

Abstract
Listeria monocytogenes is a Gram-positive bacterium and a facultative intracellular pathogen that invades mammalian cells, disrupts its internalization vacuole, and proliferates in the host cell cytoplasm. Here, we describe a novel image-based microscopy assay that allows discrimination between cellular entry and vacuolar escape, enabling high-content screening to identify factors specifically involved in these two steps. We first generated L. monocytogenes and Listeria innocua strains expressing a β-lactamase covalently attached to the bacterial cell wall. These strains were then incubated with HeLa cells containing the Förster resonance energy transfer (FRET) probe CCF4 in their cytoplasm. The CCF4 probe was cleaved by the bacterial surface β-lactamase only in cells inoculated with L. monocytogenes but not those inoculated with L. innocua, thereby demonstrating bacterial access to the host cytoplasm. Subsequently, we performed differential immunofluorescence staining to distinguish extracellular versus total bacterial populations in samples that were also analyzed by the FRET-based assay. With this two-step analysis, bacterial entry can be distinguished from vacuolar rupture in a single experiment. Our novel approach represents a powerful tool for identifying factors that determine the intracellular niche of L. monocytogenes.
INTRODUCTION
Listeria monocytogenes is a Gram-positive pathogen responsible for listeriosis, a human disease characterized by gastroenteritis in immunocompromised individuals, miscarriage in pregnant women, and meningitis in newborns (1). L. monocytogenes is able to promote its own internalization in mammalian cells due to interaction of the bacterial surface invasion proteins InlA and InlB with their host cell plasma membrane receptors, the epithelial adhesion molecule E-cadherin and the hepatocyte growth factor receptor c-Met, respectively (2). This interaction leads to the activation of signaling cascades triggering clathrin recruitment (3, 4) and cytoskeletal rearrangements for bacterial engulfment (5–8). Once inside host cells, secretion of the cholesterol-dependent pore-forming toxin listeriolysin O (LLO) and of two bacterial phospholipases, PlcA and PlcB, leads to disruption of the L. monocytogenes internalization vacuole and pathogen escape into the cytoplasm (9). An actin-based motility system dependent on activation of the Arp2/3 complex by the bacterial surface protein ActA allows L. monocytogenes to spread to neighboring cells (2, 10).
While the cell invasion process by L. monocytogenes has been extensively investigated (9), cellular components and signaling pathways leading to vacuolar escape are less well characterized. In macrophages, the small GTPase Rab5 and its interactor RABEP1 have been reported to negatively modulate L. monocytogenes survival in the vacuole (11, 12), and the heat shock protein 70 was reported to inhibit vacuolar escape (13). Furthermore, the gamma-interferon-inducible lysosomal thiol reductase (GILT) activates LLO and favors vacuolar rupture (14), while the cystic fibrosis transmembrane conductance regulator (CFTR) is exploited by L. monocytogenes to escape the phagosome (15). It has been described that under specific physiological conditions, L. monocytogenes can persist and multiply in membrane-bound compartment-denominated Listeria large phagosomes (LAPs), but the extent and relevance of this observation in cellular systems that differ from macrophages have not been explored (16).
Assays to determine L. monocytogenes vacuolar escape have relied on several methods, including the manual detection of microscopically labeled bacterial intracellular clusters (17), identification of actin comet tails present in the cytoplasm of infected cells (12), and cellular transfection with the fluorescent cell wall-binding domain (CBD) of the L. monocytogenes phage endolysin Ply118, which decorates bacteria early on disruption of their internalization compartment (18). Here, we present the development of a microscopy-based approach designed to evaluate in a single experiment factors affecting the invasion of L. monocytogenes and/or its escape from the vacuolar compartment in eukaryotic cells. We constructed a fusion protein containing the LPXTG motif of InlA coupled to a β-lactamase that is exposed at the bacterial surface. This chimeric protein is able to cleave the Förster resonance energy transfer (FRET) probe CCF4, which was previously used to monitor vacuolar rupture of Shigella or Mycobacterium tuberculosis (19). We observed that cells cytoplasmically loaded with CCF4 emit a specific fluorescent signal at 535 nm in resting conditions or when incubated with nonpathogenic Listeria innocua strains; in contrast, under conditions where vacuolar rupture by L. monocytogenes takes place, the CCF4 is cleaved by the surface β-lactamase, and fluorescence emission peaks at 450 nm. Quantification of FRET signal changes provides precise information on cytosolic access by L. monocytogenes. Furthermore, we have coupled this readout with a differential immunofluorescence staining method of extracellular versus total bacteria (20). Together, our assays allow measurement of L. monocytogenes cell invasion and vacuolar rupture in a single assay. These assays allow high-throughput analysis with spatiotemporal resolution to perform screens of host and pathogen factors that differentially alter cell invasion and vacuolar rupture.
