

Abstract
The ability of a subset of G protein-coupled receptors (GPCRs) to activate RhoA endows them with unique growth-regulatory properties. Two transcriptional pathways are activated through GPCRs and RhoA, one utilizing the transcriptional coactivator myocardin-related transcription factor A (MRTF-A) and serum response factor (SRF) and the other using the transcriptional coactivator Yes-associated protein (YAP) and TEA domain family members (TEAD). These pathways have not been compared for their relative levels of importance and potential interactions in RhoA target gene expression. GPCRs for thrombin and sphingosine-1-phosphate (S1P) on human glioblastoma cells robustly couple to RhoA and induce the matricelluar protein CCN1. Knockdown of either MRTF-A or YAP abrogates S1P-stimulated CCN1 expression, demonstrating that both coactivators are required. MRTF-A and YAP are also both required for transcriptional control of other S1P-regulated genes in various cell types and for S1P-stimulated glioblastoma cell proliferation. Interactions between MRTF-A and YAP are suggested by their synergistic effects on SRE.L- and TEAD-luciferase expression. Moreover, MRTF-A and YAP associate in coimmunoprecipitations from S1P-stimulated cells. Chromatin immunoprecipitation (ChIP) analysis of the CCN1 gene promoter demonstrated that S1P increases coactivator binding at the canonical transcription factor sequences. Unexpectedly, S1P also enhances MRTF-A binding at TEA sites. Our findings reveal that GPCR- and RhoA-regulated gene expression requires dual input and integration of two distinct transcriptional pathways.
INTRODUCTION
Stimulation of G protein-coupled receptors (GPCRs) that activate RhoA induces proliferation of human 1321N1 glioblastoma and other cells (1–7). These mitogenic GPCRs include the protease-activated thrombin receptor (PAR1) and receptors for the lysophospholipids lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) (1–7). Activation of RhoA by GPCRs or stretch also leads to rapid and robust increases in the amount of the matricellular protein CCN1 (3, 8–10), the founding member of the CCN gene family (11, 12). We previously demonstrated that CCN1 expression and resulting integrin activation are required for thrombin-stimulated proliferation of 1321N1 glioblastoma cells (3). A broader role for CCN1 signaling in tumor growth is evidenced by a wealth of published data implicating CCN1 in cancer cell proliferation, angiogenesis, survival, and invasion (10–13). Of note, the CCN1 gene has been independently used as a readout for the effects of two distinct transcriptional coactivators, myocardin-related transcription factor A (MRTF-A) and Yes-associated protein (YAP), as described below.
Mechanisms by which activated RhoA signals cells to regulate gene transcription have been elucidated over the last 2 decades (14). Treisman and others established that serum and RhoA induce serum response factor (SRF)-dependent gene transcription independently of the activation of the previously known SRF transcriptional coactivator, ternary complex factor (TCF) (15, 16). Subsequent studies demonstrated that this occurs through interaction of SRF with MRTF-A, a member of the myocardin family of transcriptional coactivators (17, 18). Basally, MRTF-A is bound to G-actin, but activated RhoA decreases the amount of free G-actin available to sequester MRTF-A, favoring its accumulation in the nucleus (17–20). Nuclear actin dynamics and polymerization also regulate the localization and transcriptional control of MRTF-A by controlling MRTF-A nuclear export (21–23). GPCRs that couple to RhoA have been shown to increase the amount of nuclear MRTF-A in smooth muscle, cardiac muscle, fibroblasts, and other cells (7, 14, 24, 25).
Recently, another transcriptional coactivator, YAP, was shown to be regulated through GPCR coupling to RhoA (26–29). These findings extend and are consistent with evidence that mechanotransduction and changes in matrix stiffness, which are transduced through RhoA, elicit YAP activation (30, 31). YAP is dephosphorylated in response to RhoA activation and translocates to the nucleus (27, 29–31). In the nucleus, YAP binds to and serves as a coactivator for the TEA domain (TEAD)-containing family of transcription factors (32, 33). While the precise mediators through which RhoA signaling leads to YAP dephosphorylation remain uncertain, these discoveries revealed a second pathway through which GPCR ligands that activate RhoA can induce transcriptional gene programs.
As indicated above, the CCN1 gene, which is regulated by GCPRs through RhoA, has been utilized as one of the major readouts for activation of MRTF-A and, independently, for signals activating the YAP pathway (25, 27, 29, 32). MRTF-A was identified as the transcriptional coactivator through which RhoA mediates cyclic strain-induced CCN1 gene expression in smooth muscle cells (34). Our lab demonstrated that CCN1 is induced through MRTF-A in response to GPCRs that activate RhoA in cardiac myocytes (25). At the same time, the CCN1 gene was used as a canonical gene readout for activation of the Hippo-YAP pathway (27, 29, 30, 32). The relative levels of importance of RhoA-mediated MRTF-A versus YAP pathways in transcriptional control of the CCN1 gene or other genes have not, to our knowledge, been examined, nor have the pathways for activation of these two RhoA-regulated transcriptional coactivators been compared in a single system. Here we address the question of whether MRTF-A and YAP exert redundant, distinct, or combinatorial effects on gene expression, using CCN1 as a model. Our studies demonstrate divergence in the pathways for regulation of YAP and MRTF-A, but most remarkably, they reveal that activation of both pathways is required for transcriptional control of RhoA-regulated genes as well as for cell proliferation. Interactive transcriptional effects and coordinate regulation of MRTF-A- and YAP-regulated genes may ensure that GPCR-mediated RhoA activation elicits cellular responses only when multiple inputs are integrated.
MATERIALS AND METHODS
Antibodies, plasmids, and other materials.
