

Abstract
Synapse development requires normal neuronal activities and the precise expression of synapse-related genes. Dysregulation of synaptic genes results in neurological diseases such as autism spectrum disorders (ASD). Mutations in genes encoding chromatin-remodeling factor Brg1/SmarcA4 and its associated proteins are the genetic causes of several developmental diseases with neurological defects and autistic symptoms. Recent large-scale genomic studies predicted Brg1/SmarcA4 as one of the key nodes of the ASD gene network. We report that Brg1 deletion in early postnatal hippocampal neurons led to reduced dendritic spine density and maturation and impaired synapse activities. In developing mice, neuronal Brg1 deletion caused severe neurological defects. Gene expression analyses indicated that Brg1 regulates a significant number of genes known to be involved in synapse function and implicated in ASD. We found that Brg1 is required for dendritic spine/synapse elimination mediated by the ASD-associated transcription factor myocyte enhancer factor 2 (MEF2) and that Brg1 regulates the activity-induced expression of a specific subset of genes that overlap significantly with the targets of MEF2. Our analyses showed that Brg1 interacts with MEF2 and that MEF2 is required for Brg1 recruitment to target genes in response to neuron activation. Thus, Brg1 plays important roles in both synapse development/maturation and MEF2-mediated synapse remodeling. Our study reveals specific functions of the epigenetic regulator Brg1 in synapse development and provides insights into its role in neurological diseases such as ASD.
INTRODUCTION
Synapses formed between axons and dendrites connect neurons and generate neural circuits that control brain functions (1). The dysregulation of synapse formation, maturation, or plasticity causes many neurodevelopmental diseases such as autism (2). Autism spectrum disorders (ASDs) are complex diseases characterized by a range of behavior abnormalities and regulated by genetic and epigenetic factors (3–5). In ASDs with various genetic or environmental causes, synaptic dysfunction is a central defect.
Many autism risk genes encode transcription factors and epigenetic regulators, which likely function to regulate the expression of synaptic genes (4, 6, 7). A gene network analysis predicted the core subunit of a SWI/SNF-like BRG1-associated factor (BAF) ATP-dependent chromatin remodeling complex, Brg1/SmarcA4, as one of the key nodes in autism pathogenesis (7). BAF complexes containing the ATPase Brg1 or Brm use energy derived from ATP hydrolysis to modulate chromatin structures and regulate transcription (8–10). Mutations in several BAF subunits are the genetic causes of Coffin-Siris syndrome and Nicolaides-Baraitser syndrome with autistic symptoms such as intellectual disability and delayed speech (11–15). In addition, de novo functional mutations of genes encoding several BAF subunits are identified repeatedly in autism patients (7, 16–18). Mutations in a gene encoding the BAF-associated protein activity-dependent neuroprotective protein (ADNP) have been identified in 1.3% of autism patients, the most frequent of all autism risk-associated mutations identified so far (18). These data suggest that BAF complexes function in normal neural development and that mutations cause autistic disorders. Previously, we identified a neuron-specific BAF complex (nBAF) that regulates neuronal gene expression and is required for neural development (19–21). The BAF53b subunit of nBAF complexes is required for activity-dependent dendrite growth and learning and memory (19, 22). However, the functions of nBAF complexes in synapse development and in ASD remain unknown.
Neuronal activity regulates the expression of many ASD-associated genes and is critical in synapse maturation and plasticity (23, 24). Neuronal activity, which triggers Ca2+ influx, initiates multiple signaling pathways that transduce the signals into the nucleus to affect gene transcription. Myocyte enhancer factor 2 (MEF2) family activity-responsive transcription factors are known to regulate ASD-associated genes important for neural development and synaptogenesis (25–27). Deletion of the key family member MEF2C in mouse brains increases synapse numbers and dendritic spines in both cortical and hippocampal neurons, which may account for the learning and memory defects and autistic phenotypes observed (25, 28). Conversely, expression of an MEF2-VP16 superactive protein causes synapse elimination (25, 29). At the molecular level, MEF2 interacts with several transcription cofactors, and Ca2+ signaling-induced exchange from the corepressor complex to coactivator complex is important for MEF2 transcription activities (30, 31). However, it is unclear how these cofactors coordinate with MEF2 to activate gene expression in response to neuronal activities.
In this report, we specifically deleted Brg1 in developing neurons and revealed essential functions of Brg1 in synapse formation, maturation, and remodeling. Brg1 specifically regulates a significant number of genes encoding synaptic proteins and proteins implicated in ASD. We found that Brg1 is required for dendritic spine/synapse elimination mediated by MEF2C and that Brg1 regulates the activity-induced expression of a number of MEF2 target genes. Our analysis showed that MEF2C is required for Brg1 recruitment to MEF2 targets upon neuronal activation. Thus, Brg1 regulates synapse formation, maturation, and MEF2-mediated synapse remodeling. Our study revealed the specific mechanisms through which the epigenetic factor Brg1 regulates synapse development and provides insights into its role in neurological diseases.
MATERIALS AND METHODS
Mice.
Brg1flox/flox (here called Brg1F/F) mice (32), Syn1-Cre mice (33), and MEF2Cflox/flox (here called MEF2CF/F) mice (34) were kindly provided by Pierre Chambon (Institut de Génétique et de Biologie Moléculaire et Cellulaire [IGBMC], France), Luis Parada (University of Texas [UT] Southwestern Medical Center), and Eric Olson (UT Southwestern Medical Center), respectively. BAF53b-Cre mice were generated by transgenic injection of a bacterial artificial chromosome (BAC) construct containing a Cre gene under the control of the neuron-specific BAF53b promoter and regulatory elements (35). These mice were maintained on a mixed genetic background at UT Southwestern Medical Center Animal Facility. All procedures were performed in accordance with the IACUC-approved protocols. In all animal experiments, both males and females were used, and there was no significant difference found between genders.
Plasmid and constructs.
The construct for expression of Cre is PMC-CreN (36). The MEF2 reporter construct MEF2 response element (MRE)-Luc contains three MRE sequences upstream of the luciferase reporter. The MEF2C, MEF2-VP16, and MEF2Δ-VP16 expression constructs in the pCDNA3 vector and the green fluorescent protein (GFP) construct containing an expression cassette for both GFP and myristoylated GFP were described previously (29) and were provided by Chris Cowan (Harvard). pSin-Brg1 (37) was used for expression of Brg1 in cultured cells.
Behavior tests.
All experiments in this study were performed in the Behavior Core Facility and approved by the IACUC at UT Southwestern Medical Center. Mice were housed with food and water available ad libitum with a 12-h light/dark cycle, and all behavior testing occurred during the light cycle. For the open-field activity test, mice were placed in the periphery of a novel open-field environment (44 cm by 44 cm with walls 30 cm high) in a dimly lit room and allowed to explore for 15 min. The animals were monitored from above by a video camera connected to a computer running video tracking software (Ethovision, version 3.0; Noldus) to determine the time, distance moved, and number of entries into two areas, the periphery (5 cm from the walls) and the center (area of 14 cm by 14 cm). The open-field arenas were wiped and allowed to dry between mice.
Immunofluorescent staining.
Immunostaining experiments were performed on paraffin sections (7-μm) or vibratome thick sections (50-μm) of brain tissues and on cultured hippocampal slices and neurons cultured on cover glasses. Antibodies used were against Brg1 (G7 or H88; Santa Cruz Biotechnology), NeuN (Abcam), GFP (Molecular Probes), and HuC/D (Molecular Probes). The images were visualized using an Olympus BX50 microscope.
Dendritic spine analyses of dentate gyrus granule neurons with Lucifer yellow injection.
Brg1F/F or Syn1-Cre Brg1F/F mice at postnatal day 21 (P21) were intracardially perfused with 1.5% paraformaldehyde (PFA) solution, and brains were postfixed in 1.5% PFA solution for 6 h and then sectioned into 300-μm thick slices. Lucifer yellow injection into single dentate gyrus (DG) neurons was performed with a microelectrode amplifier (Multiclamp 700B; Molecular Devices). Neurons in the DG granule cell layer were selected visually under the microscope and patched with the electrodes filled with Lucifer yellow solution (L-12926; Invitrogen). Secondary dendrites (50 to 200 μm from the cell body) were imaged using a Zeiss LSM780 two-photon microscope (40× water immersion lens). Spine density was measured with the NeuronStudio software package with default settings (38). Z-stack confocal images of dendrite segments were reconstructed and analyzed in NeuronStudio for dendritic spine identification and classification as mushroom-, stubby-, or thin-shaped spines. In classification of spine shapes we used the following cutoff values: aspect ratio for thin spines, 2.5; head-to-neck ratio, 1.3; and head diameter, 0.45 μm. Two segments (50 to 100 μm) per neuron (n = 16 to 22 neurons under each condition) were chosen for quantitative analysis. Dendritic spine volume was further analyzed with Imaris software (39).