MATERIALS AND METHODS
Construction of an LPXTG–β-lactamase fusion protein.
To generate a β-lactamase covalently anchored to the L. monocytogenes surface, we synthesized a chimeric gene (cBLA-inlA) containing the Hyper-SPO1 constitutive promoter (Phyper) fused to the hly 5′-untranslated region (previously shown to enhance expression of cis-associated genes) (21, 22), followed by the region encoding the signal peptide of InlA and the β-lactamase gene (codon usage optimized for expression in L. monocytogenes by using the Web server Optimizer [http://genomes.urv.es/OPTIMIZER/]). Finally, we added the region encoding the last 284 amino acids of InlA, which code for an LPXTG motif, at the 3′ end of the construct (see Fig. S1 in the supplemental material). This 1,940-bp fragment was synthetically produced by gene synthesis (Eurofins Genomics) and cloned into SmaI-SalI restriction sites of pBluescript II SK(+) to generate the plasmid pBluescriptII-cBLA-inlA. The SmaI-SalI restriction fragment was then cloned into the SmaI-SalII-digested pAD-PinlC-GFP plasmid (23). As a result, the PinlC-GFP fragment was replaced by the fragment encoding the tagged β-lactamase, yielding pAD-cBLA-inlA. The pAD-cBLA-inlA plasmid construct was verified by sequencing using primers pPL2-Rv and pPL2-Fw, and it was then transformed into L. monocytogenes EGDePrfA* (BUG 3057) and L. innocuaInlB (BUG 1531) by electroporation with a 0.1-cm cuvette using a GenePulser apparatus (Bio-Rad) set to 25 μF, 400 Ω, and 1.25 kV. Transformants were selected on brain heart infusion (BHI) medium supplemented with chloramphenicol and ampicillin at 7 and 50 μg/ml, respectively. Integration into the chromosome at the tRNA ARg-attBB site of the pPL2 vector was verified by PCR amplification using primers NC16 and PL95 (23). The newly generated strains were named L. monocytogenes EGDe PrfA*β-lact (BUG 3358) and L. innocuaβ-lact/InlB (BUG 3614).
Nitrocefin test.
One milliliter of overnight bacterial culture (optical density at 600 nm [OD600] ≈ 3) was washed 3 times with phosphate-buffered saline (PBS) and resuspended in 1 ml of PBS. Five microliters of these PBS-resuspended bacteria and 10-fold dilutions (10−1 and 10−2) were incubated with 100 μl of 0.10 mM nitrocefin (VWR) in 100 mM sodium phosphate buffer (pH 7) at 37°C for 30 min. Hydrolysis of nitrocefin produces a shift in the visible light spectrum from intact (yellow) to degraded (red) nitrocefin.
Bacterial strains and cell lines.
L. monocytogenes EGDe PrfA*β-lact was grown in BHI medium (Difco Laboratories, Detroit, MI) supplemented with 7 μg/ml of chloramphenicol at 37°C. L. innocuaβ-lact/InlB was grown in BHI medium supplemented with 7 μg/ml of chloramphenicol and 5 μg/ml of erythromycin at 37°C. Escherichia coli strains were grown in Luria-Bertani (LB) broth at 37°C. When required, chloramphenicol was used at a final concentration of 35 μg/ml for E. coli. Human epithelial cervix (HeLa) cells (ATCC CCL-2) were used for tissue culture. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% (vol/vol) fetal calf serum (Biowest). Cells were grown at 37°C with 10% CO2.
