

Abstract
Primary cilia are essential sensory and signaling organelles present on nearly every mammalian cell type. Defects in primary cilia underlie a class of human diseases collectively termed ciliopathies. Primary cilia are restricted subcellular compartments, and specialized mechanisms coordinate the localization of proteins to cilia. Moreover, trafficking of proteins into and out of cilia is required for proper ciliary function, and this process is disrupted in ciliopathies. The somatostatin receptor subtype 3 (Sstr3) is selectively targeted to primary cilia on neurons in the mammalian brain and is implicated in learning and memory. Here, we show that Sstr3 localization to cilia is dynamic and decreases in response to somatostatin treatment. We further show that somatostatin treatment stimulates β-arrestin recruitment into Sstr3-positive cilia and this recruitment can be blocked by mutations in Sstr3 that impact agonist binding or phosphorylation. Importantly, somatostatin treatment fails to decrease Sstr3 ciliary localization in neurons lacking β-arrestin 2. Together, our results implicate β-arrestin in the modulation of Sstr3 ciliary localization and further suggest a role for β-arrestin in the mediation of Sstr3 ciliary signaling.
INTRODUCTION
Primary cilia are typically solitary immotile cellular appendages that function as specialized sensory and signaling compartments (1–3). During mammalian development, primary cilia mediate critical developmental signaling pathways, including Hedgehog (Hh), Wnt, and transforming growth factor β (TGF-β) (4–6). Postnatally, primary cilia are nearly ubiquitous and are required to maintain cellular and tissue homeostasis. Primary ciliary dysfunction causes a class of human diseases, collectively referred to as ciliopathies, that present with a wide range of clinical features, including obesity, skeletal malformations, retinal degeneration, renal cystic disease, brain malformations, intellectual disability, and hypogonadism (7).
Primary cilia are restricted compartments, and specialized mechanisms exist to coordinate the selective targeting, exclusion, and retention of certain proteins (8, 9). This enrichment of select proteins is what defines the functions of cilia and determines the signaling pathways that they mediate. The importance of ciliary protein localization is highlighted by the fact that this process is disrupted in a subset of ciliopathies (10–12). Moreover, coordinated trafficking of proteins into and out of cilia is required for proper ciliary signaling. For example, in vertebrate Hh signaling, sonic Hedgehog binds to its receptor, Patched, on the ciliary membrane, causing it to exit the cilium. This allows the G protein-coupled receptor (GPCR)-like receptor Smoothened (Smo) to enter the cilium, which in turn impacts the activity of Gli transcription factors (13–16). In addition, ciliary signaling can be modulated by protein trafficking in the cilium. For example, in the photoreceptor outer segment, which is a modified primary cilium, light-induced activation of rhodopsin stimulates exit of the G-protein transducin from the cilium and entry of visual arrestin into the cilium to terminate signaling (17).
Adult neurons throughout the mammalian brain possess primary cilia that are thought to sense and signal in response to neuromodulators in the interstitial space. Neuronal cilia are enriched for certain GPCRs, including somatostatin receptor 3 (Sstr3) (18), serotonin receptor 6 (19, 20), melanin-concentrating hormone receptor 1 (Mchr1) (10, 21), dopamine receptor 1 (D1) (22), vasoactive intestinal receptor 2 (23), neuropeptide Y receptors 2 and 5 (24), and kisspeptin receptor 1 (25). These cilia are also widely enriched for the GPCR signaling element type 3 adenylyl cyclase (AC3) (26). We previously showed that the neuronal ciliary localization of Sstr3 and Mchr1 is disrupted in mouse models of the heterogeneous human ciliopathy Bardet-Biedl syndrome (BBS) (10). D1, on the other hand, accumulates in neuronal cilia in mice with BBS (22), suggesting that D1 ciliary localization is normally dynamic and D1 export from cilia is dependent on the BBS proteins. Indeed, D1 ciliary localization is increased in response to environmental cues, such as increased cyclic AMP levels, and decreased in response to agonist binding (22).
GPCR localization on the plasma membrane is a highly regulated process. Classically, agonist binding results in a conformational change in the receptor. This allows the coupling and activation of G proteins, leading to the generation of downstream signaling (27). Activated GPCRs are then phosphorylated at specific residues on their intracellular domains predominantly by G protein-coupled receptor kinases (GRKs) (28) but also by second-messenger activated kinases, such as protein kinase A or C (29). Upon receptor phosphorylation, β-arrestins are recruited and act as scaffolding proteins that interact with GPCRs and facilitate or prevent signaling to multiple effectors (30, 31). β-Arrestins also facilitate the internalization of receptors by promoting clathrin-mediated endocytosis (30). The two β-arrestin isoforms (β-arrestin 1 [βarr1] and β-arrestin 2 [βarr2]) are expressed ubiquitously and regulate most GPCRs. β-Arrestins may also impact ciliary signaling, as they localize to olfactory cilia (32, 33), to cilia on cultured NIH 3T3 (34) and RPE-1 (35) cells, and in mouse kidney sections (35). Furthermore, β-arrestins mediate the translocation of Smo to primary cilia (34). However, their role in the regulation of the ciliary localization of other GPCRs has not been determined.
Here, we show that localization of endogenous somatostatin receptor 3 to neuronal cilia is dynamic and agonist treatment causes a rapid decrease in Sstr3 ciliary localization. We further show that somatostatin (SST) treatment stimulates β-arrestin recruitment into Sstr3-positive cilia and this recruitment can be blocked by mutations in Sstr3 that impact agonist binding or phosphorylation. Importantly, agonist treatment fails to decrease Sstr3 ciliary localization in neurons lacking β-arrestin 2, suggesting that Sstr3 ciliary export is mediated by β-arrestin 2. These results show that β-arrestin modulates Sstr3 localization on neuronal cilia and indicate that Sstr3 signals on the ciliary membrane.
MATERIALS AND METHODS
Animals.
The wild-type (WT) mice used in this study were on an FVB background. The β-arrestin 2-knockout (KO) mouse line is on a mixed C57BL/6 and 129 SvJ background (36). All procedures were approved by the Institutional Animal Care and Use Committees at The Ohio State University (Animal Welfare Assurance number A3261-01) and The Scripps Research Institute (Animal Welfare Assurance number A4460-01).
Plasmid construction.
The coding sequence of mouse somatostatin receptor subtype 3 (21) was subcloned into the pDsRed2-N (Clontech, Mountain View, CA) and pcDNA3.1/myc-His (Life Technologies/Invitrogen, Grand Island, NY) vectors. The βarr1-green fluorescent protein (GFP) and βarr2-GFP constructs have been previously described (37, 38). Mutations in Sstr3 were generated using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The FK506 binding protein (FKBP) and FKBP-rapamycin binding domain (FRB) were provided by R. Briesewitz (The Ohio State University). To construct the Sstr3-DsRed-FRB fusion protein, a fusion PCR was performed to fuse the FRB-coding sequence to the C terminus of DsRed (forward primer, CAC CAC CTG TTC CTG ATG TGG CAT GAA GGC; reverse primer, GCC TTC ATG CCA CAT CAG GAA CAG GTG GTG). To clone the FKBP-GFP and FKBP-βarr2-GFP constructs, the restriction sites NheI and HindIII were first added via PCR to the 5′ and 3′ ends of FKBP, respectively. Next the NheI-FKBP-HindIII fragment was subcloned into a linearized pEGFP-N or pEGFP-N-βarr2 vector. This resulted in the insertion of the FKBP fragment upstream of the GFP- or βarr2-GFP-coding sequence. All DNA sequences were verified at the Nucleic Acid Shared Resource at The Ohio State University.
