 h, and the supernatants were tested for the release of IFN-γ as a measure of T cell recognition. All results are expressed as amounts of IFN-γ release (pg/ml), and error bars indicate standard deviations for triplicate cultures. (B) Total cell lysates were generated from the above transfections and analyzed by Western blotting using antibodies specific for BDLF3, BZLF1, and calregulin (loading control). The asterisks adjacent to the BDLF3 blot indicate monomeric and trimeric forms of BDLF3. (C) MJS-DR51 cells were first transfected with EBNA1ΔNLS, allowed to recover in culture overnight, and then divided into two groups and transfected with either BDLF3-NGFR or control-NGFR. After another 24 h, NGFR+ BDLF3+ or control NGFR+ cells were sorted with magnetic beads and used as targets for HLA-DR51-restricted, EBNA1 (SNP)-specific CD4+ T cell clones. Recognition was measured as the amount of IFN-γ release (pg/ml) by T cell clones. Error bars represent standard deviations of the means for triplicate assay replicates. Results are representative of three independent experiments. (D) Total cell lysates were generated from the above transfections and analyzed by Western blotting using antibodies specific for BDLF3, EBNA1, and calregulin (loading control).
In order to investigate the ability of BDLF3 to protect cells against recognition by CD4+ T cells, a similar method was employed. MJS cells stably expressing the HLA class II allele DR51 were transfected to express cytoplasmic EBNA1 for 48 h and either the BDLF3-NGFR or control-NGFR vector for a further 24 h (Fig. 4C). Cells were then sorted based on expression of NGFR and subsequently used as targets for CD4+ T cell clones specific for the HLA-DR51-restricted epitope SNPKFENIAEGLRVLLARSH, contained within EBNA1. As shown in Fig. 4C, the expression of BDLF3 resulted in a decrease in T cell recognition (IFN-γ release) by SNP-specific CD4+ T cell clones, from ∼1,300 pg/ml to ∼900 pg/ml, compared to that by control cells. The BDLF3-mediated reduction in EBNA1-specific CD4+ T cell recognition was not due to any change in the expression of EBNA1 target protein expression (Fig. 4D). Thus, in a pattern similar to the results seen for BDLF3 protection against CD8+ T cell recognition, the BDLF3-induced reduction in surface MHC class II molecules also correlated with protection against CD4+ T cell recognition.
These data show that BDLF3-induced reductions in cell surface MHC class I and class II are functional in protecting BDLF3-expressing cells against recognition by both CD8+ and CD4+ T cells.
BDLF3 downregulates surface MHC molecules more dramatically than total MHC molecules.
To explore the mechanism of MHC class I and class II downregulation by BDLF3, we first asked whether the total cellular pool of MHC molecules was affected or whether surface MHC molecules were selectively targeted. To this end, flow cytometry analysis of intracellular staining of fixed and permeabilized cells was used to detect the level of whole-cell MHC molecules compared to that of surface MHC molecules detected on impermeable viable cells. As expected, surface-level MFI of MHC class I and class II were both reduced approximately 50% on cells expressing BDLF3 compared to control cells (Fig. 5A, left column). This difference was found to be significant (Fig. 5B, white bars). Interestingly, there was only a slight decrease (10%) in the MFI of whole-cell MHC class I and MHC class II compared to that for control cells (Fig. 5A, right column), and this small reduction was not statistically significant when the results from three independent experiments were pooled and analyzed (Fig. 5B, gray bars). To confirm these findings by an independent method, control GFP-expressing MJS cells and BDLF3-GFP-expressing MJS cells were purified using a Mo-flow cell sorter, and the expression of total MHC-I and MHC-II was examined by Western blotting. The results showed no significant difference (see Fig. S3 in the supplemental material), confirming the results from intracellular flow cytometry data. Importantly, BDLF3 had no effect on surface or whole-cell levels of ICAM1 expression (Fig. 5A and B, bottom panels).
FIG 5.