Anti-CCN1, -YAP, -MRTF-A, -TEAD, and -SRF antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was obtained from Cell Signaling Technology (Beverly, MA). Alexa Fluor 488-conjugated secondary antibodies were obtained from Invitrogen (Carlsbad, CA). DAPI (4′,6-diamidino-2-phenylindole) was obtained from Vector Laboratories, Inc. (Burlingame, CA). Horseradish peroxidase-conjugated secondary antibodies, carbamoylcholine chloride (carbachol), and heparin-agarose beads were purchased from Sigma Chemical Company (St. Louis, MO). Dynabeads were obtained from Life Technologies. Horseradish peroxidase-conjugated light-chain-specific secondary antibodies were obtained from Abcam. Phos-tag-conjugated acrylamide was purchased from Wako Chemicals, and gels containing Phos-tag were prepared according to the manufacturer's instructions. Y-27632 and cytochalasin D were purchased from Calbiochem. C3 was purchased from Cytoskeleton Inc., and human α-thrombin was obtained from Enzyme Research Laboratories (South Bend, IN). S1P was purchased from Avanti Polar Lipids (Alabaster, AL). A control scrambled small interfering RNA (siRNA) and siRNAs targeting YAP and MRTF-A were purchased from Qiagen. FlexiTube siRNA constructs for YAP and MRTF-A were obtained from Qiagen. The SRE.L-Luc construct was generously contributed by Silvio Gutkind (National Institutes of Health/NIDCR). The pCMV-Flag-YAP and pCMV-Flag empty vectors were a kind gift from Kun-Liang Guan (University of California, San Diego, CA). The TEAD-Luc (31) and Flag-MRTF-A (17) constructs were obtained from Addgene.
Cell culture and transfection.
1321N1 human glioblastoma cells and neonatal rat ventricular myocytes (NRVMs) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 4 mM l-glutamine in a 37°C, 10% CO2 humidified environment. MCF10A cells were cultured in DMEM–F-12 (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml epidermal growth factor (EGF), 0.5 mg/ml hydrocortisone, 10 mg/ml insulin, 100 ng/ml cholera toxin, and 50 mg/ml penicillin-streptomycin. Cells were grown to 70% confluence on 6- or 10-cm plates and serum starved for 24 h prior to the start of each experiment. Cells were transfected with plasmid DNA by using FuGENE HD transfection reagent (Promega) according to the manufacturer's instructions. siRNA constructs were delivered into cells by using Dharmafect 4 (1321N1 cells) or Dharmafect 1 (NRVMs and MCF10A cells) (GE Dharmacon) according to the manufacturer's instructions.
Immunoblotting.
Following treatment with agonists and/or inhibitors, cells were washed and lysed in buffer containing 20 mM Tris-HCl (pH 7.6), 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 20 mM β-glycerophosphate, 0.5% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 mM disodium 4-nitrophenylphosphate, 100 μM sodium orthovanadate, and 10 μg/ml leupeptin. Lysates were clarified by centrifugation, 4× Laemmli buffer was added, and samples were boiled for 5 min. Proteins were resolved via SDS-PAGE analysis, and membranes were probed with primary antibodies overnight at 4°C. All primary antibodies were diluted 1:1,000, and secondary immunoglobulin G (IgG)-horseradish peroxidase was diluted 1:2,000, in 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween 20. Proteins were visualized using enhanced chemiluminescence and quantitated using gel documentation software (Alpha Innotech Corp., San Leandro, CA).
Immunofluorescence.
Immunofluorescence was conducted using serum-starved glioblastoma cells at 4 × 104 cells per well on coverslips. Following treatment with agonists and/or inhibitors, cells were washed, fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.1% Triton X-100 for 10 min. After blocking in 5% BSA in phosphate-buffered saline (PBS) for 30 min, cells were incubated with primary antibodies diluted in 5% BSA overnight at 4°C. After washing three times in PBS, Alexa Fluor 488-conjugated secondary antibody (1:100) was added and incubated for 1 h at room temperature. Slides were washed and mounted with Vectashield HardSet mounting medium with DAPI.
Quantitative PCR (qPCR).
Total RNA was extracted from agonist-treated control and knockdown glioblastoma cells by using an RNeasy kit (Invitrogen) as previously described (3). cDNA was amplified using TaqMan Universal master mix in the presence of gene-specific primers for CCN1, CTGF, ACTA2, and ANKRD1, with GAPDH as an internal control (Applied Biosystems). Data were normalized to the internal GAPDH level, and fold changes were determined according to a published protocol (35).
Chromatin obtained from chromatin immunoprecipitation (ChIP) was subjected to qPCR with primers specific to the indicated promoter regions, using Power Sybr green qPCR master mix (Applied Biosciences) and a model 7500 real-time PCR system (Applied Biosciences). Relative abundance was calculated by normalization to the input control.
Primers used were as follows: SRE1 primers, CCGAAGCTAGATGACGGAGT (forward) and GTTGCCAAGAATCGAGGTTT (reverse); SRE2 primers, TGGTTGGATAACAGAGGCAGA (forward) and GCTTCTGTTGTGGCGTCTTT (reverse); TEA1 primers, CCCCGAACTGTTTTCTTGAC (forward) and TCCATTGCACCTTTGTGTGT (reverse); TEA2 primers, CAAGCAGCTCAACGAGGACT (forward) and TTTTAAAGGGGCCAAACCA (reverse); TEA3 primers, AAATGCAAAGGAATGCAAGG (forward) and GGAATGCTCTGGACCTTTGA (reverse); and negative-control primers, ATGGTTGCCACTGGGGATCT (forward) and TGCCAAAGCCTAGGGGAAGA (reverse).
Nuclear fractionation and immunoprecipitation.
Glioblastoma cells were lysed in ice-cold buffer C, containing 10 mM HEPES (pH 7.6), 10 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 0.1% NP-40, and 1 mM phosphatase and protease inhibitors. The samples were kept on ice for 15 min and then centrifuged at 2,600 × g for 5 min. The pellets were washed twice and then lysed in high-salt radioimmunoprecipitation assay (RIPA) buffer. The samples were centrifuged at 21,000 × g for 15 min, and the supernatants, containing extracted nuclear proteins, were collected. Nuclear proteins were incubated with the appropriate antibodies overnight at 4°C. Protein A/G-conjugated beads were added for 2 h the next day. Immunoprecipitates were washed four times with lysis buffer, and SDS-PAGE was performed.
Chromatin immunoprecipitation.