Cell culture and transient transfections.
Inner medullary collecting duct 3 (IMCD-3) cells (ATCC, Manassas, VA) were maintained in Dulbecco modified Eagle medium–F-12 medium supplemented with 10% fetal bovine serum, 1.2 g/liter of sodium bicarbonate, and 0.5 mM sodium pyruvate (Life Technologies/Invitrogen). Cells (n = 5 × 106) were electroporated with 10 μg DNA and plated at a high density on glass coverslips. Cells were refed 16 to 18 h after transfection and treated and harvested at 48 h after transfection. Primary hippocampal neurons were obtained from WT and βarr2-KO neonates derived from homozygous breeding. Neurons were cultured as previously described (39). Briefly, hippocampi were dissected from passage 0 mouse pups and placed in a prewarmed (37°C), sterile solution of Leibovitz's L-15 medium (L-15; Life Technologies/Invitrogen) supplemented with 0.25 mg/ml bovine serum albumin (BSA). The hippocampal tissue was freed from any extraneous tissue and cut into small pieces. This tissue was then transferred into a solution of L-15–BSA containing 0.375 mg/ml papain (Sigma-Aldrich, St. Louis, MO) and incubated for 15 min at 37°C with 95% O2–5% CO2 blowing gently over the surface of the solution. The tissue was then washed three times in prewarmed M5-5 medium (Earle's minimal essential medium with 5% fetal bovine serum, 5% horse serum, 0.4 mM GlutaMAX, 16.7 mM glucose, 5,000 U/liter penicillin, 50 mg/liter streptomycin, 2.5 mg/liter insulin, 16 nM selenite, and 1.4 mg/liter transferrin). The tissue was then triturated in M5-5 medium with a series of Pasteur pipettes of decreasing diameters. Dissociated neurons were pelleted by centrifugation at 80 × g for 5 min, resuspended in prewarmed Neurobasal-A medium containing B-27 supplement, 0.5 mM GlutaMAX, insulin-selenite-transferrin, and gentamicin (Life Technologies/Invitrogen), and plated onto poly-d-lysine (Sigma-Aldrich)-coated coverslips. Cultured neurons were transfected using Lipofectamine LTX and Plus reagent methods (Life Technologies/Invitrogen) 6 days after plating.
Drug preparations.
Somatostatin-14 (catalog number 060-03; Phoenix Pharmaceuticals, Burlingame, CA) stock solutions were prepared at 1 mM in water and stored at 4°C. Cells were treated with a final concentration of 10 μM somatostatin for 10, 20, or 40 min, depending on the experiment. Rapamycin (catalog number 9904; Cell Signaling, Danvers, MA) stock solutions were prepared at 100 μM in dimethyl sulfoxide (DMSO) and stored at −20°C. Cells were treated with a final concentration of 100 nM rapamycin for 10 min.
Processing and immunofluorescence procedures for endogenous proteins.
Seven days after plating, neuronal cultures were treated, fixed, and processed for immunofluorescence as previously described (10). Briefly, neurons were fixed with a solution of 4% (wt/vol) paraformaldehyde and 10% (wt/vol) sucrose for 10 min at room temperature, followed by a 5-min phosphate-buffered saline (PBS) wash. The neurons were then postfixed with cold methanol at −20°C for 15 min and permeabilized with 0.3% Triton X-100 in PBS with 4% donkey serum, 0.02% sodium azide, and 10 mg/ml BSA for 6 min. After permeabilization, the cells were put in a blocking solution of PBS with 4% serum, 0.02% sodium azide, and 10 mg/ml BSA for ∼1 h at room temperature. Primary antibody incubations were carried out for 16 to 24 h at 4°C, and secondary antibody incubations were carried out for 1 h at room temperature. The neurons were washed three times for 5 min each time with PBS containing 4% serum, 0.02% sodium azide, and 10 mg/ml BSA after primary and secondary antibody incubations. Primary antibodies included rabbit anti-adenylyl cyclase 3 (antibody C-20; Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-somatostatin receptor 3 (antibody M-18; Santa Cruz Biotechnology), and rabbit anti-pan-arrestin (antibody Ab2914; Abcam, Cambridge, MA). Secondary antibodies included Alexa Fluor 488-conjugated donkey anti-goat IgG, Alexa Fluor 546-conjugated donkey anti-rabbit IgG, Alexa Fluor 488-conjugated donkey anti-rabbit immunoglobulin, and Alexa Fluor 546-conjugated donkey anti-goat immunoglobulin (Life Technologies/Molecular Probes). Nucleic acids were stained with DRAQ5 (Cell Signaling). Coverslips were mounted using Immu-Mount mounting medium (Thermo Scientific, Pittsburgh, PA). All samples were imaged on a Zeiss LSM 510 laser scanning confocal microscope at the Hunt-Curtis Imaging Facility in the Department of Neuroscience at The Ohio State University. Images of multiple consecutive focal planes (z-stack), spaced at ∼0.43-μm intervals, were captured. For all collected images, the brightness and contrast of each channel were adjusted using the Zeiss LSM Image Browser program.
Quantification and statistical analysis of neuronal cilia.
Quantification of Sstr3-positive cilia was performed on 2 or 3 coverslips for each condition from 3 independent experiments. For each coverslip, at least five fields were imaged. The number of Sstr3-positive and AC3-positive cilia in each image was counted by an individual blind to the treatment conditions. The results were expressed as the percentage of AC3-positive cilia showing Sstr3 ciliary colocalization, and the results of the statistical analysis are displayed as the mean ± standard error of the mean (SEM). Quantification of endogenous β-arrestin-positive cilia was performed on two coverslips for each condition from three independent experiments. For each coverslip, at least four fields were imaged. The number of arrestin-positive and Sstr3-positive cilia in each image was counted. The percentage of Sstr3-positive cilia showing arrestin ciliary colocalization was quantified. The results are displayed as the mean ± SEM. Statistical significance was tested using Student's t test.
Processing and immunofluorescence procedures for transiently transfected cells.
IMCD cells and cultured neurons were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.3% Triton X-100 in PBS with 4% donkey serum, 0.02% sodium azide, and 10 mg/ml BSA for 10 min. To visualize the myc-tagged Sstr3 receptors, cells were labeled with anti-myc (antibody 9E10; Santa Cruz Biotechnology), followed by incubation with Alexa Fluor 546-conjugated goat anti-mouse IgG (Life Technologies/Molecular Probes). Nuclei were visualized by DRAQ5 staining. Primary and secondary antibody incubations and sample mounting and imaging were carried out as described above.
Quantification and statistical analysis of arrestin translocation to cilia.
Quantification of βarr1-GFP and βarr2-GFP ciliary localization was performed on three independent coverslips of either IMCD cells or cultured neurons. For each coverslip, 4 to 10 fields were imaged. The number of Sstr3-expressing cilia positive for βarr1-GFP or βarr2-GFP was quantified. The results are expressed as the percentage of Sstr3-positive cilia displaying βarr1 or βarr2 ciliary colocalization. The data are expressed as the mean ± SEM.
Live cell imaging.