Hippocampal slice culture, biolistic transfection, and dendritic spine analyses.
Organotypic hippocampal slice cultures were prepared from P6 Brg1F/F mice (29). Hippocampi were dissected and sliced to a 300-μm section with a tissue slicer. Hippocampal slices were cultured on a membrane at the interface between the medium and the air. The culture medium included minimal essential medium (MEM) with horse serum, l-glutamine, CaCl2, MgSO4, dextrose, NaHCO3, HEPES (pH 7.2), and insulin. Gold bullet preparation and biolistic DNA transfection to hippocampal slices cultured for 1 day in vitro (DIV) were performed with a Helios gene gun system (Bio-Rad) according to the manufacturer's protocols. Five days after transfection, dendritic spines were imaged and analyzed as described above.
Electrophysiological measurement of synapse activities.
Organotypic hippocampal slice cultures were prepared from P6 wild-type or Brg1F/F mice. Cultures were biolistically transfected with plasmids for expression of GFP and Cre or control plasmids at 3 DIV. Ten days later, simultaneous whole-cell recordings were obtained from CA1 pyramidal neurons in slice cultures visualized using infrared-differential interference contrast (IR-DIC) and GFP fluorescence to identify transfected and nontransfected neurons. Recordings were made at 32°C in a submersion chamber perfused with artificial cerebrospinal fluid (ACSF) containing 119 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 11 mM d-glucose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM picrotoxin, 0.002 mM 2-chloro-adenosine, and 0.1% dimethyl sulfoxide (DMSO) at pH 7.28 at 305 mosM and saturated with 95% O2–5% CO2. Neurons were voltage clamped at −60 mV through whole-cell recording pipettes (∼4 to 6 MΩ) filled with an intracellular solution containing 0.2 mM EGTA, 130 mM K-gluconate, 6 mM KCl, 3 mM NaCl, 10 mM HEPES, 2 mM QX-314, 4 mM ATP-Mg, 0.4 mM GTP-Na, and 14 mM phosphocreatine-Tris, pH 7.2, adjusted using KOH, at 285 mosM.
For miniature excitatory postsynaptic current (mEPSC) measurements, the ACSF was supplemented with 1 μM tetrodotoxin (TTX). Series and input resistance were measured in a voltage clamp with a 400-ms, 10-mV step from a −60-mV holding potential (filtered at 30 kHz, sampled at 50 kHz). Cells were used for analysis only if the starting series resistance was less than 30 MΩ and was stable throughout the experiment. Input resistance ranged from 50 to 900 MΩ. Data were not corrected for junction potential. No significant difference was observed between transfected and untransfected neurons in resting membrane potentials, indicating that overall neuronal health was unaffected by expression of Cre. Synaptic currents were filtered at 3 kHz, acquired, and digitized at 10 kHz using custom software (Labview; National Instruments). mEPSCs were filtered at 1 kHz and detected off-line using an automatic detection program (MiniAnalysis; Synaptosoft, Inc.) with a detection threshold set at a value greater than at least 5-fold of the root mean square noise levels, followed by a subsequent round of visual confirmation. Significance of differences between results for transfected and untransfected neurons was determined using a paired t test.
Cortical and hippocampal neuron culture and transfection.
Embryonic day 18.5 (E18.5) hippocampal and E16.5 to E18.5 cortical cells were cultured as previously described (19). Dissociated cells were plated on poly-l-ornithine- and fibronectin-coated coverslips. Culture medium contained Dulbecco's modified Eagle's medium supplemented with F-12 medium with putrescine, 2-mercaptoethanol, transferrin, insulin, selenium, progesterone, MEM vitamin additive, and 5% fetal bovine serum (FBS). At 7 DIV, hippocampal cultures were transfected with GFP and MEF2 expression constructs using Lipofectamine 2000. Cultures were fixed and imaged at 14 DIV for dendritic spine analyses as described above. Cortical cultures were transfected with reporter and MEF2 constructs at 5 DIV and analyzed at 6 DIV. For depolarization, 50 mM KCl was added to the cultures for 1 to 6 h as described previously (25). Luciferase reporter assays were performed using a Dual-Luciferase assay kit (Promega). The mating to produce BAF53b-Cre Brg1F/F mutant embryos for primary neuron cultures was between BAF53b-Cre Brg1F/+ and Brg1F/F. Neurons from Brg1F/+, Brg1F/F, or BAF53b-Cre Brg1F/+ heterozygous littermates displayed no significant differences from each other in all experiments and were used as controls.
Reverse transcription-PCR (RT-PCR) and qPCR.
RNA from cells or ground tissues was extracted with TRIzol (Invitrogen). cDNAs were synthesized by reverse transcription using Iscript (Bio-Rad), followed by PCR or quantitative PCR (qPCR) analysis. A Bio-Rad real-time PCR system (C1000 Thermal Cycler) was used for quantitative PCR. Levels of GAPDH (glyceraldehyde-3-phosphate dehydrogenase gene) mRNA were used to normalize input RNA. Graphics shown are representative of experiments performed in triplicate. The experiments were repeated at least three times. Standard errors were calculated according to a previously described method (37). The sequences of all the primers are listed in Table S3 in the supplemental material.
RNA-seq and data analyses.
DG of P13 or P21 Brg1F/F and Syn1-Cre Brg1F/F mice were used for transcriptome sequencing (RNA-seq) analyses. One pair each of P13 and P21 control and mutant samples was used; each sample was pooled from two mice. In addition, cultured BAF53b-Cre Brg1F/F and control cortical neurons with or without KCl stimulation (each sample was pooled from three mice) were subjected to RNA-seq analyses. Total RNAs were extracted, and RNA-seq libraries prepared using an Illumina RNA-Seq Preparation kit were sequenced on a HiSeq 2500 sequencer at UT Southwestern Sequencing Core Facility. RNA-seq reads were mapped using TopHat with default settings (http://ccb.jhu.edu/software/tophat/index.shtml). The mapped reads with a Phred quality score of <20 were filtered out, whereas the duplicates were marked but not removed using SAMTOOLS (40) and Picard (http://broadinstitute.github.io/picard/). Transcript assembly and transcript abundance quantification were carried out using CUFFLINKS, and then differential expression analysis between control and Brg1 mutants was performed using CUFFDIFF (41). The differentially expressed genes with fold changes larger than 1.5 and P values of <0.05 were selected as Brg1-regulated genes. Gene ontology analysis was performed using DAVID (http://david.abcc.ncifcrf.gov/). Fisher's exact test was used to determine the significance of overlapping data sets.
ChIP experiments.
Chromatin immunoprecipitation (ChIP) experiments were performed as described previously (37, 42). Tissue disrupted by Dounce homogenization or dissociated cells were cross-linked with PFA and sonicated to fragments (200 to 500 bp). Antibodies used were against Brg1/Brm (J1) (43) or rabbit IgG control. J1 antibody has been used previously for Brg1 ChIP sequencing (ChIP-seq) analyses (44, 45). Precipitated DNA was purified and subjected to real-time PCR. The percentage of input or fold of enrichment relative to the IgG ChIP level was measured.
Immunoprecipitation and Western blotting.
Cultured cortical neurons were treated with or without KCl at 7 DIV for 1 h. Cells were harvested and lysed with coimmunoprecipitation (co-IP) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, with protease inhibitor freshly added). After centrifugation, rabbit polyclonal antibodies against Brg1/Brm (J1) were added to precleared cell extracts, and samples were incubated at 4°C overnight. Samples were incubated with protein A beads (GE Healthcare) for 1 h; beads were washed with co-IP buffer four times. Precipitated proteins were eluted by boiling in 2× sample buffer (Bio-Rad) before SDS-PAGE and Western blot analysis. For immunoblotting, cell lysates or immunoprecipitates were separated on SDS-PAGE gels. Antibodies used were against Brg1 (G7; Santa Cruz Biotechnology), MEF2C (Cell Signaling), and MEF2D (BD Biosciences). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Jackson ImmunoResearch.