ChIP was conducted using the Abcam X-ChIP protocol. Cells were fixed for 10 min in 1% formaldehyde at 90% confluence in 150-mm dishes. After cross-linking, cells were lysed in 10 mM HEPES, pH 7.9, 0.5% NP-40, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT) on ice for 10 min. Nuclei were pelleted at 2,300 × g for 5 min and were resuspended in 1% SDS, 10 mM EDTA, 50 mM Tris-Cl, pH 8.0. Chromatin was sheared into 200- to 500-bp fragments by sonication. Equal amounts of sample were immunoprecipitated with the appropriate antibody or control IgG conjugated to Dynabeads overnight at 4°C. Precipitates were washed according to the Abcam X-ChIP protocol, with the addition of a LiCl buffer wash (250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-Cl, pH 8.0), and subjected to qPCR as described above.
Cell proliferation assay.
1321N1 cells (expressing control siRNA, siYAP, or siMRTF-A) in serum-free medium were plated to 60% confluence in 6-well plates and maintained in the presence or absence of 0.3 μM S1P for 8, 24, or 48 h. Cell numbers were determined daily using a cell counter (Invitrogen). Three wells were designated for each condition in two separate experiments. Data are expressed relative to those for the untreated control at each time point.
Statistical analysis.
All results are reported as means ± standard errors of the means (SEM). Comparison of two groups with one characteristic was accomplished using unpaired Student's t test. Data from two groups with multiple characteristics were compared by using two-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison test. Data from experiments with more than two groups with one characteristic were compared by using one-way ANOVA followed by Tukey's multiple-comparison test. The D'Agostino and Pearson omnibus normality test was used to determine whether the data points were normally distributed. P values of <0.05 were considered significant.
RESULTS
Our previous studies focused on the ability of the PAR1 receptor agonist thrombin to induce robust and sustained expression of the matricellular protein CCN1 (3). Treatment of glioblastoma cells with 0.3 μM S1P also led to 2.5- to 3.5-fold increases in CCN1 protein expression, which occurred within 1 h and were sustained for at least 6 h (Fig. 1A). In contrast, carbachol (500 μM), an agonist for the Gq-coupled muscarinic cholinergic receptor (mAChR), which does not activate RhoA or stimulate proliferation in these cells (1–3), did not increase CCN1 expression. The response to S1P was blocked by treatment with C3 exoenzyme (1 μg/ml) to inhibit RhoA function, as well as by Y-27632 (10 μM), an inhibitor of the Rho-activated protein kinase (ROCK) (Fig. 1B).
FIG 1.
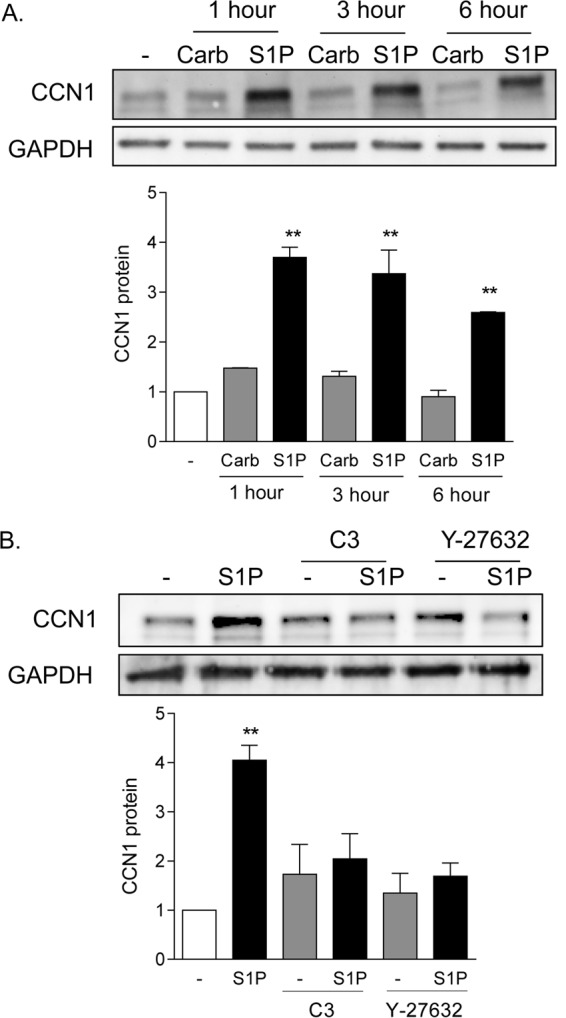
CCN1 expression is highly and rapidly increased by S1P in a Rho-dependent manner. (A) Twenty-four-hour serum-starved glioblastoma cells were treated with 500 μM carbachol (Carb) or 0.3 μM S1P (sphingosine-1-phosphate) for 1, 3, or 6 h. Total cell lysates were subject to immunoblotting for CCN1 protein and GAPDH (loading control). **, P < 0.01 versus control (n = 6). (B) Serum-starved glioblastoma cells were pretreated with 1 μg/ml C3 exoenzyme (24 h) to functionally inhibit RhoA or with 10 μM Y-27632 (1 h) to block Rho kinase (ROCK). Pretreated cells were then stimulated with 0.3 μM S1P for an additional 1 h. Total cell lysates were subject to immunoblotting for CCN1 protein and GAPDH (loading control). **, P < 0.01 versus control (n = 5).
YAP and MRTF-A are activated by S1P through RhoA signaling.
Two transcriptional coactivators, MRTF-A and YAP, have been demonstrated independently to regulate CCN1 expression through RhoA signaling (25, 27, 29, 32). To determine which of these transcriptional coactivators is regulated in glioblastoma cells treated with S1P, we examined their nuclear localization as well as activation of luciferase reporter genes driven by their cognate consensus binding sequences. As shown in Fig. 2A, MRTF-A was largely cytoplasmic in serum-starved cells, and S1P treatment led to increased nuclear MRTF-A accumulation. The accumulation of MRTF-A in the nucleus was blocked by either inhibiting RhoA function with C3 or blocking the downstream RhoA effector ROCK with Y-27632 (Fig. 2A). A TCF-independent SRF-regulated luciferase reporter (SRE.L-Luc) has been used as a measure of activation of RhoA (or, more specifically, of MRTF-A-mediated SRF activation) (17, 18, 24). S1P robustly increased SRE.L-driven luciferase expression, and this was prevented by either C3 or Y-27632 pretreatment (Fig. 2B). These data confirm that S1P transduces signals to MRTF-A and causes activation of gene expression via Rho and ROCK.