Hippocampal neurons were plated onto 35-mm glass-bottomed (no. 1.5) dishes (MatTek, Ashland, MA) that had been coated with poly-d-lysine (Sigma-Aldrich) and transfected at 6 days postplating. Neurons were processed for live cell imaging at 24 h posttransfection. Prior to live cell imaging, neuronal medium was replaced with Hibernate A low-fluorescence medium (BrainBits, Springfield, IL) supplemented with B-27 supplement, 0.5 mM GlutaMAX, and insulin-selenite-transferrin to allow manipulation and survival of neurons at ambient CO2 levels. Neurons expressing Sstr3-DsRed and βarr1-GFP or βarr2-GFP were selected for imaging. Live cell imaging was performed on 2 to 3 cells for each condition from 3 independent experiments The cells were observed with an Andor Revolution WD spinning disk confocal imaging system (Andor Technology plc, Belfast, Northern Ireland) controlled by MetaMorph software (Molecular Devices, Sunnyvale, CA) and equipped with a Nikon TiE inverted microscope (Nikon Instruments, Melville, NY), an ASI XY piezo-Z motorized stage (Applied Scientific Instrumentation, Eugene, OR), a Yokogawa CSU-W1 confocal scanning unit (Yokogawa Electric Corporation, Tokyo, Japan), and solid-state lasers. z-stacks of the red and green fluorescence were acquired sequentially at 30-s intervals for 10 min using a 0.2-μm step size, 200-ms exposures, an Andor iXon Ultra 897 back-illuminated electron-multiplying charge-coupled-device camera, and a 100× PlanApo VC oil immersion objective (numerical aperture, 1.4). Somatostatin (20 μl of a 1 mM stock solution) was added directly to the 2 ml of medium in the dish with a pipette immediately after the first time point (time zero). Focus was maintained using the Perfect Focus system on the Nikon microscope. The GFP and DsRed were excited with the 488-nm and 561-nm laser lines, respectively. The temperature and humidity were controlled using an OkoLab Bold Line stage-top incubator.
RESULTS
Ciliary localization of somatostatin receptor subtype 3 is dynamic.
To test whether Sstr3 localization to cilia is affected by agonist treatment, we generated primary cultures of wild-type (WT) mouse hippocampal neurons, which possess abundant Sstr3-positive cilia (40, 41). The neurons were then treated with somatostatin (SST) for 0, 20, or 40 min, fixed, and colabeled with antibodies to Sstr3 and AC3, which is an established marker for neuronal cilia (26). The percentages of Sstr3-positive cilia were then quantified (Fig. 1A to C). In untreated cultures, 49.5% of AC3-positive cilia were positive for Sstr3, which is consistent with previous results (41). However, the percentage of Sstr3-positive cilia was significantly less after SST treatment in a time-dependent process. After 20 and 40 min of SST treatment, the percentages of AC3-positive cilia that were also positive for Sstr3 were 34% and 22.8%, respectively (Fig. 1C). This result demonstrates that Sstr3 ciliary localization, like D1 ciliary localization (22), is dynamic and suggests that Sstr3 is exported from cilia in response to agonist-mediated activation.
FIG 1.

Somatostatin treatment causes a decrease in endogenous Sstr3 ciliary localization and localization of β-arrestin to neuronal cilia. (A and B) Representative images of hippocampal neurons from WT mice, after 7 days in culture, labeled with antibodies to Sstr3 (green) and AC3 (red). Neurons were either untreated (UT) (A) or treated with 10 μM SST for 40 min (B). SST treatment causes a reduction in Sstr3-positive cilia. (Insets) Increased magnification of individual channels in the boxed regions to aid visualization. Bars, 10 μm. (C) Percentages of AC3-positive cilia in WT cultures (n = 3) that were positive for Sstr3. Sstr3 localized to 49.5% ± 4.6% of AC3-positive cilia (n = 130), 34% ± 5.8% of AC3-positive cilia (n = 144), and 22.8% ± 8.3% of AC3-positive cilia (n = 146) in cultures treated with 10 μM SST for 0, 20, and 40 min, respectively. The percentage of Sstr3-positive cilia was significantly decreased after 40 min of SST treatment. Values are expressed as the mean ± SEM. *, significantly different results (P < 0.05). (D to I) Representative images of hippocampal neurons from WT mice, after 7 days in culture, labeled with antibodies to βarr1/2 (green) and Sstr3 (red). Neurons were treated with vehicle (Veh) (D to F) or 10 μM SST (G to I) for 20 min. β-Arrestin was not detected within cilia in vehicle-treated neurons but colocalized with Sstr3 throughout the cilium after SST treatment. (Insets) Higher-magnification images of the cilia. Nuclei were stained with DRAQ5. Bars, 10 μm (main images) and 5 μm (insets). (J) Percentages of Sstr3-positive cilia in WT cultures (n = 3) that were positive for βarr1/2. βarr1/2 localized to 0% of Sstr3-positive cilia (n = 73) in vehicle-treated cultures and 55.6% ± 4.1% of Sstr3-positive cilia (n = 127) in cultures treated with SST for 20 min. Values are expressed as the mean ± SEM.
Endogenous β-arrestin localizes to neuronal cilia in response to somatostatin treatment.
Since β-arrestins mediate Sstr3 internalization from the plasma membrane (42), we considered β-arrestins to be possible mediators of Sstr3 export from cilia. To begin to address this possibility, we examined the subcellular localization of endogenous β-arrestins in WT mouse hippocampal neurons treated with vehicle or SST for 20 min, which is a time point when Sstr3 ciliary localization is decreasing. Neurons were fixed and colabeled with an antibody to Sstr3 and an antibody that recognizes both β-arrestin 1 (βarr1) and β-arrestin 2 (βarr2). In vehicle-treated neurons, βarr1/2 localized throughout the cell body and nucleus but were not detected within cilia, as indicated by a lack of colocalization with Sstr3 (Fig. 1D to F and J). Strikingly, upon SST treatment βarr1/2 showed colocalization in approximately half of Sstr3-positive cilia (Fig. 1G to J). These results show that endogenous β-arrestin localizes to neuronal primary cilia in response to SST treatment. The presence of β-arrestin in cilia on hippocampal neurons suggests a novel role for β-arrestins in neuronal ciliary signaling.
β-Arrestin 2 selectively localizes to cilia in response to somatostatin treatment.
To facilitate the investigation of agonist-mediated β-arrestin ciliary localization, we developed a model system utilizing the ciliated inner medullary collecting duct (IMCD) cell line. IMCD cells are commonly used as a model for studying cilia, and we have previously utilized them to investigate the mechanisms of GPCR ciliary localization (21, 22). IMCD cells were transiently transfected with expression constructs encoding Sstr3 with a C-terminal myc epitope tag and βarr1 or βarr2 fused at the C terminus to green fluorescent protein (GFP) (37, 38). At 48 h after transfection, the cells were treated with vehicle or SST for 20 min, fixed, and labeled with an antibody to myc. As previously reported (21), heterologously expressed Sstr3 was targeted to cilia on IMCD cells (Fig. 2B, E, H, and K). In vehicle-treated cells, βarr1-GFP appeared to localize throughout the cytoplasm and the nucleus (Fig. 2A to C), whereas βarr2-GFP appeared to localize mainly throughout the cytoplasm and at the base of the cilium (Fig. 2G to I), consistent with the findings of a previous study showing that β-arrestin 2 is enriched in centrioles at the base of cilia in RPE-1 cells (35). Importantly, in vehicle-treated cells, βarr1-GFP and βarr2-GFP were not detected in the ciliary compartment (Fig. 2M). Strikingly, upon SST treatment βarr2-GFP showed robust ciliary localization (Fig. 2J to M), whereas βarr1-GFP failed to show ciliary localization (Fig. 2D to F and M). These findings suggest that β-arrestin 2, but not β-arrestin 1, localizes to cilia in response to Sstr3 activation.
FIG 2.