BDLF3 induces a more dramatic reduction in surface MHC class I and II than in whole-cell MHC levels. (A) MJS cells were transiently transfected with the control-GFP or BDLF3-GFP plasmid. At 24 h posttransfection, two-color flow cytometry was used to measure the levels of surface MHC class I (upper left), MHC class II (middle left), and ICAM1 (lower left) in the viable GFP+ populations of control-GFP-transfected cells (solid lines) and BDLF3-GFP transfected cells (dashed lines). The gray histograms denote background staining obtained with an isotype control antibody. In parallel, these GFP+ transfected MJS cells were analyzed for whole-cell levels of MHC class I (upper right), MHC class II (middle right), and ICAM1 (lower right), using intracellular staining of fixed and permeabilized cells. The results are representative of repeated experiments. (B) Relative mean fluorescence intensities (MFI) of MHC class I, MHC class II, and ICAM1 were calculated for BDLF3-GFP+ cells compared to control GFP+ cells. Results are combined data from three independent experiments. White bars represent surface staining, and gray bars represent whole-cell staining. Differences that reached significance (P < 0.05) by Student's paired t test are denoted by asterisks.
These data show that BDLF3 affects the levels of surface MHC molecules more dramatically than it affects whole-cell MHC molecules, suggesting that BDLF3 exerts its function predominantly on surface MHC class I and class II rather than the intracellular fraction of these molecules.
BDLF3 induces rapid internalization and delayed appearance of MHC molecules.
Since BDLF3 predominantly targets surface MHC molecules, we next examined whether it targets MHC molecules already at the cell surface or those trafficking to the cell surface. We therefore compared the kinetics of MHC class I and class II internalization and appearance at the cell surface of BDLF3-expressing cells by using flow cytometry. Representative examples are shown for MHC class I (Fig. 6A, upper panel) and class II (Fig. 6A, lower panel) internalization assays, in which the percentages of MHC class I and class II molecules remaining on the surfaces of BDLF3-expressing and control cells were measured over 60 min. Cells expressing BDLF3 showed lower levels of MHC remaining at the cell surface at each time point indicated, such that by 60 min there was 20% and 13% less surface MHC class I and MHC class II, respectively, on BDLF3-expressing cells than on control cells. These data indicate that BDLF3 induces higher rates of both MHC class I and MHC class II internalization.
FIG 6.

BDLF3 induces more rapid internalization and a delayed appearance of both MHC class I and class II molecules at the cell surface. Internalization and appearance assays were performed on MJS cells transiently expressing control-GFP or BDLF3-GFP. The GFP+ population was used to gate BDLF3-expressing cells. Internalization and appearance assays were performed on cells pretreated on ice with saturating amounts of anti-MHC class I antibody or anti-MHC class II antibody. Cells were then washed and incubated at 37°C for up to 60 min. (A) For the internalization assay, viable cells harvested at each time point were stained with an APC-conjugated goat anti-mouse IgG antibody and analyzed using flow cytometry at the indicated times; this identified the prelabeled antibody-bound MHC molecules that remained at the surface, while endocytosed labeled MHC molecules were not detected on the viable cells. The mean fluorescence intensities of staining were averaged for triplicate samples and then normalized to the values for time zero samples. (B) For the appearance assays, newly arrived MHC-I and MHC-II molecules, which were not prelabeled with unconjugated antibodies, were detected by staining with an APC-conjugated anti-MHC class I antibody or anti-MHC class II antibody. The mean fluorescence intensities of staining were averaged for triplicate samples and then normalized to the values for time zero samples. Results are representative of three independent experiments.
When a similar assay was used to measure the rates of MHC class I and class II surface appearance, BDLF3-expressing cells conversely showed decreased rates of both MHC class I and class II surface appearance at each time point compared to the rates for control cells (Fig. 6B). By 60 min, the appearance of MHC class I and class II on BDLF3-expressing cells was reduced 50% and 47%, respectively, in comparison to that on control cells. This BDLF3-mediated reduction in the rate of appearance of MHC at the surface (Fig. 6B) was noticeably larger than the accelerated rate of endocytosis (Fig. 6A).