FIG 2.

MRTF-A is activated by S1P through RhoA and ROCK signaling. (A) Twenty-four-hour serum-starved glioblastoma cells were pretreated with 1 μg/ml C3 exoenzyme (24 h) or 10 μM Y-27632 (1 h). Cells were then stimulated for 1 h with 0.3 μM S1P. MRTF-A subcellular localization was determined by immunofluorescence staining for endogenous MRTF-A (green) along with DAPI staining to show nuclei (blue). (B) Cells were transfected with a TCF-independent, SRF promoter-driven firefly luciferase reporter construct (SRE.L-Luc) for 24 h. Cells were serum starved for 24 h prior to being pretreated with 1 μg/ml C3 exoenzyme (24 h) or 10 μM Y-27632 (1 h), stimulated with 0.3 μM S1P for 8 h, and assayed for luciferase expression. Data shown are normalized to Renilla luciferase activity and expressed as fold changes over the control level. **, P < 0.01 versus control (n = 6).
Under the same conditions, S1P also increased the amount of nuclear YAP (Fig. 3A). This response was inhibited by treatment with C3, indicating that YAP activation, like that of MRTF-A, is Rho dependent. YAP translocation is not, however, dependent on ROCK, since this response was unaffected by treatment with Y-27632 (Fig. 3A). These data were confirmed using a TEAD transcription response element-regulated luciferase reporter (TEAD-Luc) as an indicator of YAP activation (31) (Fig. 3B). S1P led to robust increases in luciferase expression, which were blocked by inhibition of Rho but not by inhibition of ROCK. The YAP phosphorylation status was also measured using a Phos-tag gel and immunoblotting for total YAP. The downshift of YAP, elicited by S1P treatment and indicative of its dephosphorylation and consequent activation, was abrogated when Rho was inhibited but unaffected by inhibition of ROCK (Fig. 3C). S1P-mediated YAP dephosphorylation was, however, inhibited when cells were pretreated with cytochalasin D (Fig. 3C). Thus, some ROCK-independent aspect of actin polymerization is required.
FIG 3.

YAP is activated by S1P through RhoA signaling. (A) Twenty-four-hour serum-starved glioblastoma cells were pretreated with 1 μg/ml C3 exoenzyme (24 h) or 10 μM Y-27632 (1 h). Cells were then stimulated for 1 h with 0.3 μM S1P. YAP subcellular localization was determined by immunofluorescence staining for endogenous YAP (green) along with DAPI staining to show nuclei (blue). (B) Cells were transfected with a TEAD promoter-driven firefly luciferase reporter construct (TEAD-Luc) for 24 h. Cells were serum starved for 24 h prior to being pretreated with 1 μg/ml C3 exoenzyme (24 h) or 10 μM Y-27632 (1 h), stimulated with 0.3 μM S1P for 8 h, and assayed for luciferase expression. Data shown are normalized to Renilla luciferase activity and expressed as fold changes over the control level. **, P < 0.01 versus control (n = 6). (C) Twenty-four-hour serum-starved glioblastoma cells were pretreated with 1 μg/ml C3 exoenzyme (24 h), 10 μM Y-27632 (1 h), or 10 μM cytochalasin D (CytoD) (1 h). Cells were then stimulated for 1 h with 0.3 μM S1P, and YAP phosphorylation was determined by Phos-tag immunoblotting for YAP. Note that the downshift representing dephosphorylation is also accompanied by greater immunoreactivity, as seen by others (27, 29).
YAP and MRTF-A are both required for S1P-regulated gene expression.
To test for the involvement of YAP and MRTF-A in S1P-mediated CCN1 induction, we used siRNAs to knock down the expression of these transcriptional coactivators (Fig. 4A and B). S1P treatment led to a 4.5-fold increase in CCN1 protein in cells transfected with a control scrambled siRNA (Fig. 4C). This response was fully abolished by knockdown of either MRTF-A or YAP.
FIG 4.

Both YAP and MRTF-A are required for S1P to increase CCN1 expression. (A) Glioblastoma cells were transfected with siRNA specific to YAP for 48 h. Total cell lysates were subject to immunoblotting for YAP and GAPDH (loading control) (blots shown are representative of three experiments). MRTF-A protein levels were not affected by YAP knockdown (not shown). (B) Glioblastoma cells were transfected with siRNA specific to MRTF-A for 48 h. Total cell lysates were subject to immunoblotting for MRTF-A protein and GAPDH (loading control) (blots shown are representative of three experiments). YAP protein levels were not affected by MRTF-A knockdown (not shown). (C) Glioblastoma cells were transfected with siYAP, siMRTF-A, or both (double KD) for 48 h. Cells were serum starved for 24 h and then treated with 0.3 μM S1P for 1 h. Total cell lysates were subject to immunoblotting for CCN1 protein and GAPDH (loading control). **, P < 0.01 versus siCon (n = 5).
We extended these findings on CCN1 protein expression to demonstrate changes at the mRNA level. At the same time, we examined three other genes, encoding alpha-smooth muscle actin (ACTA2), connective tissue growth factor (CTGF), and ankyrin repeat domain-containing protein 1 (ANKRD1), all of which are established MRTF-A- and YAP-regulated genes (7, 31, 36–40). Knockdown of either YAP or MRTF-A fully abolished S1P-induced mRNA increases for all four of the genes (Fig. 5A to D). Moreover, to demonstrate the target specificity of the siRNA, we confirmed our findings by using a second siRNA construct for YAP or MRTF-A (Fig. 5E to H).
FIG 5.

Both YAP and MRTF-A are required for induction of multiple genes. (A to D) Glioblastoma cells were transfected with siYAP, siMRTF-A, or both for 48 h. Cells were serum starved for 24 h and then treated with 0.3 μM S1P for 1 h. Total cell lysates were subjected to qPCR analysis of mRNA levels for CCN1, CTGF, ANKRD1, or ACTA2. **, P < 0.01 versus siCon (n = 3). (E to H) The experiments for panels A to D were repeated with a second siRNA construct for YAP and a second siRNA construct for MRTF-A. **, P < 0.01 versus siCon (n = 3).