CCF4 assay for Listeria vacuole rupture.
HeLa cells were seeded in 96-well plates (Greiner) 1 day before infection at a density of 1 × 104 cells per well in a final volume of 100 μl. The day of infection, HeLa cells were loaded with CCF4/AM (Invitrogen) in HEPES buffer (120 mM NaCl, 7 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 5 mM glucose, and 25 mM HEPES at pH 7.3) containing 1 mM probenecid (Sigma) (which inhibits CCF4 efflux) for 2.5 h at room temperature in the dark as previously described (24). HeLa cells were washed once with 150 μl PBS–1 mM probenecid before infection. Listeria species strains were grown for 18 h in 5 ml of BHI medium at 37°C and 180 rpm. Listeria species overnight cultures grown under these conditions contained ∼3 × 109 bacteria/ml. Bacterial cultures were washed three times with PBS and finally resuspended in EM–1 mM probenecid buffer to infect host cells at the indicated multiplicity of infection (MOI). After centrifugation at 179 × g for 3 min for bacterial attachment and synchronization of invasion, plates were kept at 37°C for 1 h. Cells were washed with 150 μl PBS–1 mM probenecid, fixed with 50 μl 4% paraformaldehyde–1 mM probenecid (Electron Microscopy Sciences) for 10 min in the dark, and stained with 30 μl of 10 μM nuclei dye Draq5 (Biostatus) for 30 min. Finally, cells were washed with 150 μl PBS–1 mM probenecid and kept in 100 μl PBS–1 mM probenecid per well. For small interfering RNA (siRNA) experiments, a pool of four different oligonucleotides (Dharmacon) for the c-Met gene was used. Seven wells transfected with scrambled or c-Met siRNA were infected with L. monocytogenes EGDe PrfA*β-lact (MOIs, 0.1, 0.2, 2, 5, 10, and 20). Twelve negative-control wells, transfected with scrambled siRNA and challenged with invasive L. innocuaβ-lact/InlB, were used. Reverse siRNA transfection of HeLa cells was performed using the Lipofectamine RNA interference (RNAi) Max reagent (Life Technologies) and a final siRNA concentration of 10 nM for 72 h.
Cellular invasion assay.
Microscopic assessment of extracellular versus total bacterial populations was used to study the entry of Listeria spp. into host cells. Since the CCF4 signal is not stable for long periods (24), the same plates containing HeLa cells previously analyzed for CCF4 cleavage can be used to perform a differential staining procedure for cellular invasion. One day after the CCF4 experiment, plates were washed with 150 μl PBS and used for indirect immunofluorescence of invading Listeria spp. Extracellular Listeria spp. were labeled with a primary polyclonal rabbit anti-Listeria serum (25) and a secondary goat anti-rabbit antibody–Alexa Fluor 647 conjugate (Life Technologies). Cells were then permeabilized using 0.1%Triton X-100 for 4 min at room temperature, and total Listeria spp. were labeled with the same primary antibody and a secondary goat anti-rabbit antibody–Alexa Fluor 488 conjugate (Life Technologies). Incubation with antibodies was performed for 30 minutes. PBS buffer supplemented with 1% bovine serum albumin was used to dilute the antibodies. Four washes with PBS supplemented with 1% bovine serum albumin were performed between each step of the immunofluorescence. Nuclei were stained with Hoechst 33342 (dilution, 1/1,000) (ThermoScientific).
Imaging and statistical techniques.