RESULTS
Deleting Brg1 in hippocampal neurons impairs synapse formation and maturation.
The association of Brg1 with ASD prompted us to evaluate synapse development in Brg1 mutant neurons. To determine the cell-autonomous functions of Brg1 in synapse formation, we deleted Brg1 in individual hippocampal neurons and determined the effects on excitatory synapse activities. Using a gene gun biolistic particle delivery system, we introduced plasmids expressing Cre or control empty vector into cultured postnatal day 6 (P6) Brg1F/F (32) hippocampal slices. The cotransfection efficiency using this method is more than 95%, and we observed Cre-mediated Brg1 deletion in GFP-positive (GFP+) neurons (Fig. 1A). Since very few neurons can be transfected with this method, any effects of Brg1 deletion on postsynaptic development should be cell autonomous. We focused on CA1 pyramidal neurons as their stereotypical position and morphology make them easily identifiable in the hippocampal cultures. Ten days after transfection, miniature excitatory postsynaptic currents (mEPSCs) were measured from simultaneous recordings of transfected GFP+ and neighboring untransfected CA1 neurons. Brg1-deleted neurons displayed a decreased mEPSC frequency relative to that of untransfected neurons, whereas mEPSC amplitudes were unchanged (Fig. 1B and C). The reduction of mEPSC frequency was not caused by Cre expression but by Brg1 deletion because transfection of CA1 neurons from wild-type hippocampal slice cultures with the vector for expression of Cre had no effect on mEPSCs (Fig. 1D). mEPSC frequency is correlated with synapse number (29), whereas mEPSC amplitude represents the strength of individual synapses. These data indicate that Brg1 is required for the development of functional excitatory synapses.
FIG 1.

Deleting Brg1 in hippocampal neurons impairs synapse/dendritic spine formation and maturation. (A) Organotypic hippocampal slice cultures from P6 wild-type or Brg1F/F mice were biolistically transfected with Cre-expressing plasmids or empty vector controls together with GFP. Immunostaining showed Brg1 deletion in Cre-expressing GFP-labeled cells but not in the control cells 5 days after transfection (arrows). (B to D) In the organotypic hippocampal slice culture system, synaptic function was measured using whole-cell patch clamp recordings of Cre-transfected CA1 pyramidal neurons or neighboring untransfected neurons. Representative traces of mEPSCs are shown (B). Average mEPSC frequency and mEPSC amplitude from Cre-expressing (n = 21) or untransfected (n = 21) Brg1F/F CA1 neurons (C) and from Cre-expressing (n = 18) or untransfected (n = 18) wild-type neurons (D) are shown. (E to H) Organotypic hippocampal slice cultures from P6 Brg1F/F mice were biolistically transfected with GFP- and Cre-expressing plasmids or with a vector control. Representative pictures of dendritic spines of CA1 pyramidal neurons are shown in panel E (scale bar, 5 μm). Average dendritic spine densities (F), classifications (G), and spine volumes (H) of control (n = 16) and Brg1-deleted (n = 20) CA1 neurons are shown. Values in the graphs represent averages plus standard errors. **, P < 0.01; *, P < 0.05 (Student's t test). DAPI, 4′,6′-diamidino-2-phenylindole.
Most excitatory synapses are built on dendritic spines that contain the postsynaptic signaling machinery and receive synaptic inputs. Spine densities and morphologies faithfully reflect synapse numbers and levels of maturation (46–48). Mushroom-shaped and stubby spines with larger volumes usually indicate mature synapses, whereas long and thin spines with small volumes indicate no or immature synapses. To assess the role of Brg1 in development of dendritic spine, we measured spine densities, volumes, and shapes from CA1 neurons of cultured P6 Brg1F/F hippocampal slices biolistically transfected with constructs for expression of Cre or control and membrane-targeted GFP. Six days after transfection, two-photon confocal microscopy images of GFP+ dendritic spines were taken and analyzed with NeuronStudio software for quantification and spine classification (38, 49). We observed a significant decrease in dendritic spine densities in Brg1F/F CA1 neurons expressing Cre relative to the number of neurons transfected with empty vectors (Fig. 1E and F), indicating impaired synapse formation in the absence of Brg1. Analyses of the spine shapes showed a significant reduction in mushroom-shaped spines and an increase in thin spines in Brg1-deleted neurons compared to results in control neurons (Fig. 1G). Consistently, there was a significant decrease in spine volumes in Brg1-deleted neurons compared to volumes in control neurons (Fig. 1H). These data demonstrate that Brg1 is required for dendritic spine/synapse formation and maturation in postsynaptic neurons and indicate that Brg1 promotes synapse formation and maturation in a cell-autonomous manner.
Brg1 deletion in neurons led to neurological defects in mouse.
To determine the function of Brg1 in synapse development in vivo, we crossed Brg1 conditional knockout mice (32) with several neuron-specific Cre transgenes. A Brg1 deletion in forebrain neurons mediated by a widely used Camk2a-Cre line (50) led to hydrocephalus, mainly due to non-cell-autonomous effects (51). A newly developed BAF53b-Cre line (35) deleted Brg1 in all developing neurons, which caused lethality at birth due to respiratory defects (data not shown). Therefore, these mice cannot be used to study postnatal synaptogenesis. A Synapsin1-Cre (Syn1-Cre) transgene (33) is expressed exclusively in neurons beginning in the late embryonic stage, but its expression pattern is mosaic in most brain areas. Syn1-Cre Brg1F/F mice survive, which enabled us to study Brg1 function in synapse development in vivo. In hippocampus, Syn1-Cre is not expressed in the CA1 region but has strong expression in the dentate gyrus (DG) and CA3 regions (Fig. 2A) (33). In Syn1-Cre Brg1F/F mice, Brg1 deletion from the NeuN+ DG granule neurons was detected at P7; at P14 and P21, Brg1 deletion was clearly observed in neurons in the DG and CA3 regions (Fig. 2A). Control and Brg1-deleted dentate gyrus showed similar morphologies (Fig. 2A). The gross examination of Syn1-Cre Brg1F/F brain at different ages did not reveal obvious structural defects (Fig. 2B). The brain weights of these mice were also normal (Fig. 2C).
FIG 2.

Syn1-Cre-mediated Brg1 deletion in neurons led to neurological defects in mouse. (A) Syn1-Cre-mediated Brg1 deletion occurs in hippocampal neurons as indicated by costaining of Brg1 and the neuronal marker NeuN. Brg1 was present in all cells in Brg1F/F sections, whereas images from representative Syn1-Cre Brg1F/F mice show that Brg1 is not observed in NeuN+ DG granule neurons at P7 or in DG and CA3 regions at P14 and P21. Brg1 was intact in CA1 neurons. Scale bar, 100 μm. (B) Hematoxylin and eosin staining of coronal sections of Brg1F/F and Syn1-Cre Brg1F/F brains at different ages. Scale bar, 500 μm. (C) Brain weights of Brg1F/F and Syn1-Cre Brg1F/F mice during development (n = 3 in each group). (D) Body weights of Brg1F/F and Syn1-Cre Brg1F/F mice during development (n = 6, with matching numbers of male and females). (E) Hindlimb clasp scores of Brg1F/F and Syn1-Cre Brg1F/F mice during development (n = 6). Scoring is as follows: 0, both hindlimbs consistently splayed outward; 1, one hindlimb partially retracted; 2, both hindlimbs partially retracted; 3, both hindlimbs entirely retracted. Values in the graphs in panels D and E are shown as averages + standard errors. *, P < 0.01 (Student's t test). (F) Representative photograph of Brg1F/F and Syn1-Cre Brg1F/F pups at P7. The Brg1 mutant mouse displays a posture typical of the mutant pups indicative of difficulties righting to the face-down position. (G) Open-field activity tests were performed on adult Brg1F/F (n = 13) and Syn1-Cre Brg1F/F (n = 11) mice. No significant differences were found in either the distances moved or the preferences for the open-field areas in the full 15 min tested. (H) Foot shock test to determine the response threshold of Brg1F/F (n = 13) and Syn1-Cre Brg1F/F (n = 11) adult mice. Values in the graphs in panels G and H are shown as averages plus standard errors. **, P < 0.01 (Student's t test).
