

ABSTRACT
Adeno-associated virus 2 (AAV2) and adenovirus 5 (Ad5) are promising gene therapy vectors. Both display liver tropism and are currently thought to enter hepatocytes in vivo through cell surface heparan sulfate proteoglycans (HSPGs). To test directly this hypothesis, we created mice that lack Ext1, an enzyme required for heparan sulfate biosynthesis, in hepatocytes. Ext1HEP mutant mice exhibit an 8-fold reduction of heparan sulfate in primary hepatocytes and a 5-fold reduction of heparan sulfate in whole liver tissue. Conditional hepatocyte Ext1 gene deletion greatly reduced AAV2 liver transduction following intravenous injection. Ad5 transduction requires blood coagulation factor X (FX); FX binds to the Ad5 capsid hexon protein and bridges the virus to HSPGs on the cell surface. Ad5.FX transduction was abrogated in primary hepatocytes from Ext1HEP mice. However, in contrast to the case with AAV2, Ad5 transduction was not significantly reduced in the livers of Ext1HEP mice. FX remained essential for Ad5 transduction in vivo in Ext1HEP mice. We conclude that while AAV2 requires HSPGs for entry into mouse hepatocytes, HSPGs are dispensable for Ad5 hepatocyte transduction in vivo. This study reopens the question of how adenovirus enters cells in vivo.
IMPORTANCE Our understanding of how viruses enter cells, and how they can be used as therapeutic vectors to manage disease, begins with identification of the cell surface receptors to which viruses bind and which mediate viral entry. Both adeno-associated virus 2 and adenovirus 5 are currently thought to enter hepatocytes in vivo through heparan sulfate proteoglycans (HSPGs). However, direct evidence for these conclusions is lacking. Experiments presented herein, in which hepatic heparan sulfate synthesis was genetically abolished, demonstrated that HSPGs are not likely to function as hepatocyte Ad5 receptors in vivo. The data also demonstrate that HSPGs are required for hepatocyte transduction by AAV2. These results reopen the question of the identity of the Ad5 receptor in vivo and emphasize the necessity of demonstrating the nature of the receptor by genetic means, both for understanding Ad5 entry into cells in vivo and for optimization of Ad5 vectors as therapeutic agents.
INTRODUCTION
A better understanding of how viral vectors enter cells in vivo is critical to improve their therapeutic use. Adeno-associated virus 2 (AAV2) and adenovirus 5 (Ad5) vectors have shown promise in clinical trials for treatment of a wide variety of diseases (1, 2). Both vectors, when injected intravenously into mice, exhibit transgene expression in liver (3–5). Heparan sulfate proteoglycans (HSPGs) are the primary receptors currently thought to facilitate AAV2 and Ad5 entry into hepatocytes (6–8).
HSPGs are present both on the cell surface and in the extracellular matrix (9, 10). They consist of a protein core posttranslationally modified to contain heparan sulfate (HS) chains (11). HS biosynthesis occurs by polymerization of alternating glucuronic acid and N-acetylglucosamine residues (12–14), catalyzed by an enzyme complex composed of EXT1 and EXT2 (15). EXT1 and EXT2 are essential molecules required for HS synthesis; cells lacking either molecule do not synthesize HS (16, 49).
AAV2 binds directly to cell surface HSPGs via an HS-binding motif on the virus capsid (3, 17, 18). AAV capsid modifications that alter the cluster of positive amino acids that constitute the HS binding motif abrogate liver transduction (3, 19, 20), suggesting that the ability of the capsid to bind to HS is critical for AAV2 liver transduction in vivo. In contrast, Ad5 binding to HSPGs requires the presence of blood coagulation factor X (FX), which binds to the Ad5 hexon when the virus comes in contact with blood (7, 21–23). The interaction of Ad.FX and HS is mediated by electrostatic interactions between the heparin binding exosite of the FX serine protease domain and the sulfate groups of HS (6, 23–25). FX is required for Ad5 transduction in vivo in wild-type mice. In the absence of FX, or when viruses with mutant hexon proteins unable to bind FX are used, Ad5 liver transduction is essentially completely abrogated (7, 21–23, 26, 27).
Despite extensive evidence for a role of HS in AAV2 and Ad5 transduction in cultured cells, direct evidence for a role of HS-mediated hepatocyte transduction in vivo has not been described. In this study, we employed an in vivo genetic model to analyze directly the role of hepatocyte HS for AAV2 and Ad5 gene transfer to mouse liver.
MATERIALS AND METHODS
Mice.
All animal experiments were conducted in accordance with the guidelines of the UCLA and UCSD animal care committees. To create mice that lack heparan sulfate in hepatocytes, we crossed conditional Ext1flox/flox (Ext1f/f) mice (28) with AlbCre mice (29) (Jackson Laboratories) to create Ext1f/f;AlbCre+ (Ext1HEP) mice. All lines were on a C57BL/6J background. Genotyping was performed by PCR with tail DNA for cre (cre: forward, 5′-GTC CAA TTT ACT GAC CGT ACA CC-3′, and reverse, 5′-CGC TAT TTT CCA TGA GTG AAC GA-3′) and for the Ext1f/f allele (Ext1f/f: forward, 5′-GGA GTG TGG ATG AGT TGA AG-3′, and reverse, 5′-CAA CAC TTT CAG CTC CAG TC-3′) (Fig. 1a, 470 bp, Ext1f/f control). For verification of genetic deletion of the Ext1-exon1 in hepatocytes following Cre recombination, a second reverse primer was designed (5′-CGT CAC AAA TAC CCT TTA GTA-3′), resulting in a de novo product if recombination occurred (Fig. 1a, 692 bp, Ext1HEP mutant). Mice used for experiments were at least at 8 weeks of age and mixed sex. The total amount of heparan sulfate in livers was determined by glycan reductive isotope labeling-liquid chromatography-mass spectrometry (GRIL-LC/MS) as described previously (30). To determine heparan sulfate content in primary hepatocytes, cells were isolated from three Ext1f/f control and three Ext1HEP mutant mice as described below and pooled prior to GRIL-LC/MS analysis. Virus was injected intravenously via the tail vein with 1 × 1010 to 1.5 × 1011 particles/mouse of viral vectors as indicated for the respective experiments, and mice were euthanized 3 days (Ad) or 8 weeks (AAV) postinjection.