Acquisitions were performed using the automated confocal microscope Opera QEHS (PerkinElmer Technologies) in the following sequence: (i) vacuolar rupture detection: CCF4 (excitation/emission [ex/em] 405/535 and 405/450 nm on two separate cameras) and Draq5 (ex/em 640/690) channels; (ii) internalized bacteria: extracellular bacteria detected with an Alexa Fluor 647-conjugated secondary antibody (ex/em 640/690), and total bacteria detected with an Alexa Fluor 488-conjugated secondary antibody (ex/em 488/540) and Hoechst 33342 (ex/em 405/450). The acquisition for the immunofluorescence to differentiate extra- versus intracellular bacteria was performed 4 days after the CCF4 experiment in order to allow the escape of CCF4 from the cytoplasm and subsequent interferences. Twenty-three fields corresponding to ∼6,000 cells were acquired per well using a 10× air objective. The images were transferred to the Columbus database (PerkinElmer Technologies) for data storage, management sharing, and analysis. Columbus analysis building block routines were used for feature extraction and automated scoring of the fluorescence signal for each individual cell. Statistical analysis of the results was performed as previously described (26). The strictly standardized mean difference (SSMD) statistical tests were used for quality control (QC) analysis (26).
RESULTS
Construction of reporter Listeria species strains that constitutively express a surface β-lactamase.
In order to generate a β-lactamase covalently anchored to the L. monocytogenes surface, we synthesized a chimeric gene (pAD-cBLA-inlA) containing the sequence encoding the signal peptide of InlA to direct the protein to the plasma membrane, the β-lactamase gene, and the region encoding the last 284 amino acids of InlA, which code for an LPXTG motif that is used by the sortase A to covalently anchor proteins to the peptidoglycan (27) and allows exposure of the β-lactamase to the bacterial surrounding environment (Fig. 1A). This entire fragment was inserted at the tRNA Arg-attBB site of the pPL2 vector (23).
FIG 1.

Construction of a β-lactamase constitutively expressed at the surface of Listeria spp. (A) Modular domain construction of the gene for constitutive expression of a surface β-lactamase in Listeria. The construction consists of the constitutive promoter Phyper fused to the hly 5′-untranslated region and the β-lactamase gene (codon usage optimized for L. monocytogenes) with the signal peptide of InlA. Finally, the region encoding the last 284 amino acids of InlA, which code for an LPXTG motif to anchor the protein to the peptidoglycan, was added at the 3′ end. (B) A chromogenic cephalosporin test based on nitrocefin confirms β-lactamase activity on the surface of L. monocytogenes EGDe PrfA*β-lact and L. innocuaβ-lact/InlB strains. Hydrolysis of nitrocefin produces a shift in the visible light spectrum from intact (yellow) to degraded (red) nitrocefin. Tubes 1, 2, and 3, L. monocytogenes EGDe PrfA*β-lact and 10-fold dilutions 10−1 and 10−2; tubes 4, 5, and 6, L. innocuaβ-lact/InlB and 10-fold dilutions 10−1 and 10−2; tube 7, L. monocytogenes EGDe PrfA*; tubes 8, 9, and 10, β-lactamase diluted to 1, 0.1, and 0.01 mg/ml, respectively. The volume in all tubes was 105 μl. Bacterial concentration in tubes 1, 4, and 7 was ≈1.4 × 108 bacteria/ml.
For imaging purposes, pAD-cBLA-inlA was introduced into the strain L. monocytogenes EGDe PrfA*, which contains a Gly145Ser substitution in the transcriptional activator PrfA that causes constitutive overexpression of virulence factors and enhances cellular invasion (28), generating the strain L. monocytogenes EGDe PrfA*β-lact. Penicillin G, ampicillin, and trimethoprim-sulfamethoxazole MICs were determined for the newly generated strain L. monocytogenes EGDe PrfA*β-lact. Figure S2 in the supplemental material shows that L. monocytogenes EGDe PrfA*β-lact is as sensitive to trimethoprim-sulfamethoxazole (an effective antibiotic for listeriosis cases) as the EGDe PrfA* strain (MIC, 0.064 μg/ml). The ampicillin MICs were 0.38 μg/ml for EGDe PrfA*β-lact and 0.125 μg/ml for the EGDe PrfA* strain. The penicillin G MICs were was 0.50 μg/ml for EGDe PrfA*β-lact and 0.19 μg/ml for the EGDe PrfA* strain.
To construct a negative-control bacterium that can invade host cells but is unable to escape the internalization vacuole, we integrated the β-lactamase-containing plasmid into an L. innocua strain, which constitutively expresses InlB at its surface (29), and therefore can invade host cells by activating the InlB receptor c-Met, but does not express LLO and remains trapped in a vacuolar compartment; this strain was named L. innocuaβ-lact/InlB.