The implication of these observations for signaling in other cell types was then explored. We tested another immortalized cell line, MCF10A breast epithelial cells, as well as primary cultures of NRVMs. Cells were stimulated with S1P subsequent to knockdown of YAP or MRTF-A, and the mRNA levels of the CCN1, CTGF, and ACTA2 genes were determined (Fig. 6). Knockdown of either YAP or MRTF-A fully abolished S1P-induced increases in mRNA for these genes.
FIG 6.

Both YAP and MRTF-A are required for induction of genes in various cell types. (A to C) MCF10A breast epithelial cells were transfected with siYAP, siMRTF-A, or both for 48 h. Cells were serum starved for 24 h and then treated with 0.3 μM S1P for 1 h. Total cell lysates were subject to qPCR analysis of mRNA levels for CCN1, CTGF, or ACTA2. **, P < 0.01 versus siCon (n = 3). (D to F) Neonatal rat ventricular myocytes were transfected with siYAP, siMRTF-A, or both for 48 h. Cells were serum starved for 24 h and then treated with 0.3 μM S1P for 1 h. Total cell lysates were subject to qPCR analysis of mRNA levels for CCN1, CTGF, or ACTA2. **, P < 0.01 versus siCon (n = 3).
Taken together, the findings described above indicate that (i) MRTF-A and YAP do not serve redundant functions but are both required for agonist- and RhoA-induced expression of CCN1, (ii) this is true for S1P-induced regulation of three additional genes, and (iii) the requirement for combined activation of MRTF-A and YAP is evident in other cell lines and primary cells responsive to S1P.
YAP and MRTF-A synergize for activation of SRF- and TEAD-mediated gene expression.
To further explore the effects of YAP and MRTF-A alone and in concert, we assessed the regulation of SRE.L-Luc or TEAD-Luc in glioblastoma cells. As expected, MRTF-A expression activated the SRE.L promoter, and YAP expression did not. Remarkably, when YAP was added along with MRTF-A, SRE.L-driven promoter luciferase activity was significantly higher than that observed with MRTF-A alone (Fig. 7A). The same phenomenon was observed in the complementary experiment. Specifically, YAP activated the TEAD promoter, as expected, while MRTF-A did not. However, when MRTF-A was added along with YAP, the TEAD promoter-luciferase activity was significantly higher than that observed with YAP alone (Fig. 7B). These results imply that there is a cooperative or synergistic interaction between the two transcriptional coactivators and/or their partners.
FIG 7.

YAP and MRTF-A synergize for activation of SRF- and TEAD-mediated transcriptional activation. (A) Glioblastoma cells were transfected with SRE.L-Luc along with 100 ng, 300 ng, or 500 ng of MRTF-A or YAP alone (along with an equal amount of empty vector) or 100, 300, or 500 ng of YAP along with 100, 300, or 500 ng of MRTF-A. Cells were then serum starved for 24 h and assayed for luciferase activity. Data shown are normalized to Renilla luciferase activity and expressed as fold changes compared to the empty vector control level. *, P < 0.05 versus control (n = 6); #, P < 0.05 versus MRTF-A (n = 6). (B) Glioblastoma cells were transfected with TEAD-luciferase along with 100 ng, 300 ng, or 500 ng of MRTF-A or YAP alone (along with an equal amount of empty vector) or 100, 300, or 500 ng of YAP along with 100, 300, or 500 ng of MRTF-A. Cells were then serum starved for 24 h and assayed for luciferase activity. Data shown are normalized to Renilla luciferase activity and expressed as fold changes compared to the empty vector control level. *, P < 0.05 versus control (n = 6); #, P < 0.05 versus YAP (n = 6). We noted that while YAP and MRTF-A synergize when used together, their expression levels are not different when they are expressed together versus individually (not shown).
YAP and MRTF-A association.
Next, we carried out a series of coimmunoprecipitation experiments to directly determine whether YAP and MRTF-A interact. Since heterologous overexpression can lead to nonphysiologic associations, we studied interactions among endogenous proteins. Specifically, YAP and MRTF-A were immunoprecipitated from nuclear fractions of control or S1P-treated glioblastoma cells. The YAP immunoprecipitate contained TEAD, as expected, and this interaction was increased in S1P-treated cells. Notably, MRTF-A and SRF were also detectable in the YAP immunoprecipitate (Fig. 8A). Conversely, immunoprecipitation with MRTF-A revealed the expected stimulation-dependent interaction of MRTF-A with SRF, along with an unexpected interaction with YAP and TEAD (Fig. 8B). It therefore appears that YAP is present in a complex in which activated MRTF-A is bound to SRF and that MRTF-A is present in a complex in which activated YAP is bound to TEAD.
FIG 8.

YAP associates with SRF and MRTF-A, and MRTF-A associates with TEAD and YAP, upon S1P stimulation. (A) Glioblastoma cells were serum starved for 24 h before being stimulated with S1P for 1 h. Cells were fractionated, and the nuclear lysate was immunoprecipitated with YAP or IgG control antibody. Immunoprecipitates were immunoblotted for YAP, TEAD, MRTF-A, and SRF. Whole nuclear fractions were analyzed for lamin A/C to demonstrate nuclear purity and for RhoGDI to demonstrate a lack of cytosolic contamination. (B) Glioblastoma cells were serum starved for 24 h before being stimulated with S1P for 1 h. Cells were fractionated, and the nuclear lysate was immunoprecipitated with MRTF-A or IgG control antibody. Immunoprecipitates were immunoblotted for MRTF-A, SRF, YAP, and TEAD. Whole nuclear fractions were analyzed for lamin A/C to demonstrate nuclear purity and for RhoGDI to demonstrate a lack of cytosolic contamination.
YAP and MRTF-A bind to the CCN1 gene promoter.