Although born with normal weight, Syn1-Cre Brg1F/F mutant mice were smaller during development than control mice (Fig. 2D). Brg1 mutants displayed locomotor and behavior abnormalities beginning in the early postnatal stage. They exhibited significantly increased hindlimb clasp frequency compared to that of Brg1F/F controls which was more severe prior to weaning than after (Fig. 2E). Young Brg1 mutant pups were overactive and unbalanced. At P7, in the righting reflex test none of the Brg1 mutant pups righted after 1 min, whereas all control mice righted within 30 s (Fig. 2F and data not shown). These severe behavioral abnormalities at a young age suggest that Brg1 deletion affects neuronal and synapse development. Adult Syn1-Cre Brg1F/F mice had significantly increased hindlimb clasp (Fig. 2E) but had largely recovered from locomotor abnormalities and displayed normal activity/anxiety levels in the open-field activity test in the first 5 min (data not shown) or in the full 15 min (Fig. 2G). During a foot shock test, we observed that adult Brg1 mutant mice had significantly increased sensitivity to foot shock stimulation, as indicated by the significantly decreased jump threshold compared to that of control Brg1F/F mice (Fig. 2H). These neurological defects indicate that Brg1 deletion in developing neurons impairs neuronal development and function.
Neuronal Brg1 deletion impairs synapse maturation in vivo.
To evaluate the dendritic spine morphology in Brg1 mutant neurons in vivo, we injected a fluorescent dye, Lucifer yellow, into individual DG granule neurons in fixed hippocampal slices from P21 Brg1F/F control and Syn1-Cre Brg1F/F mice to visualize the dendritic spines. We chose P21 DG because in Syn1-Cre Brg1F/F hippocampus, Brg1 was not deleted in CA1 neurons but was completely deleted in DG granule neurons at this development stage (Fig. 2A). Analyses of two-photon confocal microscopy images of dendritic spines showed that although Brg1 mutant granule neurons displayed similar spine densities (Fig. 3A and B), there was a significant increase in thin spines in Brg1 mutant neurons (Fig. 3C), which indicates impaired synapse maturation. Similar to the individual Brg1-deleted CA1 neurons, these Brg1-deleted DG granule neurons also displayed significantly reduced dendritic spine volumes (Fig. 3D). Since spine volume correlates well with synapse maturation and activities, the synapse defects in both Brg1-deleted CA1 pyramidal neurons in hippocampal slice cultures and in DG granule neurons in vivo indicate that Brg1 is likely required for synapse development and maturation in general.
FIG 3.
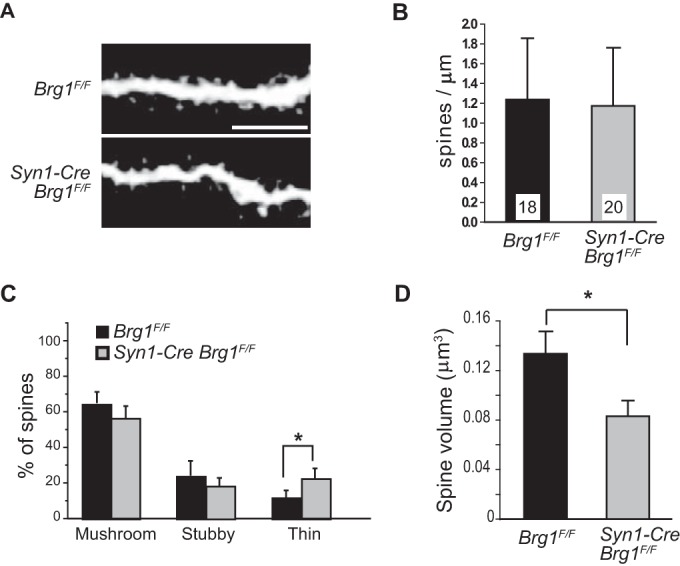
Syn1-Cre-mediated Brg1 deletion in neurons impairs synapse maturation in vivo. Individual DG granule neurons in fixed P21 Brg1F/F and Syn1-Cre Brg1F/F hippocampal slices were injected with Lucifer yellow to enable visualization. (A) Representative two-photon confocal microscopy images of dendritic spines. Scale bar, 5 μm. (B to D) Average spine densities (B), classifications of spines (C), and average spine volumes from Brg1F/F (n = 18) and Syn1-Cre Brg1F/F (n = 20) neurons (D) are shown. Values in the graphs are averages plus standard errors. *, P < 0.05 (Student's t test).
Brg1 regulates synaptic genes in developing hippocampus.
The identification of many transcription factors and epigenetic regulators as autism risk genes suggests that the regulation of the synaptic gene network is a key step to control synaptogenesis in normal development and in diseases. To determine how Brg1 regulates synapse development, we performed RNA-seq to compare the gene expression profiles in control and Syn1-Cre Brg1F/F neurons (Fig. 4; see also Table S1 in the supplemental material). We analyzed P13 and P21 DG because Brg1 is completely deleted in the Syn1-Cre Brg1F/F granule neurons at these stages (Fig. 2A). From P13 DG, we identified 1,383 differentially expressed genes (DEGs), with 868 downregulated and 515 upregulated in Brg1 mutants compared to levels in controls (fold change of >1.5; P value of <0.05). From P21 DG, we identified 1,623 DEGs; 1,187 were upregulated and 446 were downregulated in Brg1-deleted DG compared to control levels. The intersection of P13 and P21 DEGs identified 120 commonly downregulated and 148 commonly upregulated genes, which are high-confidence Brg1-regulated genes (Fig. 4A). These common Brg1-regulated genes include many genes known to encode proteins that function in neuron-specific features, such as neuron projections and channels, and in neurotransmitter release and synaptogenesis (Fig. 4B). Brg1 likely directly activates or represses a significant number of these genes since the ChIP-qPCR experiments indicate that Brg1 occupancy was enriched in the regulatory regions of these neuronal genes in P13 and P21 DG compared to levels in IgG controls and a negative Brg1 binding region (Fig. 4C). Thus, Brg1 may coordinate the expression of a transcription program that is important for synapse formation and maturation.
FIG 4.

Brg1 regulates synaptic genes in developing neurons. (A) Brg1-regulated genes in P13 and P21 DG identified with RNA-seq were compared. The Venn diagrams show the numbers of common genes activated or repressed by Brg1 in each developmental stage. (B) Examples of neuronal genes activated and repressed by Brg1 in both P13 and P21 DG. (C) Brg1 ChIP-qPCR experiments were performed using P13 and P21 DG. Enrichment of Brg1 binding to the regulatory regions of the indicated target genes compared to results with IgG ChIP was observed. A region in the CD4 gene was used as a Brg1 binding negative control. Values in the graph are shown as averages plus standard errors (n = 3). (D) List of the most enriched Brg1-regulated biological processes identified by DAVID gene ontology analysis of the RNA-seq data set obtained from both P13 and P21 DG. (E) Significant overlap between Brg1-regulated genes in developing DG and synaptic genes (52) and autism genes (SFARI gene database). Fisher's exact test was used to calculate P values.
Gene ontology analysis of the Brg1-regulated genes in both P13 and P21 DG revealed that the most enriched group of genes encode extracellular matrix-associated proteins (P = 5.9 × 10−8) (Fig. 4D). High enrichment was also observed in genes encoding growth factor binding proteins, cell adhesion proteins, cytoskeleton regulators, plasma membrane proteins, and calcium signaling pathway components (Fig. 4D), which are all closely related to synapse development. The enrichment of the target genes in these synapse-associated pathways indicates that neuronal Brg1 and nBAF complexes specifically regulate genes involved in synapse formation, maturation, and plasticity. To further understand the molecular functions of Brg1 in synaptic gene regulation, we compared the Brg1-regulated genes in developing DG with known synaptic genes (52) and with human genes linked to autism as shown in the SFARI (Simons Foundation Autism Research Initiative) gene database (www.sfari.org). The P13 DEG data set was also used for comparison as P13 is a stage when synaptic and neurological defects are apparent in Brg1 mutant mice. There are significant overlaps between Brg1-regulated genes and synaptic and autism genes (Fig. 4E). Thus, Brg1 specifically regulates synaptic genes in developing neurons; the abnormal expression of these genes may contribute to autism pathogenesis.
BAF53b-Cre-mediated pan-neuronal Brg1 deletion in cultures.