Somatostatin-mediated ciliary localization of β-arrestin 2 in IMCD cells. (A to L) Representative images of IMCD cells transfected with βarr1 fused to GFP (βarr1-GFP; green) (A to F) or βarr2 fused to GFP (βarr2-GFP; green) and myc-tagged Sstr3 (Sstr3-myc; red) (G to L). Cells were treated with vehicle (Veh) (A to C and G to I) or 10 μM SST (D to F and J to L) for 20 min, fixed, and labeled with an antibody to myc. Sstr3-myc is targeted to cilia. βarr1-GFP was not detected in cilia in vehicle- or SST-treated cells. βarr2-GFP was not detected in cilia in vehicle-treated cells but localized to cilia in SST-treated cells. (Insets) Side views of the cilia (the insets in panels A to C correspond to the dashed boxes). Nuclei were stained with DRAQ5. Bars, 10 μm. (M) Percentages of Sstr3-expressing IMCD cell cilia that were positive for βarr1-GFP or βarr2-GFP after 20 min of vehicle or 10 μM SST treatment. In vehicle-treated cultures, βarr1-GFP localized to 0% of Sstr3-positive cilia (n = 43) and βarr2-GFP localized to 0% of Sstr3-positive cilia (n = 52). In SST-treated cultures, βarr1-GFP localized to 0% of Sstr3-positive cilia (n = 51) and βarr2-GFP localized to 87.2% ± 3.4% of Sstr3-positive cilia (n = 54). Values are expressed as the mean ± SEM.
To verify and extend these results, we assessed β-arrestin ciliary localization in cultured neurons. WT mouse hippocampal neurons were transiently transfected with myc-tagged Sstr3 and either βarr1-GFP or βarr2-GFP. At 24 h posttransfection, the neurons were treated with vehicle or SST for 20 min and the percentage of Sstr3-expressing cilia positive for βarr1-GFP or βarr2-GFP was quantified. Heterologously expressed Sstr3 was targeted to neuronal cilia (Fig. 3B, E, H, and K), and in vehicle-treated neurons, neither βarr1-GFP nor βarr2-GFP was detected in the ciliary compartment (Fig. 3A to C and G to I). Upon SST treatment, however, βarr2-GFP localized to all Sstr3-positive cilia (Fig. 3J to M), whereas βarr1-GFP failed to show ciliary localization (Fig. 3D to F and M). These results indicate that βarr2-GFP selectively enters neuronal cilia upon SST treatment.
FIG 3.

Somatostatin-mediated localization of β-arrestin 2 to neuronal cilia. (A to L) Representative images of hippocampal neurons from WT mice, after 7 days in culture, transfected with βarr1 fused to GFP (βarr1-GFP; green) (A to F) or βarr2 fused to GFP (βarr2-GFP; green) and myc-tagged Sstr3 (Sstr3-myc; red) (G to L). Neurons were treated with vehicle (Veh) (A to C and G to I) or 10 μM SST (D to F and J to L) for 20 min, fixed, and labeled with an antibody to myc. Sstr3-myc is targeted to cilia. βarr1-GFP was not detected in cilia in vehicle- or SST-treated neurons. βarr2-GFP localized to cilia in SST-treated neurons. (Insets) Higher-magnification images of the cilia. Nuclei were stained with DRAQ5. Bars, 10 μm (main images) and 5 μm (insets). (M) Percentages of Sstr3-positive neuronal cilia that were positive for βarr1-GFP or βarr2-GFP after 20 min of vehicle or 10 μM SST treatment. In vehicle-treated cultures. βarr1-GFP localized to 0% of Sstr3-positive cilia (n = 25) and βarr2-GFP localized to 0% of Sstr3-positive cilia (n = 25). In SST-treated cultures βarr1-GFP localized to 0% of Sstr3-positive cilia (n = 25) and βarr2-GFP localized to 100% of Sstr3-positive cilia (n = 25).
In order to examine the dynamics of βarr2-GFP ciliary entry, we performed live cell imaging on WT mouse hippocampal neurons transiently transfected with Sstr3 fused at the C terminus to DsRed and βarr1-GFP or βarr2-GFP. Prior to treatment, Sstr3-DsRed was targeted to the cilium and βarr1-GFP and βarr2-GFP were not detected in the cilium (Fig. 4). βarr1-GFP and βarr2-GFP localized throughout the cytoplasm, and as noted in transfected IMCD cells, βarr2-GFP consistently demonstrated localization at the base of the cilium (Fig. 4B). Consistent with our results in fixed neurons, after SST treatment βarr1-GFP failed to show ciliary localization (Fig. 4A; see also Movie S1 in the supplemental material). βarr2-GFP, on the other hand, rapidly accumulated in the ciliary compartment in a time-dependent manner (Fig. 4B; see also Movies S2 and S3 in the supplemental material). βarr2-GFP ciliary localization was apparent by 4 min after SST treatment and robustly localized along the cilium at 10 min after treatment. A previous study of Sstr3 ciliary localization indicates that it takes at least 10 min for newly synthesized Sstr3-GFP to be trafficked to cilia (43). Thus, our results are consistent with a model whereby agonist treatment promotes the entry of β-arrestin 2 into the ciliary compartment, where it associates with Sstr3.
FIG 4.

Live cell confocal imaging of somatostatin-mediated β-arrestin 2 ciliary localization. Each image is a maximum-intensity projection of a z-stack that encompassed the entire cell body and cilium. (A) Representative images of a WT hippocampal neuron after 7 days in culture transfected with βarr1 fused to GFP (βarr1-GFP; green) and Sstr3 fused to DsRed (Sstr3-DsRed; red). (Top) The green channel; (bottom) both the green and red channels. The elapsed time (t; in minutes) of 10 μM SST treatment is indicated in the upper right corner of each panel. Sstr3-DsRed was targeted to the cilium, but βarr1-GFP was not detected in cilia before (time zero [t0]) or after 10 min (t10) of SST treatment. (B) Representative images of a WT hippocampal neuron after 7 days in a culture transfected with βarr2 fused to GFP (βarr2-GFP; green) and Sstr3-DsRed (red). Panels and labels are the same as those described in the legend to panel A. Before SST treatment (time zero), βarr2-GFP was enriched at the base of the cilium but was not detected within the cilium. βarr2-GFP ciliary localization was apparent after 4 min (t4) of SST treatment and was robust after 10 min (t10). Bars, 10 μm.
Somatostatin-induced β-arrestin 2 ciliary localization requires agonist binding and, possibly, phosphorylation of Sstr3.
To begin to test the molecular basis of SST-induced β-arrestin 2 ciliary localization, we leveraged our IMCD cell line model along with previous knowledge about the mechanisms of agonist binding and Sstr3 internalization from the plasma membrane. To confirm that SST-induced βarr2-GFP ciliary localization was mediated by Sstr3, we introduced into Sstr3 a point mutation (D124E) that prevents ligand binding (44). IMCD cells were then transiently transfected with βarr2-GFP and myc-tagged WT Sstr3 or Sstr3(D124E) and treated with vehicle or SST for 20 min. Importantly, Sstr3(D124E) failed to mediate βarr2-GFP ciliary localization upon SST treatment (Fig. 5A), indicating that agonist binding to Sstr3 is required for βarr2-GFP ciliary localization.
FIG 5.