It should be noted that in all experiments, the rates of TfR internalization or appearance remained similar between control and BDLF3-expressing cells (data not shown). These data indicate that BDLF3 is able to both enhance endocytosis of MHC molecules at the cell surface and interfere with the trafficking of intracellular MHC molecules to the cell surface.
BDLF3 downregulation of surface MHC molecules involves ubiquitination and the proteasomal pathway.
We next sought to identify the mechanism by which BDLF3 is able to enhance internalization and delay the appearance of surface MHC class I and MHC class II molecules. Our initial experiments were designed to identify which pathway BDLF3 might utilize in order to reduce the expression of surface MHC molecules. To this end, we incubated BDLF3-expressing cells with proteasomal and lysosomal inhibitors. In the absence of drug treatment, cells expressing BDLF3 showed lower levels of surface MHC class I and II expression than those on control cells (Fig. 7A), as expected. However, when BDLF3-expressing cells were incubated with the proteasomal inhibitor MG132, they showed no such reduction in surface MHC class I and MHC class II levels compared to control cells (Fig. 7B). Partial abrogation of the BDLF3 phenotype by MG132 was observed after 4 h of treatment, but the effect of MG132 treatment was maximal after 16 h (see Fig. S4 in the supplemental material). Similar results were seen when cells were treated with a second proteasomal inhibitor, bortezomib (see Fig. S5), whereas treatment with a lysosomal inhibitor, bafilomycin (see Fig. S6), did not prevent BDLF3-induced downregulation of surface MHC molecules. These data indicate that BDLF3-induced downregulation of surface MHC molecules is dependent upon the proteasomal pathway.
FIG 7.

Treatment of BDLF3-expressing cells with a proteasome inhibitor prevents downregulation of MHC class I and class II. MJS cells were transiently transfected with the BDLF3-GFP or control-GFP plasmid and then incubated in normal medium (A) or MG132 (5 μM)-supplemented medium (B). At 24 h posttransfection, two-color flow cytometry was used to measure surface MHC class I (upper histograms), surface MHC class II (middle histograms), and surface ICAM1 (lower histograms) in GFP+ populations of control-GFP-transfected cells (solid lines) and BDLF3-GFP-transfected cells (dashed lines). The gray histograms denote background staining obtained with an isotype control antibody. Results are representative of repeat experiments (n > 4). (C and D) MJS cells were transiently transfected with the BDLF3-GFP or control-GFP plasmid and then were incubated with MG132 (5 μM). Following drug treatment, the rates of internalization (C) of MHC-I (top panel) and MHC-II (bottom panel) and the rates of appearance (D) of MHC-I (top panel) and MHC-II (bottom panel) were measured using the same methods as those described in the legend to Fig. 6. (E and F) MJS cells were transfected with a ubiquitin expression plasmid plus either the control-NGFR or BDLF3-NGFR plasmid. The transfected cells were then divided into two groups and incubated in normal medium or medium supplemented with MG132. At 24 h posttransfection, NGFR+ BDLF3+ or control NGFR+ cells were sorted with magnetic beads, and surface MHC class I (C) or MHC class II (D) molecules were immunoprecipitated, eluted, and then immunoblotted using an anti-ubiquitin (Ub) antibody (P4D1).
Because BDLF3 downregulates surface MHC molecules through increased internalization and a delayed appearance (Fig. 6), we next examined what effect proteasomal inhibition might have on the kinetics of MHC class I and class II internalization and appearance at the cell surface of BDLF3-expressing cells. The results showed that MG132 completely abrogated the effect of BDLF3 on both the rate of internalization (Fig. 7C) and the rate of appearance (Fig. 7D), demonstrating an essential role of the proteasome in BDLF3-induced MHC molecule downregulation.