Many synaptic genes are regulated by neuronal activities that help convert transient stimuli into long-term changes in neuronal morphology and synapse activities. Neuronal BAF complexes regulate activity-dependent dendrite growth, suggesting a signaling pathway from Ca2+ influx to chromatin regulation (19). Brg1 was found to repress the basal expression of the c-fos gene (53). However, activity-induced nBAF target genes are not known, and it is not clear how nBAF complexes regulate gene activation in response to neuronal activities. To understand the function of Brg1 and nBAF complexes in activity-dependent gene regulation, we deleted Brg1 in cultured neurons. The mosaic expression pattern of Syn1-Cre in the cortex and hippocampus prevented us from using this line for neuronal culture studies. Therefore, we took advantage of the newly generated pan-neuron-specific BAF53b-Cre transgene (35) to delete Brg1 in all neurons in the culture (Fig. 5). The BAF53b subunit of the nBAF complexes is expressed exclusively in neurons and in all neurons examined (19). BAF53b-Cre BAC transgene activities were detected in all neurons by E18.5. BAF53b-Cre Brg1F/F mice died at birth due to respiratory failure. However, Brg1 proteins were not completely deleted from cortical and hippocampal neurons at birth (Fig. 5, 1 DIV). We cultured mixed cortical/hippocampal neurons from E18.5 control or BAF53b-Cre Brg1F/F mice. Using a Rosa-YFP Cre reporter (54), we observed a complete overlap between yellow fluorescent protein-positive (YFP+) cells and neuronal marker HuC/D staining in BAF53b-Cre Rosa-YFP cultures (Fig. 5, bottom panel), confirming that BAF53b-Cre is pan-neuron specific. In BAF53b-Cre Brg1F/F Rosa-YFP cultures, Brg1 proteins became undetectable in YFP+ neurons after 5 to 7 days in culture (Fig. 5, 7 DIV and 14 DIV). Brg1 was detected in all nonneuronal cells. Brg1 mutant (BAF53b-Cre Brg1F/F) and control neurons had similar viabilities and cell numbers, indicating that Brg1 is not required for neuron survival in general. We therefore used this culture system to study Brg1 function in neuronal activity-dependent gene regulation and synapse plasticity.
FIG 5.
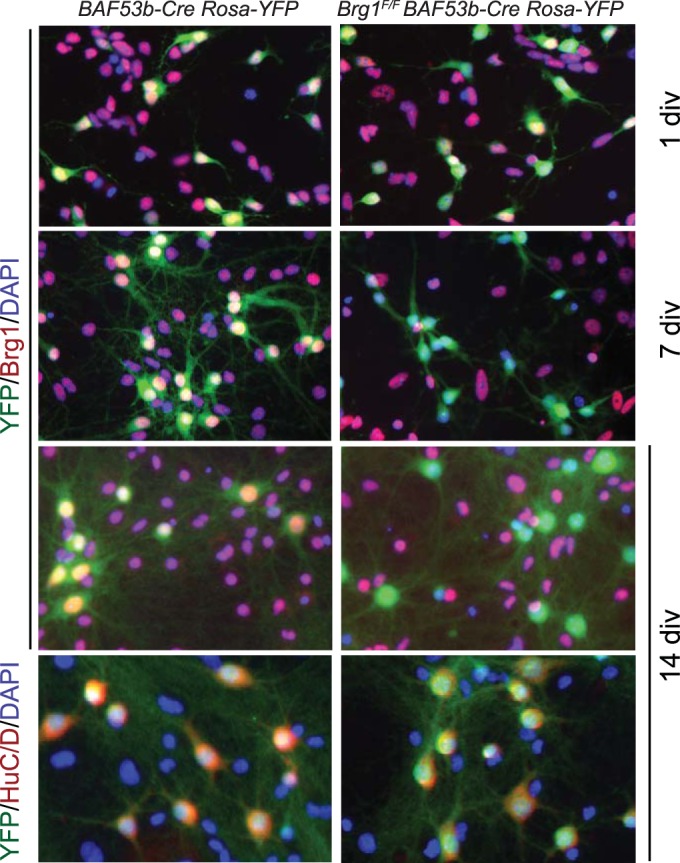
BAF53b-Cre-mediated Brg1 deletion in cultured cortical/hippocampal neurons. (A) BAF53b-Cre was used to mediate Brg1 deletion in cultured neurons. Mixed cortical and hippocampal neurons cultured from E18.5 embryos with the indicated genotypes at 1 DIV, 7 DIV, and 14 DIV were stained with antibodies against Brg1, YFP reporter, and the neuronal marker HuC/D. Note that at 1 DIV, Brg1 was not completely deleted in YFP-labeled neurons in Brg1 mutant culture, whereas at 7 DIV and 14 DIV, Brg1 was completely deleted in YFP+ Brg1 mutant neurons. At 14 DIV, the YFP reporter completely costained with all HuC/D-positive cells, confirming the pan-neuron-specific feature of BAF53b-Cre transgene. DAPI, 4′,6′-diamidino-2-phenylindole.
FIG 7.

MEF2 is required for the activity-induced recruitment of Brg1 to target genes. (A) Brg1 ChIP-qPCR experiments were performed using cultured MEF2CF/F control and Emx1-Cre MEF2CF/F cortical neurons treated with or without KCl for 1 h. Significant increases in Brg1 binding to the regulatory regions of all the activity-induced MEF2 target genes were observed. Gap43 was used as a non-activity-dependent gene control. Induction was significantly diminished in MEF2C mutant cortical cultures. **, P < 0.01 (analysis of variance with a post hoc t test). (B) Interactions between endogenous MEF2C and Brg1 were evaluated in cell lysates from cultured cortical neurons treated with or without KCl for 1 h. Samples were immunoprecipitated with Brg1 antibody or an IgG control. MEF2C and Brg1 were detected by Western blotting.
Brg1 is required for MEF2-mediated gene activation.
To determine the function of Brg1 in neuronal activity-induced gene activation, we measured gene expression profiles in the cultured control and Brg1 mutant neurons under the basal and depolarized conditions. Cultured cortical neurons were used to identify Brg1-regulated neuronal activity-induced genes because these cultures have a high content of neurons (>80%) and are suitable for molecular and biochemical experiments. Many previous studies of activity-induced gene regulation were performed using similar culture conditions (19, 26, 53). The target genes and regulatory mechanisms identified here could be applied to many other experimental systems.
BAF53b-Cre Brg1F/F mutant or control cortical cultures at 7 DIV were treated with KCl for 6 h to trigger depolarization-induced gene expression. RNA-seq experiments were performed to determine the effect of Brg1 deletion on activity-induced gene transcription. By comparing transcription profiles under basal and depolarized conditions in control neurons, we identified 1,943 DEGs (fold change of >1.5, P value of <0.05), of which 1,254 were increased by neuron depolarization (see Table S2 in the supplemental material). By comparing the RNA signals from depolarized control and Brg1 mutant neuron cultures, we found that 219 genes were downregulated after Brg1 deletion. The intersection with the activity-induced genes yielded 76 Brg1-regulated activity-induced genes (Fig. 6A). Of these 76 genes, 74 were not regulated by Brg1 under basal conditions but only at the activity-induced level. Interestingly, 15 of the 74 Brg1-regulated activity-induced genes are also target genes of the MEF2 family of transcription factors (Fig. 6A). Previously, MEF2 target genes were identified from neurons cultured under similar conditions (26). By comparing the MEF2 target genes with our activity-induced neuronal gene list, we obtained 57 activity-induced MEF2 targets; of these, 15 are also regulated by Brg1 (Fig. 6A). The overlap rate is significantly higher than the overlap between two groups of 74 and 57 genes randomly selected from the 1,254 activity-induced gene pool (P = 2.5 × 10−7). The common Brg1/MEF2 target genes include genes known to be important for synapse structure and plasticity such as BDNF, Kcna1, Homer1, Nr4a1 (Nur77), and PCDH17 (24). We confirmed the impaired induction of several Brg1/MEF2 target genes by neuronal depolarization in Brg1 mutant cortical neuron cultures (Fig. 6B). Junb, a gene that is not a MEF2 target, was used as a control. Brg1 deletion did not significantly change activity-induced Junb expression. Moreover, the fact that Brg1 regulated the activity-dependent expression of only 74 genes (<10% of all activity-induced genes) (Fig. 6A) indicates that Brg1 is required for neither activation of the upstream Ca2+ signaling nor activity-induced gene expression in general. Brg1 deletion in neurons did not impair the protein levels or activation of MEF2 proteins by dephosphorylation, as indicated by Western blotting of MEF2C and MEF2D (Fig. 6C). Thus, Brg1 is specifically required for gene activation mediated by certain transcription factors such as MEF2.
FIG 6.