FIG 1.
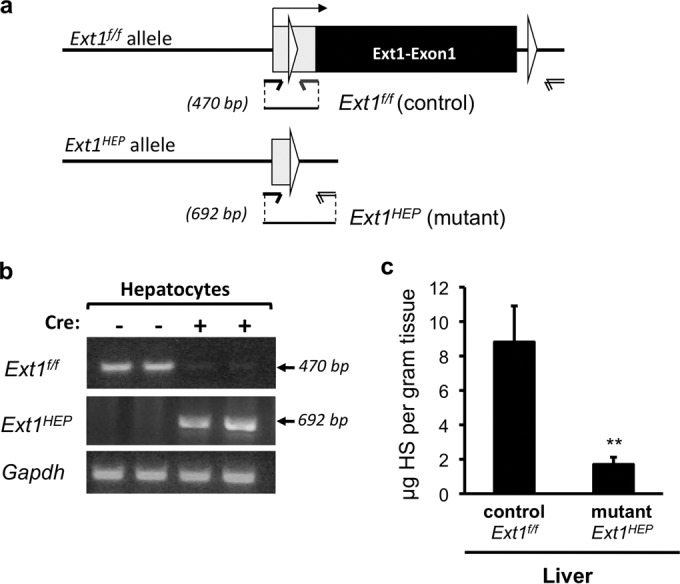
Ext1HEP mutant mice lack hepatic heparan sulfate. (a) Schematic representation indicating conditional excision of exon 1 of the Ext1 gene via Cre recombinase cleavage at loxP sites (white triangles). Upon Cre recombination, the Ext1 gene is inactivated in hepatocytes (Ext1f/f;AlbCre+ [Ext1HEP]). (b) PCR analysis of Ext1 deletion in primary hepatocytes from Ext1f/f control and Ext1HEP mutant mice, using the primer pairs indicated in panel a. (c) Quantitative glycosaminoglycan analysis in liver tissue. Heparan sulfate (HS) content was analyzed by glycan reductive isotope labeling-LC/MS. The graph illustrates data from 3 control and 3 mutant mice. Values are means ± SD. **, P < 0.01. Comparable results were obtained with two independent preparations of liver tissue.
Virus vectors.
AdLuc is a nonreplicating E1 and E3 deletion adenovirus serotype 5-based vector in which the cytomegalovirus (CMV) ubiquitous promoter drives firefly luciferase (Luc) expression (31). AdTLY477A is an adenovirus vector in which the coxsackie and adenovirus receptor (CAR) is ablated and which has a tyrosine-to-alanine point mutation at position 477 (Y477A) in the DE loop of the knob domain of the viral fiber (32). AdTLY477A contains green fluorescent protein (GFP) and firefly Luc expression cassettes, both driven from CMV promoters, in the E1 coding region (32). AdTL is identical to AdY477A except for the fiber mutation (32). AdGFP contains a CMV promoter-driven GFP transgene. AdTEA is a mutated version of AdGFP that is unable to bind FX due to a threonine-to-alanine substitution at position 425 of the hexon protein (26). Adenovirus vectors were propagated in HEK293A cells (Invitrogen) and purified by two sequential cesium chloride gradient centrifugation steps, followed by dialysis against 3% sucrose buffer. The virus particle (VP) titer (particles per milliliter) was determined by measuring absorbance at 260 nm (33). Virus infectious-unit titers (infectious units per milliliter) were determined with the Adeno-X Rapid Titer kit (Clontech, Mountain View, CA) and were typically 10- to 50-fold below particle titers. The AAV2 vector containing a CMV promoter-driven GFP transgene was obtained as a gift from Virovek Inc. (Hayward, CA).
Analysis of vector transduction.
Primary hepatocytes and Chinese hamster ovary cells (CHO K1, ATCC CCL61, and pgsD-677) (34) were cultured as described previously (25, 35, 36). Virus vector transductions of cells were performed in 24-well plates seeded with 1 × 105 cells per well. Virus particles (1 × 108/well) were diluted in serum-free Opti-MEM (Invitrogen) supplemented with 8 μg/ml of human FX (hFX) (Hematologic Technologies, Essex Junction, VT) where indicated and incubated with the cells at 37°C for 60 min. Cells were processed for analysis 2 days following transduction.
Luciferase assays, GFP immunoblotting, and GFP immunohistochemistry were performed as described previously (37). To directly visualize GFP fluorescence in frozen sections, liver tissue was prefixed overnight in 4% paraformaldehyde–phosphate-buffered saline (PBS) followed by 4 h each in 10%, 20%, and 30% sucrose at 4°C. Liver tissue was then embedded in optimal-cutting-temperature (OCT) compound and snap-frozen. Frozen sections (5 μm) were rehydrated with PBS, and 4′,6-diamidino-2-phenylindole (DAPI) was added to visualize nuclei. Bright-field and fluorescent images were captured using an inverted microscope (Nikon; Eclipse 2000 TE) and analyzed using NIS Elements imaging software.