To demonstrate that the interactions between MRTF-A and YAP suggested above can occur on the genes that they regulate, we examined binding of the transcriptional coactivators to the CCN1 gene promoter. Using the UCSC genome browser, we identified and designed primers for the two SRE and three TEA sites shown by ChIP-sequencing (ChIP-seq) to be located in the 5-kb 5′ CCN1 gene promoter (Fig. 9A). We then conducted ChIP experiments, using untreated serum-starved cells or cells treated with S1P for 1 h. Chromatin obtained from these cells was subjected to immunoprecipitation using SRF, TEAD, MRTF-A, or YAP antibodies. qPCR was carried out with primers designed for the aforementioned TEA and SRE sites on the CCN1 gene promoter (primer sequences are shown in Materials and Methods). In confirmation of the correct selection of sites and primers, we observed that SRF bound constitutively to both SRE sites (i.e., independent of S1P stimulation) (Fig. 9B). Likewise, TEAD bound to TEA sites in a constitutive manner (i.e., not increased by S1P) (Fig. 9C). In contrast, the binding of MRTF-A to SRE sites and of YAP to TEA sites was not constitutive but was basally low and markedly enhanced in response to S1P treatment (Fig. 9D and E), paralleling the S1P-mediated redistribution of these transcriptional coactivators to the nucleus.
FIG 9.

ChIP qPCR analysis of transcription factor and coactivator binding to SRE and TEA sites on the CCN1 gene promoter. (A) Schematic of SRE and TEA sites found on the CCN1 gene promoter as annotated on the UCSC genome browser, based on SRF and TEAD ChIP-seq data. (B to G) Cells were serum starved and then treated with S1P for 1 h. Chromatin was extracted by sonication of cell nuclei and immunoprecipitated with the appropriate antibodies. (B) Nuclear SRF immunoprecipitates were subjected to qPCR using primers specific for the SRE sites indicated. (C) Nuclear TEAD immunoprecipitates were subjected to qPCR using primers specific for the TEA sites indicated. (D) Nuclear MRTF-A immunoprecipitates were subjected to qPCR using primers specific for the SRE sites indicated. (E) Nuclear YAP immunoprecipitates were subjected to qPCR using primers specific for the TEA sites indicated. (F) Nuclear YAP immunoprecipitates were subjected to qPCR using primers specific for the SRE sites indicated. (G) Nuclear MRTF-A immunoprecipitates were subjected to qPCR using primers specific for the TEA sites indicated. *, P < 0.05 versus the untreated control (n = 4). Note that the three TEA sites cannot be distinguished due to their close proximity and likely mutual occurrence on chromatin fragments.
Using ChIP to look for noncanonical binding of the two coactivators revealed that MRTF-A was also present at TEA sites following S1P treatment (Fig. 9G). This observation suggests that activated MRTF-A associates with TEA sites when YAP is recruited. YAP, on the other hand, did not bind the SRE sites on the CCN1 gene promoter, regardless of stimulation (Fig. 9F).
Functional implications of YAP and MRTF-A as regulators of S1P-stimulated cell proliferation.
We previously reported that activation of the RhoA pathway leads to cell proliferation and that CCN1 is a required mediator of this response (3). Since YAP and MRTF-A are both required for CCN1 induction, we predicted that they would both be required for proliferation. Indeed, S1P stimulation resulted in a significant increase in glioblastoma cell proliferation, which was completely abolished when either YAP or MRTF-A was knocked down (Fig. 10). This finding indicates that YAP and MRTF-A are both required for S1P-stimulated cell proliferation, as they are for expression of CCN1 and other RhoA target genes.
FIG 10.
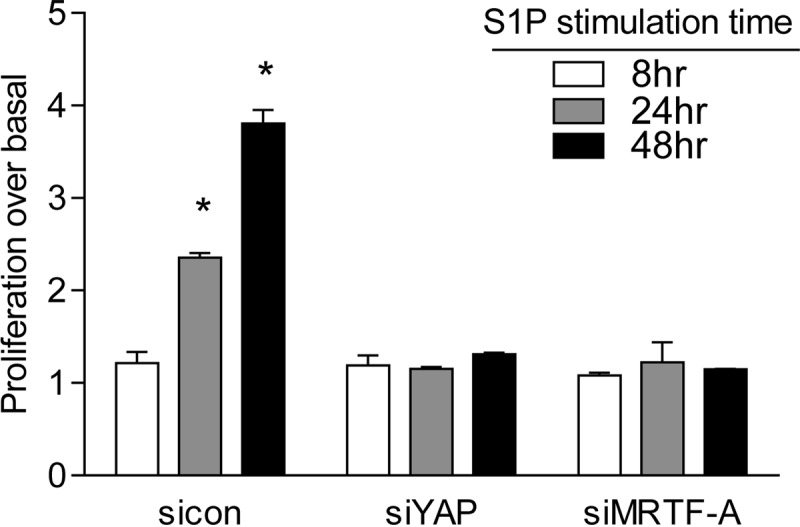
Both YAP and MRTF-A are required for S1P-stimulated cell proliferation. Glioblastoma cells were transfected with siYAP or siMRTF-A for 48 h. Cells were serum starved for 24 h and then treated with 0.3 μM S1P for 8, 24, or 48 h. Cell numbers were determined using an automatic cell counter. Data are expressed relative to the untreated control level at each time point. *, P < 0.05 versus siCon (n = 6 [two separate experiments done in triplicate]).
The findings emerging from the present study are schematized in Fig. 11.
FIG 11.

Proposed model for regulation and interactions of YAP and MRTF-A on RhoA-dependent gene regulation. In response to S1P stimulation, activation of RhoA, and nuclear accumulation of MRTF-A and YAP, these transcriptional coactivators bind and interact in an unexpected and previously undescribed manner, resulting in a dual requirement for gene regulation and cell proliferation.
DISCUSSION
We investigated two transcriptional pathways that are concomitantly activated by GPCR coupling to RhoA and which have not previously been compared for their individual roles in target gene regulation. Our studies used a human glioblastoma-derived cell line and were extended to two other cell systems. We examined effects elicited through native G protein-coupled receptors on endogenous transcription factors and target genes. Remarkably, we demonstrated that coincident activation of the transcriptional coactivators MRTF-A and YAP by S1P is required for target gene expression and cell proliferation. Furthermore, our data indicate interactions between activated MRTF-A and YAP and suggest that these occur at the level of binding to response elements on their target genes.