Mutation of agonist binding and phosphorylation sites in Sstr3 abolishes β-arrestin 2 ciliary localization in IMCD cells and neurons. (A) IMCD cells were transfected with βarr2 fused to GFP and myc-tagged WT Sstr3 or Sstr3(D124E). Cells were treated with vehicle (Veh) or 10 μM SST for 20 min, fixed, and labeled with an antibody to myc, and βarr2-GFP ciliary localization was quantified. In vehicle-treated cultures, βarr2-GFP localized to 1.2% ± 1.1% of WT (n = 76) and 0% of Sstr3(D124E)-positive cilia (n = 85). In SST-treated cultures βarr2-GFP localized to 92.4% ± 1.7% of WT (n = 85) and 1% ± 1% of Sstr3(D124E)-positive cilia (n = 106). (B) IMCD cells were transfected with βarr2 fused to GFP and myc-tagged WT or mutant Sstr3. Treatments and processing were the same as those described in the legend to panel A. In vehicle-treated cultures, βarr2-GFP localized to 0% of WT or mutant Sstr3-positive cilia (n = 31 to 49). In SST-treated cultures, βarr2-GFP localized to 90% ± 5.1% of WT Sstr3-positive cilia (n = 37), 79.2% ± 4.2% of cilia positive for the Sstr3 triple serine mutant (n = 24), 0% of cilia positive for the Sstr3 quadruple mutant (n = 29), and 0% of cilia positive for the Sstr3(T357A) mutant (n = 25). (C to F) Representative images of hippocampal neurons from WT mice after 7 days in a culture transfected with βarr2 fused to GFP (βarr2-GFP; green) and WT Sstr3 (red) (C and D) or Sstr3(T357A) (red) (E and F). Neurons were treated with vehicle (Veh) (C and E) or 10 μM SST (D and F) for 20 min and then fixed. In vehicle-treated cells, βarr2-GFP was not detected within the cilium. In SST-treated cells, βarr2-GFP localized to WT Sstr3-positive cilia but not Sstr3(T357A)-positive cilia. (Insets) Higher-magnification images of the cilia. Nuclei were stained with DRAQ5. Bars, 10 μm (main images) and 5 μm (insets). (G) Percentages of WT and mutant Sstr3-positive neuronal cilia that were positive for βarr2-GFP (n = 20 to 30 cilia for each condition and construct). In cultures treated with vehicle for 20 min, βarr2-GFP localized to 0% of WT or mutant Sstr3-positive cilia. In cultures treated with SST for 20 min, βarr2-GFP localized to 100% of WT Sstr3-positive cilia and 0% of Sstr3(T357A)-positive cilia. Values are expressed as the mean ± SEM.
Four sites (S341, S346, S351, and T357) in the C-terminal tail of Sstr3 are implicated in receptor phosphorylation, desensitization, and internalization (45). To test whether these sites are important for βarr2-GFP ciliary localization, we generated myc-tagged Sstr3 constructs containing mutations at these critical residues and evaluated the ability of the mutants to mediate agonist-dependent ciliary localization of βarr2-GFP in IMCD cells. We first generated a mutant in which all three serine residues were mutated to alanines (triple serine mutant). Mutation of these three residues disrupts basal and SST-mediated phosphorylation of the receptor and blocks receptor internalization (45). However, upon SST treatment the triple serine mutant mediated ciliary localization of βarr2-GFP as effectively as WT Sstr3 (Fig. 5B). We then tested a mutant in which all four residues were mutated to alanines (quadruple mutant). Interestingly, upon SST treatment the quadruple mutant failed to mediate βarr2-GFP ciliary localization (Fig. 5B). We then tested a mutant in which only the threonine was mutated to alanine (T357A). Remarkably, this single mutant failed to mediate βarr2-GFP ciliary localization (Fig. 5B). This suggests that phosphorylation of T357 is required for βarr2-GFP ciliary localization. We then confirmed this result in cultured neurons by transiently transfecting WT mouse hippocampal neurons with βarr2-GFP and WT Sstr3 or Sstr3(T357A) fused to DsRed. At 24 h posttransfection, the neurons were treated with vehicle or SST for 20 min and the percentage of WT or mutant Sstr3-expressing cilia positive for βarr2-GFP was quantified. Similarly to IMCD cells, in response to SST treatment βarr2-GFP localized to cilia expressing WT Sstr3 (Fig. 5C and D and G) but not to cilia expressing Sstr3(T357A) (Fig. 5E to G). Together, these results suggest that SST binds to Sstr3 on the ciliary membrane and may lead to the phosphorylation of residue T357, which facilitates β-arrestin 2 ciliary localization.
β-Arrestin 2 is recruited into cilia in response to somatostatin treatment.
Recent studies indicate that primary cilia possess a size-dependent diffusion barrier that restricts the entry of soluble proteins (43, 46, 47). We did not detect β-arrestin 2 within cilia in the absence of SST treatment, suggesting that β-arrestin 2 is normally excluded from cilia but enters the cilium upon receptor activation. An alternative possibility is that β-arrestin 2 can diffuse into cilia but is normally present at a level too low to be detected. Upon SST treatment, however, the receptor becomes phosphorylated, which increases the affinity for β-arrestin 2 and causes its retention and accumulation within cilia. To distinguish between these two possibilities, we utilized an inducible dimerization system consisting of the chemical dimerizer rapamycin and the protein moieties FK506 binding protein (FKBP) and FKBP-rapamycin binding domain (FRB) (48). These moieties can be fused to proteins and then mediate dimerization in response to rapamycin. This well-characterized system has recently been used to induce dimerization between ciliary receptors and fluorescently tagged proteins within the ciliary compartment (43, 47) (Fig. 6A). To apply this system to our experimental paradigm, we generated expression constructs containing FRB fused to the C terminus of Sstr3-DsRed and FKBP fused to the N terminus of either GFP or βarr2-GFP. IMCD cells were then transiently transfected with Sstr3-DsRed-FRB and FKBP-GFP or FKBP-βarr2-GFP and treated with vehicle, rapamycin, or SST for 10 min. This time point was chosen to minimize the contribution from the FKBP-GFP or FKBP-βarr2-GFP that entered cilia in a complex with newly synthesized Sstr3-DsRed-FRB. After treatment, the cells were fixed and the percentage of Sstr3-DsRed-FRB-expressing cilia positive for FKBP-GFP or FKBP-βarr2-GFP was quantified (Fig. 6B; see also Fig. S1 in the supplemental material). In vehicle-treated cells, FKBP-GFP and FKBP-βarr2-GFP were not detected in cilia. However, in response to rapamycin treatment, FKBP-GFP was detected in 71.6% of Sstr3-DsRed-FRB-expressing cilia. This is consistent with the findings of previous studies (43, 47) and confirms that FKBP-GFP diffuses into IMCD cell cilia and can be captured through dimerization with FRB fused to Sstr3. FKBP-βarr2-GFP was also detected in Sstr3-DsRed-FRB-expressing cilia after rapamycin treatment, although the percentage (24.8%) was much less than that of FKBP-GFP. This result indicates that FKBP-βarr2-GFP is not excluded from the cilium and suggests that it can diffuse into the cilium but is present at a lower level than FKBP-GFP. Interestingly, after SST treatment, FKBP-βarr2-GFP was detected in 87.8% of Sstr3-DsRed-FRB-expressing cilia (Fig. 6B). As SST treatment causes a much greater increase in the ciliary localization of FKBP-βarr2-GFP than rapamycin treatment, this suggests that FKBP-βarr2-GFP is actively recruited into cilia in response to SST treatment.
FIG 6.