Ad5 genome copies in liver tissues were analyzed by TaqMan quantitative PCR (qPCR) (absolute quantification), with murine Oct-4 as a control, using primers hybridizing to the L2 gene region in the Ad5 genome as previously described (38). GFP mRNA expression and AAV genomes in mouse liver were quantified by SYBR-green qPCR (threshold cycle [2−ΔΔCT] method) with murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control, using primers hybridizing within the GFP coding region and CMV promoter, respectively. Primer sequences were as follows: GFP fwd, 5′-GCA CGA CTT CTT CAA GTC CGC CAT GCC-3′; GFP rvs, 5′-GCG GAT CTT GAA GTT CAC CTT GAT GCC-3′; GAPDH fwd, 5′-CTG CGG AAA TGG TGT GAT CTT CCC CAA GGG-3′; GAPDH rvs, 5′-AGG GAG CTC CAT TCA TGT GCT AAA CAG GCC-3′; CMV fwd, 5′-ACG CCA ATA GGG ACT TTC CA-3′; and CMV rvs, 5′-TAG GGG GCG TAC TTG GCA TA-3′. PCRs were performed on ABI 7500 real-time thermocycler instrumentation using SDS software v.1.3 (Applied Biosystems).
Statistical analysis.
Data are expressed as means ± standard deviations (SD). Significance between groups was determined using an unpaired Student (two-tailed) t test. All P values of <0.05 were considered significant.
RESULTS
Conditional deletion of the enzyme EXT1 results in a reduction of heparan sulfate in hepatocytes.
To create mice with a conditional deletion of the Ext1 gene, Ext1f/f mice (28) were crossed with AlbCre mice, which express Cre from the albumin promoter (29). Conditional homozygous Ext1f/f mice expressing AlbCre (Ext1f/f;AlbCre+ mice, subsequently referred to as Ext1HEP mice) are viable and fertile; moreover, their livers showed no distinguishable difference from those of Ext1f/f control littermates by hematoxylin and eosin (H&E), F4/80, and CD31 staining (data not shown). To determine whether genetic recombination had occurred in hepatocytes of Ext1HEP mutant mice, two sets of primers were used: one that detects the floxed control allele and one that results in a product only when the floxed exon1 of the Ext1 allele is excised (Fig. 1a). PCR analysis of DNA from isolated primary hepatocytes showed robust signals for inactivated Ext1 in primary hepatocytes from mutant Ext1HEP mice expressing the Cre allele, indicating that Cre recombination and Ext1 deletion had occurred (Fig. 1b). These PCR products were not detected in hepatocytes from Cre-negative Ext1f/f control mice (Fig. 1b). Conversely, the signal for wild-type floxed Ext1 was essentially absent in hepatocytes from Ext1HEP mutant mice, indicating that Cre-driven recombination and Ext1 deletion occurred in the majority of hepatocytes from Cre-expressing Extf/f mice (Fig. 1b).
To confirm that the lack of the Ext1 alleles indeed translates into a reduction of heparan sulfate in vivo, we assessed overall disaccharide composition of glycosaminoglycan chains, both in cultured primary hepatocytes and in whole liver tissue. Liver tissue from mutant Ext1HEP mice displayed an ∼5-fold reduction of heparan sulfate compared to the heparan sulfate content of liver tissue from Ext1f/f control mice (Fig. 1c). Heparan sulfate in primary hepatocytes from Ext1HEP mice was reduced ∼8-fold compared to HS in cultured hepatocytes from Ext1f/f control mice (0.32 μg of HS/mg of protein versus 0.04 μg of HS/mg of protein).
Both adeno-associated virus 2 transduction and adenovirus 5 transduction are reduced in cultured hepatocytes from Ext1HEP mutant mice.
Chinese hamster ovary (CHO) cells genetically modified to lack Ext1 (CHO pgsD-677 cells) showed greatly reduced GFP transgene expression compared to that of wild-type CHO K1 cells 2 days after transduction with an AAV vector encoding GFP (Fig. 2a). To compare these results with those for hepatocytes from Ext1HEP mice, we isolated and cultured primary hepatocytes from Ext1f/f control and Ext1HEP mutant mice. AAV-GFP transduction resulted in GFP and mRNA expression in primary hepatocytes from Ext1f/f control mice (Fig. 2b). In contrast, no GFP was detectable in cell extracts from Ext1HEP hepatocytes, and GFP mRNA expression was ∼20-fold reduced (Fig. 2b).
FIG 2.

AAV2 and Ad5 transduction in Ext1-deficient cells in culture. (a) Analysis of GFP transgene expression following AAV transduction of control CHO K1 or EXT1-deficient CHO pgsD-677 cells. Cells were transduced with an AAV2 vector encoding GFP (AAV-GFP) for 1 h in reduced serum-medium. GFP expression was analyzed in cell extracts by immunoblotting (IB) 48 h after transduction (left side). Total RNA was analyzed for GFP mRNA expression by quantitative real-time RT-PCR. GFP mRNA levels are normalized to GAPDH mRNA levels in the same samples (right side). (b) Analysis of GFP protein and RNA expression in primary hepatocyte cultures isolated from Ext1f/f control or Ext1HEP mutant mice 48 h following transduction with AAV-GFP as in panel a. GFP mRNA levels are normalized to mouse GAPDH mRNA levels in the same samples. (c) Transduction of wild-type CHO K1 cells and EXT1-deficient mutant CHO pgsD-677 cells with AdTL, an Ad5 vector carrying a GFP transgene expression cassette and a luciferase transgene expression cassette. The cells were transduced in serum-reduced medium or in serum-reduced medium supplemented with 8 μg/ml of FX. Forty-eight hours posttransduction, cell lysates were analyzed for luciferase transgene expression. (d) Primary cultured hepatocytes from Ext1f/f control or Ext1HEP mutant mice were transduced with AdTLY477A, an Ad5 vector in which CAR binding is ablated, carrying the same GFP and luciferase transgene expression cassettes as AdTL, in the presence or absence of FX. Forty-eight hours following transduction, the cells were analyzed for GFP (by immunofluorescence) and for luciferase transgene expression. RLU, relative light units. Values are means ± SD relative to the value for the control (wild-type CHO K1 cells or control Ext1f/f hepatocytes). *, P < 0.05; **, P < 0.01.