Given the essential role that the proteasome plays in the BDLF3-induced reduction of surface MHC molecules and knowing that ubiquitination is an important component of the proteasomal pathway, we next assessed whether BDLF3 induces ubiquitination of surface MHC molecules. To this end, MJS cells were transfected with various expression vectors, including BDLF3, control, and ubiquitin vectors (Fig. 7E and F). These cells were then incubated with or without the proteasomal inhibitor MG132. At 24 h posttransfection, surface MHC class I or MHC class II molecules were immunoprecipitated from BDLF3-expressing or control cells, and resulting immunoblots were probed with ubiquitin-specific antibodies. As shown in Fig. 7, polyubiquitinated high-molecular-weight bands appeared in immunoblots for immunoprecipitated MHC class I (Fig. 7E) and MHC class II (Fig. 7F) from BDLF3-expressing cells treated with MG132. These ubiquitin-reactive bands were less pronounced for both control cells treated with MG132 and BDLF3-expressing cells not treated with MG132.
DISCUSSION
This study reveals the identity and mechanism of the novel immune evasion protein BDLF3, which induces downregulation of not only cell surface MHC class I but also MHC class II, to the extent that antigen recognition by both CD8+ and CD4+ virus-specific T cells is functionally impaired. The BDLF3 protein was first identified a number of years ago as the glycoprotein gp150, which is located at the cell membrane and in the virion, is not essential for EBV replication, and hitherto had no known function (18–20). Our study now allows a function to be assigned to BDLF3.
The identification of BDLF3 as an immune evasion protein has an important impact on our knowledge of the T cell response to lytic EBV antigens and the protection of EBV-infected cells from recognition by these T cells. The EBV lytic cycle involves the synchronous expression of more than 60 viral proteins, many of which elicit strong CD4+ and CD8+ T cell responses, but various immune evasion mechanisms enable EBV to persist as a lifelong infection. For CD8+ T cell responses to lytic cycle antigens, there is a pattern of immunodominance that correlates with the efficiency of antigen presentation during the lytic cycle. An earlier study by our group revealed that the known immune evasion proteins BGLF5, BNLF2a, and BILF1 act in cooperation to afford protection to EBV-infected cells against lytic cycle-specific CD8+ T cell recognition. However, the ultimate protection that is seen in the late phase of the lytic cycle could not be explained fully by the action of these known evasion genes (16). The identification of BDLF3, which is expressed during the late stage of the lytic cycle, as a potent inhibitor of the MHC class I antigen presentation pathway makes it a prime candidate as the missing link immune evasion protein responsible for protecting EBV-infected cells from CD8+ T cell responses during the late stage of the lytic cycle.
Another important feature of BDLF3 is its ability to induce MHC class II downregulation and evade CD4+ T cell recognition. Evidence is accumulating in the literature to show that viruses target multiple points on the MHC class II antigen presentation pathway, including suppression of CIITA (28, 29), diversion or degradation of DR molecules during membrane transport (30), and direct targeting of the CD74 (invariant chain) chaperone of DR (8). Two viral genes expressed during the EBV lytic cycle have been reported to manipulate the MHC class II antigen presentation pathway. BZLF1 induces a marked downregulation of surface CD74 to impair antigen presentation and CD4+ T cell recognition (8), and gp42 sterically inhibits interactions between the T cell receptor on the CD4+ T cell and MHC-II–peptide complexes (10). The identification of BDLF3 as a novel MHC class II evasion protein from EBV indicates that, similarly to interference with MHC class I antigen presentation, interference with MHC class II antigen presentation very likely involves the cooperative action of multiple evasion genes.
The fact that the BDLF3 protein is an EBV late lytic cycle protein suggests that it helps to provide enhanced protection of virus-producing cells prior to release of mature virions. In addition, as BDLF3 can be detected in the EBV virion (31), this raises the possibility that BDLF3 can act immediately after new infections of B cells to modulate recognition by existing EBV-specific CD4+ T cells. Considering the important role of MHC class II molecules and gp42 in EBV infection, and also the observation that BDLF3 knockout EBV particles can infect epithelial cells better than B cells (18), we can propose a potential role for BDLF3 in EBV infection. BDLF3 expression results in decreased MHC class II expression at the surfaces of lytically replicating cells, reducing the amount of MHC class II available to bind gp42. We therefore predict that BDLF3 knockout virions may contain less envelope gp42 than wild-type EBV, with a consequent enhanced ability to infect epithelial cells.