The ciliary localization of FKBP-βarr2-GFP is greater in response to somatostatin treatment than rapamycin treatment. (A) Schematic of rapamycin-induced dimerization in the ciliary compartment. FKBP-GFP diffuses into and out of the cilium. In the absence of rapamycin, the level of FKBP-GFP in the cilium is low. In the presence of rapamycin, FKBP dimerizes with FRB fused to Sstr3, which is enriched in the ciliary membrane, thereby trapping FKBP-GFP in the ciliary compartment. (B) IMCD cells were transfected with Sstr3 fused to DsRed and FRB (Sstr3-DsRed-FRB) and FKBP fused to GFP (FKBP-GFP) or βarr2-GFP (FKBP-βarr2-GFP). Cells were treated with DMSO (vehicle [Veh]), 100 nM rapamycin (Rap), or 10 μM SST for 10 min and fixed, and the GFP ciliary localization was quantified. In DMSO-treated cultures, FKBP-GFP and FKBP-βarr2-GFP localized to 0% of Sstr3-positive cilia (n = 33 to 38). In rapamycin-treated cultures, FKBP-GFP localized to 71.6% ± 4.8% of Sstr3-positive cilia (n = 41) and FKBP-βarr2-GFP localized to 24.8% ± 5.3% of Sstr3-positive cilia (n = 44). In SST-treated cultures, FKBP-GFP localized to 3.7% ± 3.7% of Sstr3-positive cilia (n = 33) and FKBP-βarr2-GFP localized to 87.8% ± 4% of Sstr3-positive cilia (n = 45). The percentage of FKBP-βarr2-GFP ciliary colocalization was significantly greater after SST treatment than rapamycin treatment. Values are expressed as the mean ± SEM. ***, significantly different results (P < 0.001).
The somatostatin-mediated decrease in Sstr3 ciliary localization requires β-arrestin 2.
Recruitment of β-arrestin 2 into cilia upon SST treatment may have many functional consequences, including regulation of Sstr3 signaling and internalization. To begin to explore the potential functional consequences, we assessed whether β-arrestin 2 is required for the dynamic localization of Sstr3. Hippocampal neurons were generated from WT and β-arrestin 2-knockout (KO) mice and treated with vehicle or SST for 40 min. Neurons were then fixed and colabeled with anti-Sstr3 and anti-AC3, and the percentages of Sstr3-positive cilia were quantified (see Fig. S2 in the supplemental material). In WT cultures, 60.4% of AC3-positive cilia were positive for Sstr3 after vehicle treatment, and, as expected, the percentage of Sstr3-positive cilia decreased significantly to 41.6% after 40 min of SST treatment (Fig. 7). In β-arrestin 2-KO cultures, 50.8% of AC3-positive cilia were positive for Sstr3 after vehicle treatment, indicating that βarr2 is not required for Sstr3 ciliary localization. However, there was no reduction in the percentage of Sstr3-positive cilia (53.1%) after 40 min of SST treatment (Fig. 7). These findings indicate that the agonist-mediated dynamic ciliary localization of Sstr3 is dependent on β-arrestin 2 and suggest that β-arrestin 2 mediates Sstr3 export from cilia.
FIG 7.
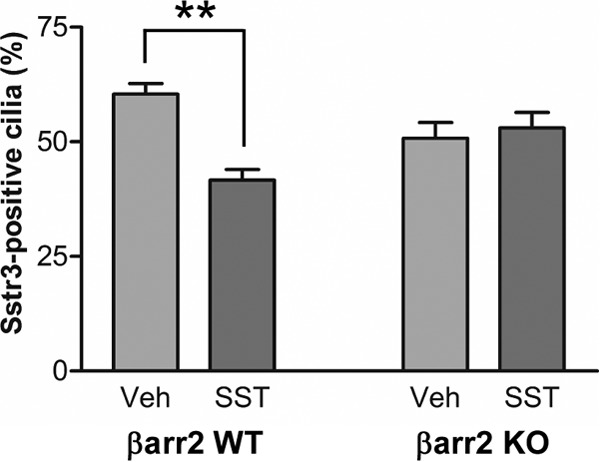
Agonist-mediated decreases in endogenous Sstr3 neuronal ciliary localization require βarr2. Hippocampal neurons from WT and βarr2-KO mice after 7 days in culture were treated with vehicle (Veh) or 10 μM SST for 40 min, fixed, and labeled with antibodies to Sstr3 and AC3 (n = 3 experiments per genotype). In vehicle- and SST-treated WT cultures, Sstr3 localized to 60.4% ± 2.4% of AC3-positive cilia (n = 369) and 41.6% ± 2.3% of AC3-positive cilia (n = 388), respectively. In vehicle- and SST-treated βarr2-KO cultures, Sstr3 localized to 50.8% ± 3.4% of AC3-positive cilia (n = 333) and 53% ± 3.4% of AC3-positive cilia (n = 324), respectively. The percentage of Sstr3-positive cilia was significantly decreased after 40 min of SST treatment in WT neuronal cultures but did not change in βarr2-KO neuronal cultures. The percentages of Sstr3-positive cilia in vehicle-treated WT and βarr2-KO neuronal cultures were not significantly different. Values are expressed as the mean ± SEM. **, significantly different results (P < 0.005).
DISCUSSION
We have found that the SST-mediated activation of Sstr3 induces the ciliary localization of β-arrestin, thereby implicating β-arrestin in neuronal ciliary signaling. Based on the roles of β-arrestins in GPCR signaling on the plasma membrane, we can postulate several functional consequences of β-arrestin ciliary recruitment. First, β-arrestin may desensitize Sstr3 by uncoupling the receptor from a G protein. However, it is not known whether G proteins localize to cilia on hippocampal neurons and couple to ciliary GPCRs. Second, β-arrestins may induce GPCR signaling in the primary cilium. β-Arrestins function as scaffolds for numerous intracellular signaling proteins, including Src, extracellular signal-regulated kinases 1 and 2, p38, AKT, and phosphatidylinositol 3-kinase (49), and have the capability of signaling independently of G-protein coupling. Thus, β-arrestins may act as direct signal transducers of Sstr3 on the ciliary membrane through diverse signaling cascades. Third, β-arrestins can also act as adaptors for components of clathrin-dependent endocytosis machinery and promote receptor internalization via clathrin-coated pits. At the base of primary cilia there is an invagination of the plasma membrane that forms a specific membrane domain, called the ciliary pocket (50, 51). Recent studies have revealed that the ciliary pocket is enriched in active and dynamic clathrin-coated pits (52, 53). Our finding that the SST-mediated decrease in Sstr3 ciliary localization is disrupted in β-arrestin 2-KO neurons is consistent with a role for β-arrestin 2 in promoting clathrin-mediated endocytosis of Sstr3 at the ciliary pocket. Although receptor internalization is normally associated with the attenuation of signaling, in some cases GPCR signaling can be sustained or even enhanced upon internalization into endosomes (54, 55). Recent studies show that D1 receptors mediate rapid G protein- and adenylyl cyclase-mediated signaling from endocytic vesicles (56), and, using conformational biosensors, GPCRs have been shown to engage the heterotrimeric G-protein complex once it is internalized into the vesicles (57). It is attractive to speculate that the interaction between Sstr3 and β-arrestin 2 may serve to orient the receptor with its signaling partners. Intriguingly, a recent study demonstrated that proper transforming growth factor β (TGF-β) signaling requires clathrin-dependent endocytosis of TGF-β receptors at the ciliary pocket (58). Thus, it is possible that Sstr3 export from cilia is a mechanism for increasing Sstr3 signaling. Overall, β-arrestin potentially impacts Sstr3 signaling in cilia through numerous mechanisms.