Unlike for AAV, Ad5 transduction of cells via HSPGs requires FX (6). Transduction of CHO K1 cells by AdTL, an adenovirus serotype 5 vector encoding cytomegalovirus (CMV) promoter-driven GFP and firefly luciferase transgene cassettes, was increased in the presence of FX (Fig. 2c). However, as observed for AAV, AdTL.FX transduction was abrogated in mutant CHO pgsD-677 cells lacking heparan sulfate (Fig. 2c).
In addition to FX-HSPG-mediated cell entry, Ad5 vectors can also use the coxsackie and adenovirus receptor (CAR) to enter cells in culture (5, 39, 40). Since cell entry through CAR can mask FX-mediated transduction in cultured hepatocytes (data not shown), we characterized Ad5 transduction of primary hepatocytes from Ext1f/f control and Ext1HEP mutant mice by an Ad5 vector, AdTLY477A, in which CAR binding is ablated (32). AdTLY477A transduction of hepatocytes from Ext1f/f control cells, in the presence of FX, led to greatly enhanced GFP transgene expression 2 days after transduction and to an ∼20-fold increase in luciferase activity (Fig. 2d). In contrast, FX could not enhance AdTLY477A-mediated GFP transgene expression or luciferase activity in mutant Ext1HEP hepatocytes (Fig. 2d). These results suggest that both Ad.FX and AAV transduction of primary hepatocytes in culture require HS.
Hepatocyte heparan sulfate is required for AAV2 transduction of mouse liver in vivo.
To directly determine the role of hepatocyte heparan sulfate for AAV2 transduction of mouse liver in vivo, Ext1f/f control mice and Ext1HEP mutant mice were injected intravenously with 1 × 1011 AAV-GFP VP/mouse. Immunohistochemistry for GFP showed that the number of GFP-expressing hepatocytes was significantly lower in livers of Ext1HEP mutant mice than in the livers of Ext1f/f control mice (Fig. 3a). Consistent with this finding, immunoblot analysis of liver extracts showed reduced GFP content in livers of Ext1HEP mutant mice (Fig. 3b, Cre+) compared to that in livers of Ext1f/f control mice (Fig. 3b, Cre−) and an ∼5-fold reduction of GFP mRNA expression (Fig. 3c). Relative AAV vector DNA was also significantly lower in livers of Ext1HEP mutant mice, suggesting that the reduction in GFP transgene expression is due to reduced viral transduction (Fig. 3d). Overall, these results show that hepatocyte heparan sulfate is required for efficient AAV transduction of mouse liver in vivo.
FIG 3.

AAV transduction in Ext1f/f control and Ext1HEP mutant mice. Shown is an analysis of liver transduction 8 weeks after intravenous injection of 1 × 1011 VP/mouse of an AAV2 vector encoding green fluorescent protein (AAV-GFP) in Ext1f/f control or Ext1HEP mutant mice. (a) GFP immunohistochemistry in livers from Ext1f/f control or Ext1HEP mutant mice. GFP is visible as brown staining in individual hepatocytes (red arrows). The graph depicts quantification of GFP-positive cells in liver sections. (b) GFP content in liver extracts from Ext1f/f control (Ext1f/f Cre−) or Ext1HEP mutant (Ext1f/f Cre+) mice. Liver extracts were assayed for GFP and GAPDH by immunoblotting. “+” indicates the presence of the AlbCre transgene, leading to the targeted deletion of the Ext1 gene to create Ext1HEP mutant mice; “−” indicates the absence of the AlbCre transgene, resulting in Ext1f/f mice with an intact Ext1 gene. (c) Total liver RNA was analyzed for GFP mRNA expression by quantitative real-time RT-PCR and normalized to the mouse liver GAPDH mRNA of the same samples. (d) AAV vector genomes in livers from AAV-injected Ext1f/f control or Ext1HEP mutant mice were quantified after DNA extraction by quantitative real-time PCR, using primers within the CMV promoter of the transgene expression cassette, and normalized to liver GAPDH DNA values of the same sample. Values are means ± SD. Differences in number of GFP-expressing cells per field, GFP mRNA expression, and vector genomes between control and mutant mice were compared by Student's t test. *, P < 0.05; n = 3. Scale bar represents 50 μm.
Hepatocyte heparan sulfate is not required for adenovirus transduction of mouse liver in vivo.
To evaluate the role of heparan sulfate in adenovirus hepatocyte transduction, we examined the ability of Ad5 to transduce liver in Ext1HEP mice. To examine virus titers across a broad dose range, Ext1f/f control and Ext1HEP mutant mice were injected intravenously with an Ad5 vector encoding a GFP transgene (AdGFP) at 1 × 1010, 3 × 1010, or 1 × 1011 VP/mouse. Transgene expression was assessed 3 days later. As expected, both the number of cells demonstrating GFP staining in liver sections (Fig. 4a) and the GFP content in liver extracts (Fig. 4b) increased with increasing viral dose in livers from AdGFP-injected Ext1f/f control (Cre−) mice. However, equivalent GFP transgene expression was observed in livers from Ext1HEP mutant (Cre+) mice (Fig. 4a and b).