The chromogenic cephalosporin test based on nitrocefin was used to detect β-lactamase activity on the Listeria species strains generated in the present study. A red color for undiluted overnight cultures or orange color for the 10−1 diluted cultures clearly indicated the presence of β-lactamase activity in these strains (Fig. 1B).
Monitoring L. monocytogenes access to the host cytosol using a FRET-based assay.
To quantify the access to the host cytoplasm of the newly generated Listeria species strains, HeLa cells were preloaded with the FRET probe CCF4 as previously described to track vacuolar rupture by Shigella and Mycobacterium (24). On HeLa cell infection with the invasive nonpathogenic L. innocuaβ-lact/InlB strain for 1 h, the CCF4 probe displayed a 535-nm fluorescent emission (green) when excited at 405 nm (Fig. 2A), indicating that the probe was not cleaved. In contrast, infection with the pathogenic L. monocytogenes EGDe PrfA*β-lact resulted in most cells in a shift of the CCF4 emission toward 450 nm (blue), revealing that the FRET probe had been cleaved by the β-lactamase on the surface of bacteria that access the host cytosol (Fig. 2B).
FIG 2.

Tracking of L. monocytogenes vacuolar escape using a FRET-based microscopic assay. Cellular infection with L. innocuaβ-lact/InlB (A) or L. monocytogenes EGDe PrfA*β-lact (B). HeLa cells were loaded with CCF4-AM for 2.30 h and infected with bacterial strains for 1 h. After paraformaldehyde fixation for 10 min, nuclei were stained with Draq5, and cells were imaged using an Opera QEHS confocal microscope with a 10× objective. Pictures were obtained with the following merged channels: the intact CCF4 probe peaks at 535 nm (green), and the cleaved CCF4 probe peaks at 450 nm (blue). As shown, L. monocytogenes EGDe PrfA*β-lact can escape its vacuolar compartments and induce the cleavage of the CCF4 probe (B). Bar = 10 μm. Panels at the bottom show Draq5 staining of images A and B.
In order to determine whether the CCF4 assay and the newly generated Listeria species strains expressing the surface β-lactamase are suitable to study factors involved in the infectious process, we first transfected HeLa cells in 96-well plates with scrambled or specific siRNAs targeting the InlB cellular receptor c-Met and then infected them using different MOIs (0.1, 0.2, 2, 5, 10, and 20). Images from replicate wells were automatically acquired and analyzed using a customized image analysis algorithm (26). We observed a strong reduction in the 450 nm/535 nm fluorescence ratio of the c-Met siRNA-treated cells compared to the scrambled siRNA-treated cells that was dependent on the MOI used (Fig. 3A), indicating that inactivation of the cellular c-Met-mediated entry pathway, as expected, inhibits bacterial access to the cytoplasm and cleavage of the CCF4 FRET probe and correlates with the bacterial load. To determine which multiplicity of infection was the most suitable to perform an siRNA screen, we then calculated the SSMD on the pair of controls for each MOI condition as a QC assessment of the assay. SSMD calculation provides powerful metrics of the magnitude of differences between two populations and has been widely used for both QC and hit selection in RNAi screening since its introduction by Zhang in 2007 (30, 31). The calculated values showed that MOIs of 5, 10, and 20 yielded very strong to extremely strong QC criteria (Fig. 3B), indicating that these conditions were suitable for performing a robust RNAi screen on this assay. To avoid undesirable effects caused by cytotoxic factors secreted by L. monocytogenes, we decided to use an MOI of 10 for all subsequent experiments.
FIG 3.