Brg1 is required for MEF2-mediated gene activation. (A) Analyses of Brg1-regulated activity-dependent gene expression using RNA-seq data sets obtained from BAF53b-Cre Brg1F/F Brg1 mutant and control cultured cortical neurons at 7 DIV were treated with or without KCl for 6 h. The Venn diagrams show the intersections of Brg1-activated genes and activity-induced genes (left panel) and the significant overlap between Brg1-regulated and MEF2-regulated activity-induced genes (right panel). (B) Impaired expression of activity-induced MEF2 target genes in Brg1 mutant neuronal cultures as indicated by RT-qPCR analyses. Junb was used as a non-MEF2 target gene control. (C) Western blot showing MEF2 proteins in control and Brg1 mutant neuron cultures in the absence and presence of KCl treatment for 1 h. (D) Brg1 is required for MEF2C-mediated reporter gene activation. MRE-Luc and plasmids expressing MEF2C, MEF2-VP16, or a vector control were cotransfected into cultured cortical neurons at 5 DIV. Luciferase assays were performed 24 h later. KCl was added to a group of cells 6 h before harvesting. (E) MEF2-VP16 or a vector control was transfected into cultured control or Brg1 mutant cortical neurons at 5 DIV. KCl was added at 7 DIV for 6 h. MEF2-VP16 rescued impaired MEF2 target gene Kcna1 expression in Brg1 mutant cultures in response to KCl depolarization as indicated by RT-qPCR measurement. Values in the graphs are shown as averages plus standard errors. Significance was determined using a t test or analysis of variance with a post hoc t test. ** P < 0.01 (n = 3).
The MEF2 family of transcription factors contains four members that have high homology in their DNA binding domains. MEF2C is the major form expressed in the cortex and has been shown to play a predominant role in neuronal synapse development and function (55). A fusion of the MEF2C DNA-binding and dimerization domain with the VP16 activation domain serves as an activator of MEF2 target genes and bypasses the need for MEF2-specific coactivators (25). To determine whether Brg1 is required for MEF2-mediated transcription activation, we examined the effect of Brg1 deletion on the expression of an MEF2-activated reporter gene. Although the reporters are different from endogenous genes, plasmids could incorporate nucleosomes and have been used successfully to test the transcription regulator functions of Brg1 (53). A luciferase reporter with three MEF2 response elements (MRE-Luc) (25) was cotransfected with plasmids for expression of MEF2C or MEF2-VP16 or a control plasmid into cultured BAF53b-Cre Brg1F/F or control neurons. As expected, in control neurons, MRE-Luc was minimally expressed in the resting stage but was induced by either depolarization or coexpression of MEF2C or MEF-VP16 (Fig. 6D). In Brg1 mutant neurons, both depolarization-induced and MEF2C-induced reporter expression was significantly impaired. Interestingly MEF2-VP16 activated MRE-Luc to the same levels in the presence and absence of Brg1 (Fig. 6D). The different requirements for Brg1 in activation of MRE-Luc by exogenous MEF2C and MEF2-VP16 suggest that Brg1 functions as a coactivator of MEF2C and that this requirement is bypassed by MEF2-VP16. Importantly, defective depolarization-induced expression of endogenous MEF2 targets such as Kcna1 caused by Brg1 deletion could be rescued by expression of MEF2-VP16 (Fig. 6E). Therefore, Brg1 is required for MEF2-mediated gene activation.
MEF2C is required for the activity-dependent recruitment of Brg1 to target genes.
MEF2 regulates activity-dependent target genes by exchanging cofactors from corepressors to coactivators upon neuron activation. Since Brg1 is a potential coactivator for MEF2-activated gene expression, we examined the dynamic binding of Brg1 to MEF2 targets. Cultured cortical neurons at 7 DIV were depolarized by a 1-h KCl treatment. Brg1 ChIP was performed, and the signals in the regulatory regions of activity-dependent MEF2 target genes in resting and depolarized neurons were compared. Depolarization of the neurons significantly induced the binding of Brg1 to these genes (Fig. 7A). GAP43 is a neuronal Brg1 target gene that is not induced by neuronal activities and is not an MEF2 target; Brg1 was not further recruited to the GAP43 promoter upon depolarization. Since Brg1 is required for the depolarization-induced activation of these MEF2 target genes, the activity-dependent recruitment of Brg1 likely coordinates with MEF2 to direct the activation of these genes in response to neuronal depolarization.
To determine whether MEF2 is required for the activity-dependent recruitment of Brg1 to the target genes, we performed Brg1 ChIP in control (MEF2CF/F) (34) and MEF2C mutant (Emx1-Cre MEF2CF/F) cortical neuron cultures under resting and depolarized conditions. Loss of MEF2C significantly diminished the activity-induced Brg1 binding to the regulatory regions of the target genes examined (Fig. 7A). Thus, MEF2C is required for the activity-induced Brg1 binding to target genes. In cultured cortical neurons, endogenous MEF2C coimmunoprecipitated with Brg1 under basal and KCl-depolarized conditions; depolarization led to an increase in the MEF2C-Brg1 coimmunoprecipitation efficiency (Fig. 7B). This suggests that MEF2C interacts with Brg1 or other tightly associated BAF subunits to facilitate the recruitment of nBAF complexes to target genes in response to neuronal activities; nBAF then directs the activation of these genes.
Brg1 is required for MEF2-mediated dendritic spine elimination.
The function of MEF2 in synapse remodeling and plasticity is most clearly demonstrated by data indicating that MEF2 activator overexpression in neurons reduces synapse and dendritic spine densities (25, 29). To determine whether Brg1 is required for MEF2-mediated dendritic spine/synapse elimination, we transfected plasmids for expression of MEF2C, MEF2-VP16, or a control MEF2-VP16 mutant without DNA-binding ability (MEF2Δ-VP16) together with GFP into BAF53b-Cre Brg1F/F or control hippocampal neuron cultures. In control neurons, both MEF2-VP16 and MEF2C significantly reduced dendritic spine densities compared to results with MEF2Δ-VP16 (Fig. 8A and B). In Brg1 mutant neurons, MEF2-VP16 reduced the dendritic spine densities, but the ability of MEF2C to eliminate dendritic spines was impaired (Fig. 8A and B), indicating that Brg1 is required for MEF2C-mediated synapse elimination in dissociated neuron cultures.
FIG 8.

Brg1 is required for MEF2C-induced dendritic spine elimination. (A and B) Brg1 is required for MEF2C-mediated dendritic spine elimination in cultured hippocampal neurons. BAF53b-Cre Brg1F/F mutant and control hippocampal neuron cultures were transfected with plasmids expressing GFP and MEF2C, MEF2-VP16, or control MEF2Δ-VP16 at 7 DIV. GFP-labeled neurons were imaged at 14 DIV (A), and dendritic spine densities were measured and compared (B). (C to E) Organotypic hippocampal slice cultures from P6 Brg1F/F mice were biolistically transfected with plasmids for expression of Cre, GFP, and MEF2C or a control at 1 DIV. GFP-labeled CA1 neurons were imaged after 5 days (C), and the dendritic spine densities (D) and classifications (E) were analyzed. Scale bar, 5 μm. Values in the graphs are averages + standard errors. **, P < 0.01; *, P < 0.05 (analysis of variance with a post hoc t test). The numbers of neurons examined in each group are shown in the bar graph. (F) A model illustrating the function of Brg1 in synapse development and plasticity. During synaptic development, nBAF complexes are recruited by specific transcription factors to regulate expression of a significant number of genes required for synapse formation and maturation (left panel). In response to neuronal activity-triggered Ca2+ signaling, activated MEF2 proteins recruit nBAF complexes to MEF2 targets and regulate the activity-induced genes required for synapse elimination and plasticity (right panel). These two scenarios are not mutually exclusive and may reflect the different developmental stages of neurons.
To exclude the potential for non-cell-autonomous effects of Brg1 deletion, we overexpressed MEF2C in individual Brg1-deleted CA1 neurons in cultured hippocampal slices. In cultured Brg1F/F hippocampal slices, plasmids for expression of GFP and MEF2C and/or Cre and corresponding controls were cotransfected using a biolistic delivery system. GFP-labeled CA1 pyramidal neurons were imaged after 5 days for dendritic spine analyses. In control neurons, MEF2C expression significantly reduced dendritic spine densities. In CA1 neurons where Brg1 was deleted by Cre induction, dendritic spine densities were significantly lower than those in control neurons; moreover, MEF2C failed to further reduce spine densities in the Brg1 mutant neurons (Fig. 8C and D). The expression of MEF2C did not significantly change the ratio between the thin and mushroom-shaped or stubby spines in either control or Brg1-deleted neurons (Fig. 8E). These experiments indicate that Brg1 is required for MEF2C-mediated dendritic spine/synapse elimination in hippocampal neurons in both dissociated and slice cultures.