Several lines of evidence presented here demonstrate a dual requirement and heretofore undescribed interaction between MRTF-A and YAP in GPCR-stimulated gene expression. First, while there is abundant evidence that YAP, through its interaction with TEAD, regulates expression of the CCN1 gene, we showed that activating YAP alone is insufficient to elicit CCN1 protein or mRNA expression. This is evidenced by the observation that siRNA-mediated downregulation of MRTF-A fully inhibits S1P-induced CCN1 gene expression even though YAP activation is unchanged. Likewise, siRNA-mediated knockdown of YAP abrogates CCN1 expression, indicating that MRTF-A alone is not sufficient for CCN1 induction in response to S1P. Similar observations were made when thrombin was used to induce CCN1 through PAR1 receptors and RhoA (not shown).
Recent studies using RNA and ChIP-seq in genome-wide studies of serum-induced gene expression revealed that RhoA signaling through MRTF-A is the predominant transcriptional pathway by which serum regulates SRF-dependent gene expression (41). Of additional interest, it was noted that the gene array signatures for MRTF-A–SRF showed significant overlap with those previously published for the YAP-TEAD pathway. We used the UCSC genome browser to search for the SRF and TEAD binding sites defined by ChIP-seq and noted the close proximity of these binding sites in the promoter regions of the ACTA2, CTGF, and ANKRD1 genes, all of which are established MRTF-A- and YAP-regulated genes (7, 29, 36–38, 41, 42). Accordingly, we extended our study on agonist-mediated regulation of CCN1 to include these additional genes. Using siRNA to knock down either or both coactivators, we demonstrated that S1P-induced increases in their mRNA levels require the coordinate activation of YAP and MRTF-A. Moreover, we demonstrated that the same genes are induced through the combined actions of YAP and MRTF-A in MCF10A cells and primary rat cardiomyocytes, as well as in 1321N1 glioblastoma cells. These collective findings may define a universal principle governing gene regulation through GPCR and RhoA signaling.
The concept that there are functional interactions between MRTF-A and YAP signaling pathways is supported by our studies with luciferase reporter genes. Remarkably, while overexpression of YAP does not induce SRE.L-luciferase gene expression, it does so when MRTF-A is present, resulting in enhanced SRE.L-luciferase expression. Likewise, MRTF-A does not induce TEAD-luciferase expression except when YAP is present. Thus, cross talk at the level of the cognate transcription factor response element occurs only when its transcriptional coactivator is present, as would be the case when the cell responds to GPCR and RhoA activation. Using another experimental approach, coimmunoprecipitation, we obtained further evidence that interactions between these transcriptional coactivators are elicited upon their activation. Specifically, in cells treated with S1P (which promotes activation and nuclear accumulation of the transcriptional coactivators), we observed an association of YAP not only with its cognate transcription factor, TEAD, but also with MRTF-A (and SRF). Similarly, MRTF-A was associated not only with its cognate transcription factor, SRF, but also with YAP (and TEAD).
Our studies using ChIP qPCR to examine endogenous transcription factor and coactivator binding to defined regulatory sites in the CCN1 gene promoter delineate potential sites of interaction of the transcriptional coactivators. These experiments revealed that when the two pathways are stimulated by S1P treatment, MRTF-A as well as YAP is localized at TEAD binding sites. It is interesting that MRTF-A was also found at some sites that did not bind SRF in genome-wide studies of serum-responsive genes (41), an observation that might be explained by the ability of MRTF-A to associate with YAP-occupied TEAD binding sites. While we cannot describe the precise nature of the unanticipated interactions between MRTF-A and YAP and their transcriptional partners on the native CCN1 gene, our ChIP data lend further credibility to the hypothesis that the pathways act in concert, not alone. Other recent papers suggest interactions between these gene-regulatory factors, showing that TEAD or YAP can compete with myocardin (a member of the same family as MRTF-A) for binding to SRF and eliciting SRF signaling in vascular smooth muscle (39, 40). As a more universal concept, our data support the hypothesis that there is convergence of two RhoA-regulated transcriptional pathways and that this is functionally important in determining the ultimate extent of gene expression.
Beyond the level of gene expression, we show here that a critical functional response, cell proliferation, is also stimulated by S1P only when both MRTF-A and YAP can be activated. The implication of this finding is that more than a single signaling pathway needs to be turned on to cause the cell to make the important decision to increase its rate of proliferation. Such a fail-safe mechanism would be facilitated by divergence in the upstream signals leading to MRTF-A and YAP activation, as appears to be the case here, where MRTF-A activation is regulated through Rho kinase (ROCK), while YAP is regulated through a ROCK-independent, not yet fully delineated set of upstream signals. Divergence in the activation mechanisms for MRTF-A and YAP would allow for their independent regulation and, thus, for stimulus-dependent activation of cellular responses mediated through YAP versus MRTF-A target genes. On the other hand, as demonstrated here, there are multiple genes for which coregulation is likely to be needed. These in turn may be genes meant to be activated only when multiple cellular inputs confirm that the environment is suitable for cells to proliferate. Conversely, dysregulation at steps in the signaling process that subvert the normal control of YAP or MRTF-A activation may underlie pathophysiological changes in gene expression and downstream responses, such as glioblastoma tumor cell growth or fibrosis.
ACKNOWLEDGMENTS
We thank Silvio Gutkind and Kun-Liang Guan for their generous gifts of plasmids used in this work. Jung-Soon Mo (Guan lab), Chia-An Yen (Ren lab), and Julian Rosenberg (Mack lab) also provided helpful technical assistance.