FIG 4.

Adenovirus transduction in Ext1f/f control and Ext1HEP mutant mice. (a) GFP transgene expression in livers of Ext1f/f control or mutant Ext1HEP mice 3 days after intravenous injection with increasing doses (1 × 1010, 3 × 1010, or 1 × 1011VP/mouse) of an Ad5 vector expressing GFP. GFP content was visualized by immunofluorescence staining of paraffin-embedded liver sections. Corresponding fields stained with DAPI are displayed to visualize nuclei. (b) Liver extracts were analyzed by immunoblotting for GFP and GAPDH. Liver extracts from two representative mice per group are shown. (c and d) Liver transduction using a low dose (2 × 1010 VP/mouse) and a high dose (8 × 1010 VP/mouse) of AdLuc, an Ad5 vector encoding firefly luciferase. Luciferase transgene expression (c) and adenovirus vector genomes (d) in Ext1f/f control and Ext1HEP mutant mice were analyzed 3 days after intravenous injection. Ad5 genomes were detected in liver by quantitative PCR. Values are means ± SD. Differences in either transgene expression or vector genomes between control and mutant mice were compared by Student's t test. ns, not significant (P > 0.05); n = 4 mice per group.
To analyze transgene expression in a more quantifiable way, we injected mice with 2 × 1010 or 8 × 1010 VP/mouse of AdLuc, an Ad5 vector expressing firefly luciferase. Luciferase activity in mouse livers was not significantly different at either dose in mutant Ext1HEP and Ext1f/f control mice (Fig. 4c). As observed with luciferase transgene expression, adenovirus genomes in liver increased with increasing virus dose, but no detectable quantifiable difference in viral genomes was observed between control Ext1f/f mice and mutant Ext1HEP mice (Fig. 4d).
Although HS is significantly reduced in hepatocytes of Ext1HEP mice, other cell types in the liver may still express HS. It is possible that Ad5 may transduce other liver cell types in Ext1HEP mice. To determine whether the cell types that express the Ad5 transgene differ in Ext1f/f and Ext1HEP mouse livers, liver sections were stained by immunohistochemistry after injection of 8 × 1010 AdGFP VP/mouse (Fig. 5). GFP staining was visible throughout the cytoplasm of transduced hepatocytes in liver sections from both mutant Ext1HEP and control Ext1f/f mice injected with AdGFP (Fig. 5). These results suggest that Ad5 liver transductions in vivo are similar in Ext1f/f control and Ext1HEP mutant mice.
FIG 5.

Histological analysis of liver sections from adenovirus-transduced Ext1f/f control and Ext1HEP mutant mice. Immunohistochemistry results for GFP in liver sections from Ext1f/f control and Ext1HEP mutant mice 3 days following intravenous injection with 8 × 1010 AdGFP VP/mouse are shown. GFP in cells is visible as dark-brown staining throughout the cytoplasm. Red arrows indicate stained (GFP-positive) and unstained (GFP-negative) cells side by side. Hematoxylin counterstaining was used to visualize nuclei in blue (black arrows). White arrows indicate sinusoids and bile ducts. Scale bar indicates 50 μm.
CAR cannot serve as an alternative adenovirus receptor for mice deficient in hepatocyte heparan sulfate.
To exclude the possibility that CAR might serve as an alternative receptor for Ad5 entry into hepatocytes in the absence of HS, we examined the ability of an Ad5 vector in which CAR binding is ablated, AdTLY477A (32), to transduce livers of Ext1f/f and Ext1HEP mice. Mice of both genotypes were injected with 5 × 1010 particles/mouse of AdTLY477A or with the parental AdTL control adenovirus from which AdTLY477A was derived (32). No significant difference in luciferase activity was observed in livers of Ext1f/f control mice and Ext1HEP mutant mice 3 days after either AdTL or AdTLY477A vector administration (Fig. 6a). In addition, vector genome content in the livers of control Ext1f/f mice and mutant Ext1HEP mice were indistinguishable (Fig. 6b). These data suggest that CAR does not serve as a receptor for hepatocyte adenovirus transduction in vivo, regardless of whether HS is expressed by hepatocytes.
FIG 6.

The roles of CAR binding and FX binding in Ad5 liver transduction in Ext1f/f control and Ext1HEP mutant mice. Shown is luciferase transgene expression (a) and viral genomes (b) in the livers of Ext1f/f control and Ext1HEP mutant mice 3 days after intravenous injection of 5 × 1010 VP/mouse of AdTLY477A, a luciferase-GFP-expressing Ad5 vector in which CAR binding is ablated, or the corresponding unmodified AdTL control vector. (c) Histological assessment of AdGFP- and AdTEA-mediated GFP expression in frozen sections of Ext1f/f control and Ext1HEP mutant mouse livers 3 days after intravenous injection with 1.5 × 1011 viral particles/mouse. AdTEA is a GFP-expressing Ad5 vector in which FX binding is ablated. DAPI-stained fields are displayed to visualize nuclei. (d) Immunoblot analysis of GFP expression in the livers of Ext1f/f and Ext1HEP mice injected with AdGFP or AdTEA adenovirus vectors. GFP expression in liver extracts from two representative mice per group is shown. (e) Viral genome content in liver tissue of Ext1f/f and Ext1HEP mice injected with AdGFP or AdTEA. DNA was extracted from the same livers as in panels c and d, and viral genomes were quantified using quantitative real-time PCR. Individual groups were compared by Student's t test. Values are displayed as means ± SD. **, P < 0.01; ***, P < 0.001. n = 4.