DISCUSSION
In this study, using different Cre systems that are most suitable for the required studies, we found that the ASD-associated chromatin remodeler Brg1 regulates the expression of genes involved in synapse development and plasticity during development and in response to neuronal activities. During neuronal development, Brg1 is required for synapse formation and maturation in several types of neurons. In response to neuronal activities, Brg1 is recruited to MEF2 target genes to control synapse elimination (Fig. 8F). Brg1 deletion in neurons of mice led to severe behavioral defects especially during development. Our studies thus identified potential molecular and cellular mechanisms underlying Brg1 functions in neurological diseases such as autism.
Brg1 is critical for synaptic gene regulation and synapse development.
Brg1 and BAF complexes regulate gene activation or repression by modulating chromatin structures either directly by ATP-dependent chromatin remodeling or indirectly by recruiting other epigenetic regulators (8–10). The recruitment of BAF complexes to target genes mostly requires sequence-specific transcription factors. Functional studies of BAF subunits in different developmental tissues at different stages revealed distinct cell-type-specific functions of BAF complexes. The diverse subunit compositions of BAF complex in different cell types may provide interactions with tissue-specific transcription factors to target the complex to loci that are required for specific developmental programs. Previous studies have shown that BAF complexes in embryonic stem cells, neural progenitors, and neurons have distinct functions in each cell type (19, 20, 56). There are few overlapping target loci of Brg1/BAF in different tissues (44, 57).
In this study, we showed that synaptic genes are specific Brg1 targets in developing neurons. A large number of Brg1-regulated genes have known functions in neuronal and synapse development. We speculate that Brg1 supports a transcription program that coordinates synapse formation and maturation. However, it is not clear which transcription factors are involved in activating or repressing these genes. Our observation that Brg1 is a MEF2 coactivator enables us to speculate that MEF2 is one of the transcription factors that mediate Brg1 functions in synaptic gene regulation and in synapse maturation during development. Interestingly, we observed that Brg1 regulates both synapse formation and MEF2C-mediated synapse elimination. The seemingly paradoxical role of Brg1 in synapse development reflects the diverse functions of Brg1 in neuron development and the dynamic synapse developmental processes. At early postnatal stage, synapse/dendritic spines are actively formed and eliminated. Synapse formation peaks around the second week postnatally in mouse brain, whereas synapse elimination peaks as neurons mature in the third week after birth, partially due to the increased activities of MEF2 family transcription factors (58–61). Therefore, proteins that play diverse functions during synapse development may appear to cause the opposite effects on synapse numbers, depending on the developmental stages when the genes are altered and on MEF2 activities at the specific time point. For example, it has been reported that the autism-associated fragile X mental retardation protein (FMRP) bidirectionally regulates synaptogenesis as a function of developmental age and MEF2 activity (60). Therefore, we propose that Brg1 is required for synapse formation and maturation by regulating synaptic structural genes. In addition, Brg1 also regulates MEF2C-mediated activity-induced gene expression and synapse elimination. The two scenarios described in the proposed model about nBAF functions during development and in activated neurons are not mutually exclusive and reflect diverse functions of Brg1 during different synapse developmental stages (Fig. 8F).
In addition to synaptic genes, Brg1 deletion in dentate gyrus also led to the increased expression of several growth factors such as Igf1 and Igf2 (data not shown), which may compensate for the impaired synapse formation. Therefore, the subtle defects in synaptic spine densities in Syn1-Cre Brg1F/F DG neurons in vivo (Fig. 3) relative to those in Cre-expressing individual CA1 neurons in hippocampal cultures (Fig. 1) could be due to the non-cell-autonomous compensatory mechanisms. Alternatively, the different defects could be caused by the different developmental stages of the neurons examined. However, we could not exclude the possibility that the different defects are due to the regional differences between dentate gyrus granule neurons and CA1 neurons. Nevertheless, our experiments clearly demonstrated that Brg1 is required for synapse maturation for both neuron types and that it is required cell autonomously for synapse formation in CA1 neurons.
nBAF complexes function in activity-dependent gene activation.
The ability of neurons to convert transient stimuli into long-term changes in brain function underlies long-lasting neural plasticity; activity-dependent gene expression plays a central role in this process. Much evidence suggests that the cooperation between transcription factors and chromatin regulators controls the rapid response of neurons to stimuli as well as long-lasting changes in neuron function (24, 62, 63). Despite the understanding that chromatin regulation is important in neuronal plasticity, the biochemical and molecular mechanisms remain largely unclear.
Analyses of BAF53b knockout neurons demonstrated that nBAF complexes regulate activity-dependent dendrite growth, suggesting that signaling resulting from Ca2+ influx leads to chromatin regulation (19). In this study, by deleting Brg1 specifically in neurons and by comparing the Brg1-regulated genes under basal and depolarized conditions, we identified a specific group of genes that are regulated by Brg1 during activation of neurons. These genes significantly overlap activity-dependent MEF2 targets. We demonstrated the requirement of Brg1 for the expression of MEF2C targets and the requirement of MEF2C for Brg1 recruitment in response to neuronal depolarization. These results indicate that Brg1 functions as an essential coactivator of MEF2C-mediated transcription. This regulation is consistent with our observation that Brg1 is essential for MEF2C-mediated synapse elimination, which may also contribute to nBAF complex functions in neurodevelopmental diseases. However, in addition to MEF2, Brg1 may regulate additional transcription factors or pathways that are important for synapse development. Brg1 mutant mice display early defects in synapse formation and maturation, whereas MEF2C mutant neurons have defects in synapse elimination, which is a relatively late stage in synapse development. Deleting Brg1 using inducible Cre lines in neurons at later developmental stages after synapse formation may reveal its requirement for synapse elimination in vivo and additional functions in mature neurons.
One remaining question is how nBAF complexes, including Brg1 and its 10 tightly associated subunits, are recruited to MEF2 targets in response to Ca2+ signaling activation. Several modifications to MEF2 lead to an exchange of corepressors for coactivators (30, 31). One possibility is that Ca2+ signaling induces MEF2 modification changes that facilitate the recruitment of Brg1. This is supported by our observation of an increase of MEF2C-Brg1 coimmunoprecipitation efficiency in neurons in response to KCl treatment. However, the interaction between MEF2C and Brg1 is moderate. Therefore, additional mechanisms may also contribute to the recruitment of Brg1 in response to neuronal activation. It is possible that changes in the local chromatin environment induced by MEF2 activation help recruit nBAF complexes through several histone modification binding domains in various BAF subunits (8).
In this study, we found that the core BAF subunit Brg1 is critical for synapse development, maturation, and plasticity. The close functional connections of Brg1 to the ASD-associated MEF2 proteins discovered here provide molecular and cellular mechanisms for the role of BAF complexes in neurodevelopmental diseases. In Brg1 mutant mice, the mutant phenotypes are most severe during development and become less severe in adults. It is possible that mice develop compensating mechanisms that enable recovery from the defects. Interestingly, in adult neurons, the Brg1 homolog Brm is highly expressed, and this might compensate for Brg1 deletion. Mutations in Brm, but not in Brg1, have been linked to schizophrenia (64). Thus, during different developmental stages, specific BAF complexes may be required for different aspects of neuronal development and function. The understanding of the functions and mechanisms of epigenetic regulators in ASD and other neurological disorders may provide new treatment strategies for these diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Qiu Wang for technical support and mouse colony maintenance. We are grateful to Andrew Yoo, Lei Chen, and Gerald Crabtree (Stanford University) for generating the BAF53b-Cre transgenic mouse. We thank Pierre Chambon (IGBMC, France), Luis Parada (UT Southwestern Medical Center), and Eric Olson (UT Southwestern Medical Center) for providing transgenic or knockout mice, and Kacey Rajkovich, Makoto Taniguchi, and Christopher Cowan for providing reagents. The behavior tests were performed in the Behavior Core Facility in UT Southwestern Medical Center. The RNA-seq experiments with the basic analyses were performed in the UT Southwestern Medical Center Sequencing Facility .
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00534-15.