Our data indicate that the mechanism of BDLF3 interference with the appearance of MHC molecules at, and their internalization from, the cell surface involves ubiquitination of MHC molecules and a proteasome-dependent pathway. The targeting of MHC molecules for ubiquitination has been described for other viral immune evasion proteins, but the mechanism of action of BDLF3 is clearly distinct. An example is the K3 and K5 proteins encoded by Kaposi's sarcoma-associated herpesvirus (KSHV), which function as two membrane-bound E3 ubiquitin ligases and have been shown to facilitate the rapid endocytosis and subsequent degradation of MHC class I by inducing ubiquitination (32–37). Unlike K3 and K5, there is no evidence to show that BDLF3 itself is an E3 ubiquitin ligase; therefore, it is very likely that BDLF3 functions via a different mechanism. Indeed, in terms of MHC class I downregulation, BDLF3 can affect all MHC class I alleles we studied, whereas K5 affects HLA-A and -B but has a weak effect on HLA-C, while K3 downregulates all HLA class I alleles (33). More importantly, KSHV K3 and K5 can downregulate a range of other surface proteins, including B7-2, CD54 (ICAM-1), CD1d, CD31 (PECAM-1), IFN-γR1, MICA/B, BST-2, ALCAM, and Syntaxin-4 (38–42). In contrast, we found that BDLF3 targets MHC-I and MHC-II but not CD54 (Fig. 2) or MICA/B (data not shown). Another distinguishing feature of BDLF3 is that while the surface levels of MHC class I and class II were reduced about 50%, there were minimal decreases of whole-cell MHC class I and MHC class II (Fig. 5A; see Fig. S3 in the supplemental material). The observation that BDLF3 can induce a 50% reduction in surface MHC molecules, compared to a reduction of less than 10% of the whole-cell MHC molecules, reflects the fact that there is a relatively large reservoir of MHC class I and MHC class II molecules inside cells. Therefore, the 10% reduction in whole-cell MHC classes I and II could represent the complete degradation of the 50% of MHC classes I and II that is lost from the surface in the presence of BDLF3.
While BDLF3 does not function as a ubiquitin E3 ligase, it nevertheless downregulates surface MHC class I and MHC class II through inducing ubiquitination. How might this be? One possible explanation is that it may recruit other cellular E3 ubiquitin ligase proteins, such as members of the membrane-associated RING-CH (MARCH) proteins, the cellular orthologues of K3 and K5. These proteins have been implicated in the regulation of cell surface molecules, including MHC class I, MHC class II, ICAM, and transferrin receptor (36, 43–46). Indeed, the overexpression of MARCH-IV and MARCH-IX proteins induces ubiquitination and rapid internalization of MHC class I molecules (43). Numerous MARCH proteins have been identified that target MHC class I or MHC class II, although at the time of this writing, no single known MARCH protein induces the ubiquitination of both MHC class I and class II without affecting other surface markers that are left unaffected by BDLF3 (47). Our preliminary experiments have not been able to demonstrate coimmunoprecipitation of MHC molecules with BDLF3 (data not shown). Thus, BDLF3 may recruit an as yet unidentified MARCH family protein, or perhaps several of these proteins. If so, then the specificity of BDLF3 would be due to the target molecules of these recruited proteins. Future work will be aimed at resolving these possibilities.
Considering the data that we obtained for BDLF3 and the features that distinguish the effects of BDLF3 from those of previously characterized immune evasion proteins, we postulate that its mechanism of action is broadly as follows. Because BDLF3 reduces the rate of appearance of MHC molecules at the cell surface to a greater extent than it increases the rate of endocytosis (Fig. 6), we suppose that the reduced rate of appearance must be due at least in part to an effect on de novo-synthesized MHC molecules trafficking to the surface. Therefore, BDLF3 targets both de novo-synthesized and recycling endocytosed MHC molecules for ubiquitination. These ubiquitinated MHC molecules are directed to proteasomal degradation or, in the presence of proteasomal inhibitors, accumulate at the cell surface. The finer details of the biochemical mechanisms and the identities of the ubiquitin ligases involved remain to be resolved.