An interesting result from our study is that in the absence of SST treatment, heterologously expressed βarr1-GFP and βarr2-GFP appear to be excluded from cilia on either IMCD cells or neurons. However, we did detect FKBP-βarr2-GFP in cilia after rapamycin-induced dimerization with Sstr3-DsRed-FRB, indicating that FKBP-βarr2-GFP can passively diffuse into the ciliary compartment. A potential explanation to reconcile these results is that under basal conditions the amount of protein within cilia is too low to be detected, presumably due to molecular crowding effects within the cilium that limit the volume accessible to βarr1-GFP or βarr2-GFP. Such a mechanism has been proposed to explain the apparent exclusion of visual arrestin from dark-adapted rod photoreceptor outer segments (59). It is only upon trapping and accumulation in the ciliary compartment through dimerization or binding to an activated receptor that β-arrestin levels become high enough to be detected. Interestingly, a previous study found constitutive ciliary localization of Cherry-tagged β-arrestin 2 in RPE-1 cells (35). One possible explanation for this difference is that the accessible volume in cilia on RPE-1 cells is greater than that in cilia on IMCD cells.
A striking result from our dimerization experiments was that SST treatment resulted in a much larger increase in FKBP-βarr2-GFP ciliary localization than rapamycin treatment. This suggests that SST treatment not only allows Sstr3-DsRed-FRB to capture freely diffusing FKBP-βarr2-GFP but also stimulates entry of FKBP-βarr2-GFP into the cilium. Thus, it is likely that activation of Sstr3 generates an undefined signal within the ciliary compartment that is transmitted to the base of the cilium and stimulates βarr2-GFP transport into cilia. Although the mechanism mediating βarr2 transport into cilia was not directly tested in this study, a likely candidate is intraflagellar transport (IFT). IFT is a bidirectional transport process within cilia that is required for the formation, maintenance, and functioning of all cilia (60). IFT consists of the transport of protein complexes, or particles, along the ciliary microtubule core by the molecular motors kinesin-II (anterograde) and dynein (retrograde). β-Arrestins have been shown to interact with the kinesin-II subunit Kif3a and mediate the translocation of Smoothened to primary cilia (34). One possibility is that Sstr3 signaling induces an interaction between β-arrestin 2 and Kif3a at the base of the cilium, which facilitates β-arrestin 2 transport into the cilium. Our live cell studies show that β-arrestin 2 rapidly becomes distributed along the length of the cilium. This progression may be the result of IFT-mediated transport along the microtubules or the lateral diffusion of Sstr3 in the ciliary membrane that carries along bound β-arrestin 2. Ciliary receptors, such as Sstr3, demonstrate rapid diffusion along the entire ciliary membrane (61), which can drive the progression of bound soluble proteins from the proximal region to the distal tip of the cilium (43). Thus, β-arrestin 2 movement within the ciliary compartment may be due to active transport, passive diffusion, or a combination of both.
Interestingly, in both IMCD cells and neurons, SST treatment resulted in the ciliary localization of βarr2-GFP but not βarr1-GFP. This is surprising, considering that the results of previous studies with HEK cells demonstrated that the SST-induced activation of Sstr3 results in the rapid redistribution of both βarr1-GFP and βarr2-GFP from the cytoplasm to the plasma membrane, although βarr2-GFP showed a more marked redistribution (62). One potential explanation is that βarr1-GFP cannot access the ciliary compartment as freely as βarr2-GFP. Alternatively, βarr1-GFP may not bind to the Sstr3 that is activated on the ciliary membrane. We also found that mutation of a single putative phosphorylation site (T357) in Sstr3 was sufficient to disrupt βarr2-GFP ciliary localization. Mutation of this residue has previously been shown to reduce basal and SST-mediated phosphorylation of the receptor and block internalization (45). This suggests that upon SST treatment the Sstr3 on the ciliary membrane is phosphorylated by a kinase in the ciliary compartment. Further studies will be required to determine the identity of this kinase.
As β-arrestins regulate signaling and the internalization of most GPCRs, it is likely that β-arrestins are recruited into cilia in response to activation of other GPCRs. Indeed, we have found that agonist treatment of IMCD cells expressing Mchr1 or D1 induces βarr2-GFP ciliary localization (J. A. Green and K. Mykytyn, unpublished results). Further studies will be required to determine whether activation of these and other ciliary GPCRs mediates recruitment of β-arrestins into neuronal cilia and whether β-arrestins mediate their ciliary export.
In summary, our data are consistent with a model whereby agonist binding to Sstr3 leads to the phosphorylation of T357 and a signal that stimulates β-arrestin 2 recruitment into the cilium. In the cilium, β-arrestin 2 binds to the phosphorylated receptor, potentially inhibiting and/or stimulating signaling, and mediates the export of Sstr3 from the cilium. These results provide direct evidence for receptor signaling on neuronal cilia, implicate β-arrestins in the regulation of ciliary GPCR signaling, and suggest a further link between β-arrestins and ciliopathies. Understanding the molecular mechanisms that regulate GPCR ciliary localization and signaling is critical for providing novel strategies to manage or treat the disorders associated with ciliopathies.
Supplementary Material
Funding Statement
The funders had no role in the study design, the data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00765-15.
REFERENCES
- 1.Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK. 2009. The primary cilium as a complex signaling center. Curr Biol 19:R526–R535. doi: 10.1016/j.cub.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green JA, Mykytyn K. 2010. Neuronal ciliary signaling in homeostasis and disease. Cell Mol Life Sci 67:3287–3297. doi: 10.1007/s00018-010-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satir P, Pedersen LB, Christensen ST. 2010. The primary cilium at a glance. J Cell Sci 123:499–503. doi: 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goetz SC, Anderson KV. 2010. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lienkamp S, Ganner A, Walz G. 2012. Inversin, Wnt signaling and primary cilia. Differentiation 83:S49–S55. doi: 10.1016/j.diff.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Schou KB, Pedersen LB, Christensen ST. 2015. Ins and outs of GPCR signaling in primary cilia. EMBO Rep 16:1099–1113. doi: 10.15252/embr.201540530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hildebrandt F, Benzing T, Katsanis N. 2011. Ciliopathies. N Engl J Med 364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Gonzalo FR, Reiter JF. 2012. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol 197:697–709. doi: 10.1083/jcb.201111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nachury MV, Seeley ES, Jin H. 2010. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol 26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. 2008. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A 105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF. 2011. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, Martens JR. 2007. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci U S A 104:15917–15922. doi: 10.1073/pnas.0704140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. 2005. Vertebrate Smoothened functions at the primary cilium. Nature 437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 14.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. 2005. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein Polaris for processing and function. PLoS Genet 1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Kato M, Beachy PA. 2009. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A 106:21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohatgi R, Milenkovic L, Scott MP. 2007. Patched1 regulates Hedgehog signaling at the primary cilium. Science 317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 17.Najafi M, Calvert PD. 2012. Transport and localization of signaling proteins in ciliated cells. Vision Res 75:11–18. doi: 10.1016/j.visres.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Hollt V. 1999. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 89:909–926. doi: 10.1016/S0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- 19.Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D. 2000. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res 872:271–275. doi: 10.1016/S0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- 20.Hamon M, Doucet E, Lefevre K, Miquel MC, Lanfumey L, Insausti R, Frechilla D, Del Rio J, Verge D. 1999. Antibodies and antisense oligonucleotide for probing the distribution and putative functions of central 5-HT6 receptors. Neuropsychopharmacology 21:68S–76S. doi: 10.1038/sj.npp.1395368. [DOI] [PubMed] [Google Scholar]