REFERENCES
- 1.Choquet D, Triller A. 2013. The dynamic synapse. Neuron 80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Zoghbi HY. 2003. Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 3.Spooren W, Lindemann L, Ghosh A, Santarelli L. 2012. Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharmacol Sci 33:669–684. doi: 10.1016/j.tips.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 4.van Bokhoven H. 2011. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet 45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- 5.Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. 2010. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry 167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- 6.Ben-David E, Shifman S. 2013. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry 18:1054–1056. doi: 10.1038/mp.2012.148. [DOI] [PubMed] [Google Scholar]
- 7.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma'ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, et al. 2014. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu JI, Lessard J, Crabtree GR. 2009. Understanding the words of chromatin regulation. Cell 136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns BR. 2007. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol 14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu JI. 2012. Diverse functions of ATP-dependent chromatin remodeling complexes in development and cancer. Acta Biochim Biophys Sin (Shanghai) 44:54–69. doi: 10.1093/abbs/gmr099. [DOI] [PubMed] [Google Scholar]
- 11.Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. 2012. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 12.Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. 2012. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- 13.Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, McKee S, Magee A, de Man SA, Moreau Y, Morice-Picard F, Obersztyn E, Pilch J, Rosser E, Shannon N, Stolte-Dijkstra I, Van Dijck P, Vilain C, Vogels A, Wakeling E, Wieczorek D, Wilson L, Zuffardi O, van Kampen AH, Devriendt K, Hennekam R, Vermeesch JR. 2012. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44:445–449, S441. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- 14.Hersh JH, Bloom AS, Weisskopf B. 1982. Childhood autism in a female with Coffin-Siris syndrome. J Dev Behav Pediatr 3:249–252. doi: 10.1097/00004703-198212000-00016. [DOI] [PubMed] [Google Scholar]
- 15.Gana S, Panizzon M, Fongaro D, Selicorni A, Memo L, Scandurra V, Vannucci C, Bigozzi M, Scordo MR. 2011. Nicolaides-Baraitser syndrome: two new cases with autism spectrum disorder. Clin Dysmorphol 20:38–41. doi: 10.1097/MCD.0b013e32833edaa9. [DOI] [PubMed] [Google Scholar]
- 16.Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, Nielsen M, den Hollander NS, Ruivenkamp CA, Isidor B, Le Caignec C, Zannolli R, Mucciolo M, Renieri A, Mari F, Anderlid BM, Andrieux J, Dieux A, Tommerup N, Bache I. 2012. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet 82:248–255. doi: 10.1111/j.1399-0004.2011.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, Yntema HG, Bakshi M, Wilson M, Witherspoon KT, Malmgren H, Nordgren A, Anneren G, Fichera M, Bosco P, Romano C, de Vries BB, Kleefstra T, Kooy RF, Eichler EE, Van der Aa N. 2014. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet 46:380–384. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. 2007. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 20.Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. 2007. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Son EY, Crabtree GR. 2014. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am J Med Genet C Semin Med Genet 166C:333–349. doi: 10.1002/ajmg.c.31416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel-Ciernia A, Matheos DP, Barrett RM, Kramar EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, Jia Y, Tran A, Dang R, Post RJ, Chabrier M, Babayan AH, Wu JI, Crabtree GR, Baldi P, Baram TZ, Lynch G, Wood MA. 2013. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci 16:552–561. doi: 10.1038/nn.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West AE, Greenberg ME. 2011. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol 3:a005744. doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert DH, Greenberg ME. 2013. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. 2006. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 26.Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. 2008. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. 2006. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 28.Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN. 2008. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A 105:9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. 2010. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potthoff MJ, Olson EN. 2007. MEF2: a central regulator of diverse developmental programs. Development 134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 31.Guasconi V, Puri PL. 2009. Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell Biol 19:286–294. doi: 10.1016/j.tcb.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumi-Ichinose C, Ichinose H, Metzger D, Chambon P. 1997. SNF2β-BRG1 is essential for the viability of F9 murine embryonal carcinoma cells. Mol Cell Biol 17:5976–5986. doi: 10.1128/MCB.17.10.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. 2001. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. 2007. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Zhan X, Cao M, Yoo AS, Zhang Z, Chen L, Crabtree GR, Wu JI. 2015. Generation of BAF53b-Cre transgenic mice with pan-neuronal Cre activities. Genesis 53:440–448. doi: 10.1002/dvg.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Achatz G, Nitschke L, Lamers MC. 1997. Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science 276:409–411. doi: 10.1126/science.276.5311.409. [DOI] [PubMed] [Google Scholar]
- 37.Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. 2011. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc Natl Acad Sci U S A 108:12758–12763. doi: 10.1073/pnas.1018510108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. 2008. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanger SA, Yao X, Gross C, Bassell GJ. 2011. Automated 4D analysis of dendritic spine morphology: applications to stimulus-induced spine remodeling and pharmacological rescue in a disease model. Mol Brain 4:38. doi: 10.1186/1756-6606-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, Shpargel K, Magnuson T, Li Q, Wang R, Wang C, Ge K, Wu J. 2014. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun 5:5425. doi: 10.1038/ncomms6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khavari PA, Peterson CL, Tamkun JW, Mendel DB, Crabtree GR. 1993. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature 366:170–174. doi: 10.1038/366170a0. [DOI] [PubMed] [Google Scholar]
- 44.Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. 2009. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A 106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, Chan JR, Wu J, Lu QR. 2013. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152:248–261. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhatt DH, Zhang S, Gan WB. 2009. Dendritic spine dynamics. Annu Rev Physiol 71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- 47.Nimchinsky EA, Sabatini BL, Svoboda K. 2002. Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- 48.Star EN, Kwiatkowski DJ, Murthy VN. 2002. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- 49.Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL III, Bezprozvanny I. 2014. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. 1999. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. doi: 10.1016/S0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 51.Cao M, Wu JI. 2015. Camk2a-Cre-mediated conditional deletion of chromatin remodeler Brg1 causes perinatal hydrocephalus. Neurosci Lett 597:71–76. doi: 10.1016/j.neulet.2015.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Eichborn J, Dunkel M, Gohlke BO, Preissner SC, Hoffmann MF, Bauer JM, Armstrong JD, Schaefer MH, Andrade-Navarro MA, Le Novere N, Croning MD, Grant SG, van Nierop P, Smit AB, Preissner R. 2013. SynSysNet: integration of experimental data on synaptic protein-protein interactions with drug-target relations. Nucleic Acids Res 41:D834–D840. doi: 10.1093/nar/gks1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu Z, Ghosh A. 2008. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60:775–787. doi: 10.1016/j.neuron.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, Monteggia LM. 2012. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One 7:e34863. doi: 10.1371/journal.pone.0034863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, Crabtree GR. 2009. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci U S A 106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Attanasio C, Nord AS, Zhu Y, Blow MJ, Biddie SC, Mendenhall EM, Dixon J, Wright C, Hosseini R, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Afzal V, Ren B, Bernstein BE, Rubin EM, Visel A, Pennacchio LA. 2014. Tissue-specific SMARCA4 binding at active and repressed regulatory elements during embryogenesis. Genome Res 24:920–929. doi: 10.1101/gr.168930.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holtmaat A, Svoboda K. 2009. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 59.Hua JY, Smith SJ. 2004. Neural activity and the dynamics of central nervous system development. Nat Neurosci 7:327–332. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- 60.Zang T, Maksimova MA, Cowan CW, Bassel-Duby R, Olson EN, Huber KM. 2013. Postsynaptic FMRP bidirectionally regulates excitatory synapses as a function of developmental age and MEF2 activity. Mol Cell Neurosci 56:39–49. doi: 10.1016/j.mcn.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuo Y, Yang G, Kwon E, Gan WB. 2005. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature 436:261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]
- 62.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. 2008. Decoding the epigenetic language of neuronal plasticity. Neuron 60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsieh J, Gage FH. 2005. Chromatin remodeling in neural development and plasticity. Curr Opin Cell Biol 17:664–671. doi: 10.1016/j.ceb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Koga M, Ishiguro H, Yazaki S, Horiuchi Y, Arai M, Niizato K, Iritani S, Itokawa M, Inada T, Iwata N, Ozaki N, Ujike H, Kunugi H, Sasaki T, Takahashi M, Watanabe Y, Someya T, Kakita A, Takahashi H, Nawa H, Muchardt C, Yaniv M, Arinami T. 2009. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet 18:2483–2494. doi: 10.1093/hmg/ddp166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.