The acquisition of immune evasion proteins has played a critical role in the evolution of viruses. It is interesting that an EBV homolog, the marmoset lymphocryptovirus (maLCV), which naturally infects New World nonhuman primates, lacks BDLF3. This may be relevant to the fact that serological studies revealed maLCV infection in marmosets to be much less ubiquitous than EBV infection in humans (48, 49). It might therefore be speculated that the acquisition of BDLF3 immune evasion functions was a later evolutionary event that contributed to the success of EBV in successfully colonizing the vast majority of the human population.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andrew Hislop for helpful discussions during this study.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02183-15.
REFERENCES
- 1.Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Ressing ME, Horst D, Griffin BD, Tellam J, Zuo J, Khanna R, Rowe M, Wiertz EJ. 2008. Epstein-Barr virus evasion of CD8(+) and CD4(+) T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol 18:397–408. doi: 10.1016/j.semcancer.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Rowe M, Zuo J. 2010. Immune responses to Epstein-Barr virus: molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect 12:173–181. doi: 10.1016/j.micinf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers-Lalic D, Croft NP, Neefjes JJ, Rickinson AB, Wiertz EJ. 2007. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med 204:1863–1873. doi: 10.1084/jem.20070256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe M, Glaunsinger B, van Leeuwen D, Zuo J, Sweetman D, Ganem D, Middeldorp J, Wiertz EJ, Ressing ME. 2007. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc Natl Acad Sci U S A 104:3366–3371. doi: 10.1073/pnas.0611128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeidler R, Eissner G, Meissner P, Uebel S, Tampe R, Lazis S, Hammerschmidt W. 1997. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 90:2390–2397. [PubMed] [Google Scholar]
- 7.Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, Wiertz EJ, Rowe M. 2009. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog 5:e1000255. doi: 10.1371/journal.ppat.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuo J, Thomas WA, Haigh TA, Fitzsimmons L, Long HM, Hislop AD, Taylor GS, Rowe M. 2011. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog 7:e1002455. doi: 10.1371/journal.ppat.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ressing ME, van Leeuwen D, Verreck FA, Keating S, Gomez R, Franken KL, Ottenhoff TH, Spriggs M, Schumacher TN, Hutt-Fletcher LM, Rowe M, Wiertz EJ. 2005. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J Virol 79:841–852. doi: 10.1128/JVI.79.2.841-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ressing ME, van Leeuwen D, Verreck FA, Gomez R, Heemskerk B, Toebes M, Mullen MM, Jardetzky TS, Longnecker R, Schilham MW, Ottenhoff TH, Neefjes J, Schumacher TN, Hutt-Fletcher LM, Wiertz EJ. 2003. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A 100:11583–11588. doi: 10.1073/pnas.2034960100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo J, Thomas W, van Leeuwen D, Middeldorp JM, Wiertz EJ, Ressing ME, Rowe M. 2008. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J Virol 82:2385–2393. doi: 10.1128/JVI.01946-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ, Hislop AD, Rowe M. 2011. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J Virol 85:1604–1614. doi: 10.1128/JVI.01608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffin BD, Gram AM, Mulder A, Van Leeuwen D, Claas FH, Wang F, Ressing ME, Wiertz E. 2013. EBV BILF1 evolved to downregulate cell surface display of a wide range of HLA class I molecules through their cytoplasmic tail. J Immunol 190:1672–1684. doi: 10.4049/jimmunol.1102462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croft NP, Shannon-Lowe C, Bell AI, Horst D, Kremmer E, Ressing ME, Wiertz EJ, Middeldorp JM, Rowe M, Rickinson AB, Hislop AD. 2009. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog 5:e1000490. doi: 10.1371/journal.ppat.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horst D, van Leeuwen D, Croft NP, Garstka MA, Hislop AD, Kremmer E, Rickinson AB, Wiertz EJ, Ressing ME. 2009. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J Immunol 182:2313–2324. doi: 10.4049/jimmunol.0803218. [DOI] [PubMed] [Google Scholar]