- 21.Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K. 2008. Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 19:1540–1547. doi: 10.1091/mbc.E07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, Mykytyn K. 2011. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci 68:2951–2960. doi: 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soetedjo L, Glover DA, Jin H. 2013. Targeting of vasoactive intestinal peptide receptor 2, VPAC2, a secretin family G-protein coupled receptor, to primary cilia. Biol Open 2:686–694. doi: 10.1242/bio.20134747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Loktev AV, Jackson PK. 2013. Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep 5:1316–1329. doi: 10.1016/j.celrep.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Koemeter-Cox AI, Sherwood TW, Green JA, Steiner RA, Berbari NF, Yoder BK, Kauffman AS, Monsma PC, Brown A, Askwith CC, Mykytyn K. 2014. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A 111:10335–10340. doi: 10.1073/pnas.1403286111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop GA, Berbari NF, Lewis JS, Mykytyn K. 2007. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J Comp Neurol 505:562–571. doi: 10.1002/cne.21510. [DOI] [PubMed] [Google Scholar]
- 27.Hanyaloglu AC, von Zastrow M. 2008. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol 48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 28.Marchese A, Paing MM, Temple BR, Trejo J. 2008. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol 48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly E, Bailey CP, Henderson G. 2008. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol 153(Suppl 1):S379–S388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shenoy SK, Lefkowitz RJ. 2011. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shukla AK, Xiao K, Lefkowitz RJ. 2011. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawson TM, Arriza JL, Jaworsky DE, Borisy FF, Attramadal H, Lefkowitz RJ, Ronnett GV. 1993. Beta-adrenergic receptor kinase-2 and beta-arrestin-2 as mediators of odorant-induced desensitization. Science 259:825–829. doi: 10.1126/science.8381559. [DOI] [PubMed] [Google Scholar]
- 33.Menco BP. 2005. The fine-structural distribution of G-protein receptor kinase 3, beta-arrestin-2, Ca2+/calmodulin-dependent protein kinase II and phosphodiesterase PDE1C2, and a Cl(−)-cotransporter in rodent olfactory epithelia. J Neurocytol 34:11–36. doi: 10.1007/s11068-005-5045-9. [DOI] [PubMed] [Google Scholar]
- 34.Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, Wang J, Chen W, Lefkowitz RJ. 2008. Beta-arrestin-mediated localization of Smoothened to the primary cilium. Science 320:1777–1781. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molla-Herman A, Boularan C, Ghossoub R, Scott MG, Burtey A, Zarka M, Saunier S, Concordet JP, Marullo S, Benmerah A. 2008. Targeting of beta-arrestin2 to the centrosome and primary cilium: role in cell proliferation control. PLoS One 3:e3728. doi: 10.1371/journal.pone.0003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. 1999. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 37.Barak LS, Ferguson SS, Zhang J, Caron MG. 1997. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem 272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- 38.Barak LS, Warabi K, Feng X, Caron MG, Kwatra MM. 1999. Real-time visualization of the cellular redistribution of G protein-coupled receptor kinase 2 and beta-arrestin 2 during homologous desensitization of the substance P receptor. J Biol Chem 274:7565–7569. doi: 10.1074/jbc.274.11.7565. [DOI] [PubMed] [Google Scholar]
- 39.Domire JS, Mykytyn K. 2009. Markers for neuronal cilia. Methods Cell Biol 91:111–121. doi: 10.1016/S0091-679X(08)91006-2. [DOI] [PubMed] [Google Scholar]
- 40.Berbari NF, Bishop GA, Askwith CC, Lewis JS, Mykytyn K. 2007. Hippocampal neurons possess primary cilia in culture. J Neurosci Res 85:1095–1100. doi: 10.1002/jnr.21209. [DOI] [PubMed] [Google Scholar]
- 41.Green JA, Gu C, Mykytyn K. 2012. Heteromerization of ciliary G protein-coupled receptors in the mouse brain. PLoS One 7:e46304. doi: 10.1371/journal.pone.0046304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreuzer OJ, Krisch B, Dery O, Bunnett NW, Meyerhof W. 2001. Agonist-mediated endocytosis of rat somatostatin receptor subtype 3 involves beta-arrestin and clathrin coated vesicles. J Neuroendocrinol 13:279–287. [DOI] [PubMed] [Google Scholar]
- 43.Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, Nachury MV. 2013. An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J Cell Biol 203:129–147. doi: 10.1083/jcb.201212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schafer J, Baumeister H, Meyerhof W, Nehring R, Richter D. 1996. Characterization of cloned somatostatin receptor subtypes. Localization of rat SSTRs in gastrointestinal tissues, SSTR1 gene promoter studies, and mutational analyses of SSTR3. Ann N Y Acad Sci 805:601–606. [DOI] [PubMed] [Google Scholar]
- 45.Roth A, Kreienkamp HJ, Meyerhof W, Richter D. 1997. Phosphorylation of four amino acid residues in the carboxyl terminus of the rat somatostatin receptor subtype 3 is crucial for its desensitization and internalization. J Biol Chem 272:23769–23774. doi: 10.1074/jbc.272.38.23769. [DOI] [PubMed] [Google Scholar]
- 46.Kee HL, Dishinger JF, Blasius TL, Liu CJ, Margolis B, Verhey KJ. 2012. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol 14:431–437. doi: 10.1038/ncb2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin YC, Niewiadomski P, Lin B, Nakamura H, Phua SC, Jiao J, Levchenko A, Inoue T, Rohatgi R. 2013. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat Chem Biol 9:437–443. doi: 10.1038/nchembio.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komatsu T, Kukelyansky I, McCaffery JM, Ueno T, Varela LC, Inoue T. 2010. Organelle-specific, rapid induction of molecular activities and membrane tethering. Nat Methods 7:206–208. doi: 10.1038/nmeth.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeFea KA. 2011. Beta-arrestins as regulators of signal termination and transduction: how do they determine what to scaffold? Cell Signal 23:621–629. doi: 10.1016/j.cellsig.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Benmerah A. 2013. The ciliary pocket. Curr Opin Cell Biol 25:78–84. doi: 10.1016/j.ceb.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 51.Rohatgi R, Snell WJ. 2010. The ciliary membrane. Curr Opin Cell Biol 22:541–546. doi: 10.1016/j.ceb.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, Emmerson C, Romeo K, Bourdoncle P, Schmitt A, Saunier S, Spassky N, Bastin P, Benmerah A. 2010. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci 123:1785–1795. doi: 10.1242/jcs.059519. [DOI] [PubMed] [Google Scholar]
- 53.Rattner JB, Sciore P, Ou Y, van der Hoorn FA, Lo IK. 2010. Primary cilia in fibroblast-like type B synoviocytes lie within a cilium pit: a site of endocytosis. Histol Histopathol 25:865–875. [DOI] [PubMed] [Google Scholar]
- 54.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 55.Sorkin A, von Zastrow M. 2009. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol 10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. 2011. Endocytosis promotes rapid dopaminergic signaling. Neuron 71:278–290. doi: 10.1016/j.neuron.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. 2013. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST. 2013. TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep 3:1806–1814. doi: 10.1016/j.celrep.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 59.Najafi M, Maza NA, Calvert PD. 2012. Steric volume exclusion sets soluble protein concentrations in photoreceptor sensory cilia. Proc Natl Acad Sci U S A 109:203–208. doi: 10.1073/pnas.1115109109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pedersen LB, Rosenbaum JL. 2008. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 61.Ye F, Breslow DK, Koslover EF, Spakowitz AJ, Nelson WJ, Nachury MV. 2013. Single molecule imaging reveals a major role for diffusion in the exploration of ciliary space by signaling receptors. eLife 2:e00654. doi: 10.7554/eLife.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tulipano G, Stumm R, Pfeiffer M, Kreienkamp HJ, Hollt V, Schulz S. 2004. Differential beta-arrestin trafficking and endosomal sorting of somatostatin receptor subtypes. J Biol Chem 279:21374–21382. doi: 10.1074/jbc.M313522200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.