Blood coagulation factor X remains essential for efficient adenovirus liver transduction in Ext1HEP mice.
To determine whether FX is required for Ad5 transduction of Ext1-deficient hepatocytes in vivo, we used AdTEA, a GFP-expressing mutant adenovirus in which FX binding is ablated (26). Ext1f/f and Ext1HEP mice were injected with 1.5 × 1011 VP/mouse of AdTEA, or with AdGFP, and GFP expression was analyzed 3 days later. Analysis of GFP fluorescence in frozen liver sections (Fig. 6c) and immunoblotting for GFP in liver extracts (Fig. 6d) demonstrated that AdTEA administration did not lead to detectable GFP expression either in livers of Ext1f/f mice that express hepatocyte HS or in livers of hepatocyte HS-deficient Ext1HEP mice. In addition, Ad5 genomes in mouse liver were greatly reduced in AdTEA-injected mice of both genotypes compared with AdGFP-injected animals (Fig. 6e). In summary, eliminating the ability of adenovirus to bind to FX reduced liver transduction equally in Ext1f/f and Ext1HEP mice, demonstrating that FX is required for adenovirus transduction both in the presence and in the absence of hepatocyte heparan sulfate.
DISCUSSION
Effective targeting to the desired cells or tissues remains among the major hurdles for virus-mediated gene therapy. Resolution of viral targeting and transduction efficacy questions requires more complete knowledge of the respective receptors and transduction mechanisms involved in viral entry into cells in vivo.
Adeno-associated virus 2 is currently thought to enter hepatocytes in vivo through HSPGs. Adenovirus 5 was initially thought to enter hepatocytes in vivo through CAR. However, experiments that involved mutation of the CAR binding site on the Ad5 fiber demonstrated that this hypothesis was incorrect (32, 41). Subsequent cell culture studies and in vivo experiments led to the currently accepted paradigm that Ad5 hepatocyte transduction in vivo requires Ad-HSPG bridging by FX (6, 7, 23, 42).
To determine directly the role of hepatocyte HSPG in AAV2 and Ad5 liver transduction in vivo, we used a mouse model of hepatocyte-specific HS deficiency in which EXT1, a key enzyme in HS biosynthesis, is conditionally eliminated. HS expression was necessary for AAV2 liver transduction, suggesting that HSPGs function as the receptor for AAV2 uptake in vivo. In contrast to the case with AAV2, Ad5 liver transductions in Ext1HEP mutant mice and Ext1f/f mice were similar. These findings demonstrate that adenovirus, unlike AAV, does not require hepatocyte HS for transduction of mouse liver in vivo. The most likely interpretation of these data is that hepatocyte HSPGs do not serve as the primary receptors for Ad5 liver transduction in vivo.
To exclude the possibility that adenovirus uses CAR to enter hepatocytes in the absence of heparan sulfate, we investigated the ability of the vector AdY477A, in which CAR binding is ablated, to transduce Ext1HEP and Ext1f/f mice. AdY477A elicited equivalent liver transduction of both Ext1f/f and mutant Ext1HEP mice, confirming that CAR is not used for hepatocyte entry in vivo. AdTEA—a virus that is unable to bind FX—could not transduce livers of either Ext1f/f or Ext1HEP mutant mice, demonstrating that unlike heparan sulfate, FX is required for hepatocyte transduction.
A substantial amount of experimental evidence indicates that Ad.FX can bind directly to heparin and to HS (6, 23, 24, 43). Therefore, not surprisingly, heparin injected intravenously into mice immediately prior to Ad5 injection blocks liver uptake (6). However, heparin inhibition of Ad5 liver transduction in vivo is likely to be the result of heparin binding to and sequestering of Ad.FX in the blood and, consequently, preventing Ad.FX from binding to its physiological receptor. In another approach, heparinase I, which enzymatically removes HS from the cell surface, administered intravenously into mice prior to Ad5 injection led to ∼5-fold reduced virus transduction relative to that in control mice (42). However, it is likely that systemic in vivo enzymatic treatment could release heparan-containing fragments that compete with FX for binding to adenovirus.
In another approach, Waddington et al. (23) demonstrated that preincubation of FX with NAPc2 or Ixolaris, naturally occurring anticoagulants that bind to and mask the FX heparin-binding exosite, prior to injection into warfarin-treated mice led to a reduction in FX liver rescue of Ad5 transduction. While these observations are consistent with the hypothesis that HSPGs may play a role in Ad.FX hepatocyte transduction in vivo, NAPc2 and Ixolaris have additional effects on coagulation that are distinct from blocking the heparin binding exosite of FX (44–46).
Xu et al. (47) suggest that FX is necessary to shield Ad5 from natural antibody and complement inactivation but may not be required for Ad5 liver transduction in vivo. In either immunodeficient Rag1−/− mice or mice deficient in complement component C1q or C4, an Ad5 viral mutant in which FX binding is ablated showed only a modest reduction in transduction. While suggesting that FX is not required for Ad5 transduction under certain conditions, and therefore may not be the molecule that bridges the virus to hepatocellular receptors, these studies do not address the identity of the Ad5 hepatocyte receptor.
HS content in isolated hepatocytes from Ext1HEP mutant mice was reduced by ∼90%. It could be argued that this residual low level of HS is nonetheless sufficient to promote Ad.FX binding and entry in vivo. However, Ad.FX transduction was abrogated in primary Ext1HEP hepatocytes in culture, suggesting that the residual HS on these cells cannot promote Ad.FX transduction.