- 16.Quinn LL, Zuo J, Abbott RJ, Shannon-Lowe C, Tierney RJ, Hislop AD, Rowe M. 2014. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog 10:e1004322. doi: 10.1371/journal.ppat.1004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pudney VA, Leese AM, Rickinson AB, Hislop AD. 2005. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J Exp Med 201:349–360. doi: 10.1084/jem.20041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borza CM, Hutt-Fletcher LM. 1998. Epstein-Barr virus recombinant lacking expression of glycoprotein gp150 infects B cells normally but is enhanced for infection of epithelial cells. J Virol 72:7577–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurilla MG, Heineman T, Davenport LC, Kieff E, Hutt-Fletcher LM. 1995. A novel Epstein-Barr virus glycoprotein gp150 expressed from the BDLF3 open reading frame. Virology 209:108–121. doi: 10.1006/viro.1995.1235. [DOI] [PubMed] [Google Scholar]
- 20.Nolan LA, Morgan AJ. 1995. The Epstein-Barr virus open reading frame BDLF3 codes for a 100-150 kDa glycoprotein. J Gen Virol 76:1381–1392. doi: 10.1099/0022-1317-76-6-1381. [DOI] [PubMed] [Google Scholar]
- 21.Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. 2010. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A 107:2165–2170. doi: 10.1073/pnas.0909448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JP, Demmer-Dieckmann M, Meo T, Hadam MR, Riethmuller G. 1981. Surface antigens of human melanoma cells defined by monoclonal antibodies. I. Biochemical characterization of two antigens found on cell lines and fresh tumors of diverse tissue origin. Eur J Immunol 11:825–831. [DOI] [PubMed] [Google Scholar]
- 23.Ben-Bassat H, Goldblum N, Mitrani S, Goldblum T, Yoffey JM, Cohen MM, Bentwich Z, Ramot B, Klein E, Klein G. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int J Cancer 19:27–33. doi: 10.1002/ijc.2910190105. [DOI] [PubMed] [Google Scholar]
- 24.Sabbah S, Jagne YJ, Zuo J, de Silva T, Ahasan MM, Brander C, Rowland-Jones S, Flanagan KL, Hislop AD. 2012. T-cell immunity to Kaposi sarcoma-associated herpesvirus: recognition of primary effusion lymphoma by LANA-specific CD4+ T cells. Blood 119:2083–2092. doi: 10.1182/blood-2011-07-366476. [DOI] [PubMed] [Google Scholar]
- 25.Rabin H, Hopkins RF III, Ruscetti FW, Neubauer RH, Brown RL, Kawakami TG. 1981. Spontaneous release of a factor with properties of T cell growth factor from a continuous line of primate tumor T cells. J Immunol 127:1852–1856. [PubMed] [Google Scholar]
- 26.Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, Ziegler A. 1978. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens—new tools for genetic analysis. Cell 14:9–20. doi: 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- 27.Young LS, Lau R, Rowe M, Niedobitek G, Packham G, Shanahan F, Rowe DT, Greenspan D, Greenspan JS, Rickinson AB, Farrell PJ. 1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J Virol 65:2868–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Qian L, Chen C, Shi M, Yu M, Hu M, Song L, Shen B, Guo N. 2009. Down-regulation of MHC class II expression through inhibition of CIITA transcription by lytic transactivator Zta during Epstein-Barr virus reactivation. J Immunol 182:1799–1809. doi: 10.4049/jimmunol.0802686. [DOI] [PubMed] [Google Scholar]
- 29.Zuo J, Hislop AD, Leung CS, Sabbah S, Rowe M. 2013. Kaposi's sarcoma-associated herpesvirus-encoded viral IRF3 modulates major histocompatibility complex class II (MHC-II) antigen presentation through MHC-II transactivator-dependent and -independent mechanisms: implications for oncogenesis. J Virol 87:5340–5350. doi: 10.1128/JVI.00250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 5:1039–1043. doi: 10.1038/12478. [DOI] [PubMed] [Google Scholar]