REFERENCES
- 1.Aragay AM, Collins LR, Post GR, Watson AJ, Feramisco JR, Brown JH, Simon MI. 1995. G12 requirement for thrombin-stimulated gene expression and DNA synthesis in 1321N1 astrocytoma cells. J Biol Chem 270:20073–20077. doi: 10.1074/jbc.270.34.20073. [DOI] [PubMed] [Google Scholar]
- 2.Post GR, Collins LR, Kennedy ED, Moskowitz SA, Aragay AM, Goldstein D, Brown JH. 1996. Coupling of the thrombin receptor to G12 may account for selective effects of thrombin on gene expression and DNA synthesis in 1321N1 astrocytoma cells. Mol Biol Cell 7:1679–1690. doi: 10.1091/mbc.7.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh CT, Radeff-Huang J, Matteo R, Hsiao A, Subramaniam S, Stupack D, Brown JH. 2008. Thrombin receptor and RhoA mediate cell proliferation through integrins and cysteine-rich protein 61. FASEB J 22:4011–4021. doi: 10.1096/fj.08-113266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridley AJ, Hall A. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 5.Mutoh T, Rivera R, Chun J. 2012. Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol 165:829–844. doi: 10.1111/j.1476-5381.2011.01622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seasholtz TM, Radeff-Huang J, Sagi SA, Matteo R, Weems JM, Cohen AS, Feramisco JR, Brown JH. 2004. Rho-mediated cytoskeletal rearrangement in response to LPA is functionally antagonized by Rac1 and PIP2. J Neurochem 91:501–512. doi: 10.1111/j.1471-4159.2004.02749.x. [DOI] [PubMed] [Google Scholar]
- 7.Lockman K, Hinson JS, Medlin MD, Morris D, Taylor JM, Mack CP. 2004. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J Biol Chem 279:42422–42430. doi: 10.1074/jbc.M405432200. [DOI] [PubMed] [Google Scholar]
- 8.Han JS, Macarak E, Rosenbloom J, Chung KC, Chaqour B. 2003. Regulation of Cyr61/CCN1 gene expression through RhoA GTPase and p38MAPK signaling pathways. Eur J Biochem 270:3408–3421. doi: 10.1046/j.1432-1033.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 9.Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A, Drexler H. 2004. Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation 109:2227–2233. doi: 10.1161/01.CIR.0000127952.90508.9D. [DOI] [PubMed] [Google Scholar]
- 10.Young N, Pearl DK, Van Brocklyn JR. 2009. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res 7:23–32. doi: 10.1158/1541-7786.MCR-08-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun JI, Lau LF. 2011. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10:945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau LF. 2011. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci 68:3149–3163. doi: 10.1007/s00018-011-0778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menendez JA, Vellon L, Mehmi I, Teng PK, Griggs DW, Lupu R. 2005. A novel CYR61-triggered ‘CYR61-alphavbeta3 integrin loop’ regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene 24:761–779. doi: 10.1038/sj.onc.1208238. [DOI] [PubMed] [Google Scholar]
- 14.Yu OM, Brown JH. 2015. G protein-coupled receptor and RhoA-stimulated transcriptional responses: links to inflammation, differentiation, and cell proliferation. Mol Pharmacol 88:171–180. doi: 10.1124/mol.115.097857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill CS, Wynne J, Treisman R. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81:1159–1170. doi: 10.1016/S0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 16.Sahai E, Alberts AS, Treisman R. 1998. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J 17:1350–1361. doi: 10.1093/emboj/17.5.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. 2003. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol 23:6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miralles F, Posern G, Zaromytidou AI, Treisman R. 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113:329–342. doi: 10.1016/S0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 19.Cen B, Selvaraj A, Prywes R. 2004. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem 93:74–82. doi: 10.1002/jcb.20199. [DOI] [PubMed] [Google Scholar]
- 20.Guettler S, Vartiainen MK, Miralles F, Larijani B, Treisman R. 2008. RPEL motifs link the serum response factor cofactor MAL but not myocardin to Rho signaling via actin binding. Mol Cell Biol 28:732–742. doi: 10.1128/MCB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baarlink C, Wang H, Grosse R. 2013. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 340:864–867. doi: 10.1126/science.1235038. [DOI] [PubMed] [Google Scholar]
- 22.Rajakyla EK, Viita T, Kyheroinen S, Huet G, Treisman R, Vartiainen MK. 2015. RNA export factor Ddx19 is required for nuclear import of the SRF coactivator MKL1. Nat Commun 6:5978. doi: 10.1038/ncomms6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vartiainen MK, Guettler S, Larijani B, Treisman R. 2007. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316:1749–1752. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- 24.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. 2007. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther 6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 25.Zhao X, Ding EY, Yu OM, Xiang SY, Tan-Sah VP, Yung BS, Hedgpeth J, Neubig RR, Lau LF, Brown JH, Miyamoto S. 2014. Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J Mol Cell Cardiol 75C:152–161. doi: 10.1016/j.yjmcc.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q, Gutkind JS. 2014. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25:831–845. doi: 10.1016/j.ccr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mo JS, Yu FX, Gong R, Brown JH, Guan KL. 2012. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev 26:2138–2143. doi: 10.1101/gad.197582.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J, Chen X, Zhang L, Wang CY, Bastian BC, Zhang K, Guan KL. 2014. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25:822–830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL. 2012. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. 2013. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 31.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 32.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao B, Kim J, Ye X, Lai ZC, Guan KL. 2009. Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of Yes-associated protein. Cancer Res 69:1089–1098. doi: 10.1158/0008-5472.CAN-08-2997. [DOI] [PubMed] [Google Scholar]
- 34.Hanna M, Liu H, Amir J, Sun Y, Morris SW, Siddiqui MA, Lau LF, Chaqour B. 2009. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem 284:23125–23136. doi: 10.1074/jbc.M109.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 36.Hwang SM, Jin M, Shin YH, Ki Choi S, Namkoong E, Kim M, Park MY, Park K. 2014. Role of LPA and the Hippo pathway on apoptosis in salivary gland epithelial cells. Exp Mol Med 46:e125. doi: 10.1038/emm.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haak AJ, Tsou PS, Amin MA, Ruth JH, Campbell P, Fox DA, Khanna D, Larsen SD, Neubig RR. 2014. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther 349:480–486. doi: 10.1124/jpet.114.213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. 2010. Myocardin-related transcription factor-A controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu F, Wang X, Hu G, Wang Y, Zhou J. 2014. The transcription factor TEAD1 represses smooth muscle-specific gene expression by abolishing myocardin function. J Biol Chem 289:3308–3316. doi: 10.1074/jbc.M113.515817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. 2012. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem 287:14598–14605. doi: 10.1074/jbc.M111.329268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R. 2014. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28:943–958. doi: 10.1101/gad.239327.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinson JS, Medlin MD, Lockman K, Taylor JM, Mack CP. 2007. Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am J Physiol Heart Circ Physiol 292:H1170–H1180. doi: 10.1152/ajpheart.00864.2006. [DOI] [PubMed] [Google Scholar]