If HSPGs are not the principal physiological receptor for Ad.FX liver transduction, we are faced with the question of how Ad.FX enters hepatocytes in vivo. Hepatocytes in Ext1HEP mutant mice still express dermatan sulfate (DS) and chondroitin sulfate (CS), the other two major types of sulfated glycans. The lack of Ext1 leads to an increase in CS expression (48, 49), which could potentially serve as the receptor for Ad.FX. However, mutant Ext1−/− CHO pgsD-677 cells, although lacking heparan sulfate, contain three times the amount of CS and DS present in wild-type CHO K1 cells (34), and FX cannot increase Ad5 transduction of Ext1−/− CHO cells (6, 25). These data suggest that Ad.FX is unlikely to use CS or DS for cell entry.
In addition to hepatocytes, endothelial cells that surround the liver sinusoids also express proteoglycans. Both hepatocytes and endothelial cells express HSPGs on their cell surfaces and secrete heparan sulfate. It is possible that HS expressed by endothelial cells contributes to Ad5 liver transduction. However, mice that express undersulfated HS in endothelial cells, due to a targeted deletion of the Ndst1 gene (50), are still fully competent for Ad5 transduction of liver in vivo (data not shown).
We conclude that Ad5 vectors, after intravenous injection, are able to transduce mouse livers with substantially reduced HS content. Our data are consistent with the involvement of other receptors, and/or blood factors, and suggest that further investigation is needed to fully understand Ad5 liver transduction.
ACKNOWLEDGMENTS
We thank Jose S. Gil (UCLA) for help with the real-time PCR protocols for viral genome copies and Ding Xu (UCSD) for help with heparan sulfate analysis. We also thank Sotirios Tetradis (UCLA) for helpful comments on the manuscript and experimental design.
We declare no conflict of interest.
Funding Statement
This work was supported by NIH grants P50 CA086306-08 (H.R.H.) and GM33063 and HL107150 (J.D.E.). A.K.Z. is the recipient of an American Society of Hematology (ASH) Scholar Award. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Mingozzi F, High KA. 2011. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 2.Nienhuis AW. 2013. Development of gene therapy for blood disorders: an update. Blood 122:1556–1564. doi: 10.1182/blood-2013-04-453209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kern A, Schmidt K, Leder C, Muller OJ, Wobus CE, Bettinger K, Von der Lieth CW, King JA, Kleinschmidt JA. 2003. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol 77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koeberl DD, Alexander IE, Halbert CL, Russell DW, Miller AD. 1997. Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc Natl Acad Sci U S A 94:1426–1431. doi: 10.1073/pnas.94.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci U S A 94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradshaw AC, Parker AL, Duffy MR, Coughlan L, van Rooijen N, Kahari VM, Nicklin SA, Baker AH. 2010. Requirements for receptor engagement during infection by adenovirus complexed with blood coagulation factor X. PLoS Pathog 6:e1001142. doi: 10.1371/journal.ppat.1001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, Kemball-Cook G, Ni S, Lieber A, McVey JH, Nicklin SA, Baker AH. 2006. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 108:2554–2561. doi: 10.1182/blood-2006-04-008532. [DOI] [PubMed] [Google Scholar]
- 8.Summerford C, Samulski RJ. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 72:1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 10.Kim SH, Turnbull J, Guimond S. 2011. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol 209:139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- 11.Esko JD, Kimata K, Lindahl U. 2009. Proteoglycans and sulfated glycosaminoglycans, p 229–248. In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (ed), Essentials of glycobiology, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 12.Busse-Wicher M, Wicher KB, Kusche-Gullberg M. 2014. The exostosin family: proteins with many functions. Matrix Biol 35:25–33. doi: 10.1016/j.matbio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Esko JD, Selleck SB. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 14.Nadanaka S, Kitagawa H. 2008. Heparan sulphate biosynthesis and disease. J Biochem 144:7–14. doi: 10.1093/jb/mvn040. [DOI] [PubMed] [Google Scholar]
- 15.Esko JD, Lindahl U. 2001. Molecular diversity of heparan sulfate. J Clin Invest 108:169–173. doi: 10.1172/JCI200113530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, Tufaro F. 1998. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet 19:158–161. doi: 10.1038/514. [DOI] [PubMed] [Google Scholar]
- 17.Zhang F, Aguilera J, Beaudet JM, Xie Q, Lerch TF, Davulcu O, Colon W, Chapman MS, Linhardt RJ. 2013. Characterization of interactions between heparin/glycosaminoglycan and adeno-associated virus. Biochemistry 52:6275–6285. doi: 10.1021/bi4008676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Opie SR, Warrington KH Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. 2003. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol 77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam HJ, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. 2010. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perabo L, Goldnau D, White K, Endell J, Boucas J, Humme S, Work LM, Janicki H, Hallek M, Baker AH, Buning H. 2006. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol 80:7265–7269. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alba R, Bradshaw AC, Parker AL, Bhella D, Waddington SN, Nicklin SA, van Rooijen N, Custers J, Goudsmit J, Barouch DH, McVey JH, Baker AH. 2009. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood 114:965–971. doi: 10.1182/blood-2009-03-208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov DM. 2008. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A 105:5483–5488. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SM, Greig JA, Denby L, Custers J, Morita T, Francischetti IM, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Havenga MJ, Nicklin SA, Baker AH. 2008. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Duffy MR, Bradshaw AC, Parker AL, McVey JH, Baker AH. 2011. A cluster of basic amino acids in the factor X serine protease mediates surface attachment of adenovirus/FX complexes. J Virol 85:10914–10919. doi: 10.1128/JVI.05382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaiss AK, Lawrence R, Elashoff D, Esko JD, Herschman HR. 2011. Differential effects of murine and human factor X on adenovirus transduction via cell-surface heparan sulfate. J Biol Chem 286:24535–24543. doi: 10.1074/jbc.M111.241562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doronin K, Flatt JW, Di Paolo NC, Khare R, Kalyuzhniy O, Acchione M, Sumida JP, Ohto U, Shimizu T, Akashi-Takamura S, Miyake K, MacDonald JW, Bammler TK, Beyer RP, Farin FM, Stewart PL, Shayakhmetov DM. 2012. Coagulation factor X activates innate immunity to human species C adenovirus. Science 338:795–798. doi: 10.1126/science.1226625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vigant F, Descamps D, Jullienne B, Esselin S, Connault E, Opolon P, Tordjmann T, Vigne E, Perricaudet M, Benihoud K. 2008. Substitution of hexon hypervariable region 5 of adenovirus serotype 5 abrogates blood factor binding and limits gene transfer to liver. Mol Ther 16:1474–1480. doi: 10.1038/mt.2008.132. [DOI] [PubMed] [Google Scholar]
- 28.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. 2003. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science 302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- 29.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD, Esko JD. 2008. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem 283:33674–33684. doi: 10.1074/jbc.M804288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang Q, Yamamoto M, Curiel DT, Herschman HR. 2004. Noninvasive imaging of transcriptionally restricted transgene expression following intratumoral injection of an adenovirus in which the COX-2 promoter drives a reporter gene. Mol Imaging Biol 6:395–404. doi: 10.1016/j.mibio.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Alemany R, Curiel DT. 2001. CAR-binding ablation does not change biodistribution and toxicity of adenoviral vectors. Gene Ther 8:1347–1353. doi: 10.1038/sj.gt.3301515. [DOI] [PubMed] [Google Scholar]
- 33.Mittereder N, March KL, Trapnell BC. 1996. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol 70:7498–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lidholt K, Weinke JL, Kiser CS, Lugemwa FN, Bame KJ, Cheifetz S, Massague J, Lindahl U, Esko JD. 1992. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci U S A 89:2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seglen PO. 1976. Incorporation of radioactive amino acids into protein in isolated rat hepatocytes. Biochim Biophys Acta 442:391–404. doi: 10.1016/0005-2787(76)90313-0. [DOI] [PubMed] [Google Scholar]
- 36.Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. 2009. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest 119:3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaiss AK, Zuber J, Chu C, Machado HB, Jiao J, Catapang AB, Ishikawa TO, Gil JS, Lowe SW, Herschman HR. 2014. Reversible suppression of cyclooxygenase 2 (COX-2) expression in vivo by inducible RNA interference. PLoS One 9:e101263. doi: 10.1371/journal.pone.0101263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallaher SD, Gil JS, Dorigo O, Berk AJ. 2009. Robust in vivo transduction of a genetically stable Epstein-Barr virus episome to hepatocytes in mice by a hybrid viral vector. J Virol 83:3249–3257. doi: 10.1128/JVI.01721-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 40.Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. 1998. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol 72:7909–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicklin SA, Wu E, Nemerow GR, Baker AH. 2005. The influence of adenovirus fiber structure and function on vector development for gene therapy. Mol Ther 12:384–393. doi: 10.1016/j.ymthe.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. 2005. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol 79:7478–7491. doi: 10.1128/JVI.79.12.7478-7491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonsson MI, Lenman AE, Frangsmyr L, Nyberg C, Abdullahi M, Arnberg N. 2009. Coagulation factors IX and X enhance binding and infection of adenovirus types 5 and 31 in human epithelial cells. J Virol 83:3816–3825. doi: 10.1128/JVI.02562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Francischetti IM, Valenzuela JG, Andersen JF, Mather TN, Ribeiro JM. 2002. Ixolaris, a novel recombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood 99:3602–3612. doi: 10.1182/blood-2001-12-0237. [DOI] [PubMed] [Google Scholar]
- 45.Monteiro RQ, Rezaie AR, Ribeiro JM, Francischetti IM. 2005. Ixolaris: a factor Xa heparin-binding exosite inhibitor. Biochem J 387:871–877. doi: 10.1042/BJ20041738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami MT, Rios-Steiner J, Weaver SE, Tulinsky A, Geiger JH, Arni RK. 2007. Intermolecular interactions and characterization of the novel factor Xa exosite involved in macromolecular recognition and inhibition: crystal structure of human Gla-domainless factor Xa complexed with the anticoagulant protein NAPc2 from the hematophagous nematode Ancylostoma caninum. J Mol Biol 366:602–610. doi: 10.1016/j.jmb.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 47.Xu Z, Qiu Q, Tian J, Smith JS, Conenello GM, Morita T, Byrnes AP. 2013. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat Med 19:452–457. doi: 10.1038/nm.3107. [DOI] [PubMed] [Google Scholar]
- 48.Le Jan S, Hayashi M, Kasza Z, Eriksson I, Bishop JR, Weibrecht I, Heldin J, Holmborn K, Jakobsson L, Soderberg O, Spillmann D, Esko JD, Claesson-Welsh L, Kjellen L, Kreuger J. 2012. Functional overlap between chondroitin and heparan sulfate proteoglycans during VEGF-induced sprouting angiogenesis. Arterioscler Thromb Vasc Biol 32:1255–1263. doi: 10.1161/ATVBAHA.111.240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. 2005. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 132:5055–5068. doi: 10.1242/dev.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Fuster M, Sriramarao P, Esko JD. 2005. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol 6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]