- 31.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coscoy L, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci U S A 97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J Virol 74:5300–5309. doi: 10.1128/JVI.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishido S, Goto E, Matsuki Y, Ohmura-Hoshino M. 2009. E3 ubiquitin ligases for MHC molecules. Curr Opin Immunol 21:78–83. doi: 10.1016/j.coi.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Haque M, Ueda K, Nakano K, Hirata Y, Parravicini C, Corbellino M, Yamanishi K. 2001. Major histocompatibility complex class I molecules are down-regulated at the cell surface by the K5 protein encoded by Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. J Gen Virol 82:1175–1180. doi: 10.1099/0022-1317-82-5-1175. [DOI] [PubMed] [Google Scholar]
- 36.Lehner PJ, Hoer S, Dodd R, Duncan LM. 2005. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol Rev 207:112–125. doi: 10.1111/j.0105-2896.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 37.Brulois K, Toth Z, Wong LY, Feng P, Gao SJ, Ensser A, Jung JU. 2014. Kaposi's sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J Virol 88:9335–9349. doi: 10.1128/JVI.00873-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartee E, McCormack A, Fruh K. 2006. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog 2:e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timms RT, Duncan LM, Tchasovnikarova IA, Antrobus R, Smith DL, Dougan G, Weekes MP, Lehner PJ. 2013. Haploid genetic screens identify an essential role for PLP2 in the downregulation of novel plasma membrane targets by viral E3 ubiquitin ligases. PLoS Pathog 9:e1003772. doi: 10.1371/journal.ppat.1003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez DJ, Gumperz JE, Ganem D. 2005. Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest 115:1369–1378. doi: 10.1172/JCI200524041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, Wills M, Lehner PJ. 2008. Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc Natl Acad Sci U S A 105:1656–1661. doi: 10.1073/pnas.0707883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, Means R, Lang S, Jung JU. 2007. Downregulation of gamma interferon receptor 1 by Kaposi's sarcoma-associated herpesvirus K3 and K5. J Virol 81:2117–2127. doi: 10.1128/JVI.01961-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. 2004. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol 78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, Hotta H, Ishido S. 2006. A novel family of membrane-bound E3 ubiquitin ligases. J Biochem 140:147–154. doi: 10.1093/jb/mvj160. [DOI] [PubMed] [Google Scholar]
- 45.Ohmura-Hoshino M, Matsuki Y, Aoki M, Goto E, Mito M, Uematsu M, Kakiuchi T, Hotta H, Ishido S. 2006. Inhibition of MHC class II expression and immune responses by c-MIR. J Immunol 177:341–354. doi: 10.4049/jimmunol.177.1.341. [DOI] [PubMed] [Google Scholar]
- 46.Hoer S, Smith L, Lehner PJ. 2007. MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett 581:45–51. doi: 10.1016/j.febslet.2006.11.075. [DOI] [PubMed] [Google Scholar]
- 47.Nathan JA, Lehner PJ. 2009. The trafficking and regulation of membrane receptors by the RING-CH ubiquitin E3 ligases. Exp Cell Res 315:1593–1600. doi: 10.1016/j.yexcr.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 48.Fogg MH, Carville A, Cameron J, Quink C, Wang F. 2005. Reduced prevalence of Epstein-Barr virus-related lymphocryptovirus infection in sera from a new world primate. J Virol 79:10069–10072. doi: 10.1128/JVI.79.15.10069-10072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang F. 2013. Nonhuman primate models for Epstein-Barr virus infection. Curr Opin Virol 3:233–237. doi: 10.1016/j.coviro.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.