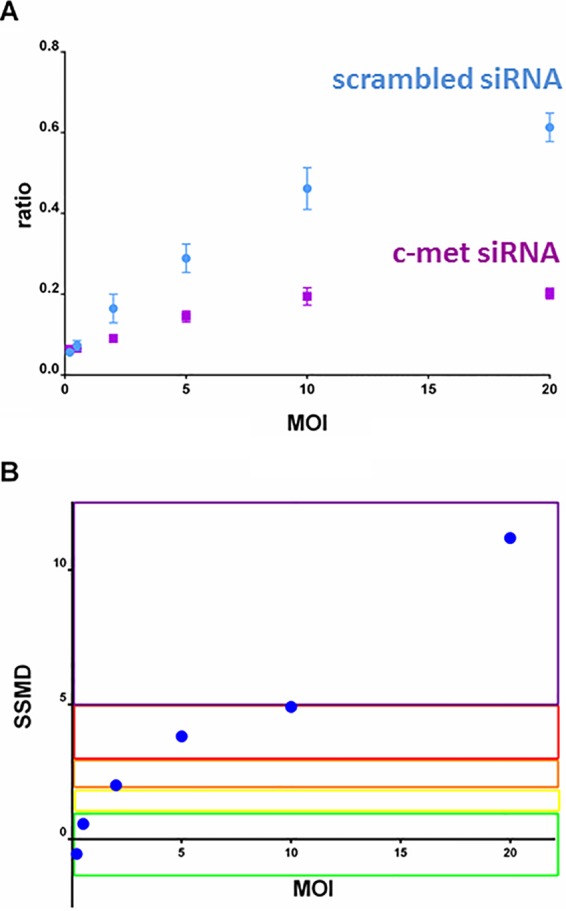
Quality control by MOI for the efficiency of the CCF4 assay. HeLa cells transfected with scrambled or InlB cellular receptor c-Met siRNAs were infected using a dynamic range of MOIs (0.1, 0.2, 2, 5, 10, and 20). Seven wells per MOI were automatically acquired and analyzed to extract the 450 nm/535 nm ratio for each condition. (A) The 450 nm/535 nm ratio as a function of MOI for c-Met (purple) and scrambled (blue) siRNA-treated cells. Each plot corresponds to the mean ratio between the intensity in the 450- and 535-nm channels on 405 nm excitation (n = 7 for each experimental condition). Vertical bars represent the standard deviation. (B) Quality control for the efficiency of the CCF4 assay for each MOI. For quality control, SSMD statistical tests were used to quantify the magnitude of difference between negative and positive controls for each MOI. This test yields a score for each pair of controls per assay condition. The SSMD scores to qualify the robustness of an assay were defined by Zhang (30, 31) as follows: SSMD ≥ 5 for extremely strong (purple), 5 > SSMD ≥ 3 for very strong (red), 3 > SSMD ≥ 2 for strong (orange), 2 > SSMD ≥ 1.645 for fairly strong (yellow), 1.645 > SSMD ≥ 1.28 for moderate (green), and SSMD < 1.28 for weak or no effects.
Differentiating between L. monocytogenes cell entry and cytosolic access by combinatorial indirect immunofluorescence and CCF4 vacuolar rupture measurements.
The reduction in the 450 nm/535 nm fluorescence ratio after gene silencing may be the consequence of a reduction in bacterial entry efficiency (as shown above) or in vacuolar escape. In order to establish an assay that can differentiate between these two critical steps in infection, we combined the vacuolar rupture assay with inside-outside staining of fluorescently labeled Listeria. This distinguished extracellular and total L. monocytogenes bacterial numbers in cells transfected with scrambled siRNA (Fig. 4A) or with siRNA against c-Met (Fig. 4B). For this purpose, we simultaneously visualized both bacterial populations (the extracellular bacteria and the total bacterial number) in plates previously analyzed for CCF4 cleavage after cellular infection with L. monocytogenes using an MOI of 10. In order to avoid interferences caused by CCF4 emission during the microscopy acquisition of the extracellular/total bacterial staining, 96-well plates fixed with 4% paraformaldehyde without probenecid were kept at 4°C for 4 days, resulting in a decreased CCF4 cell load. Differential staining was performed as previously published (20, 32). Images of ∼6,000 cells per well were automatically acquired and analyzed with a specific image analysis algorithm to segment bacteria. c-Met-silenced cells presented a significantly reduced number of intracellular L. monocytogenes cells compared with that found in scrambled siRNA-treated cells with a similar MOI (Fig. 4C). Calculation of the SSMD QC criterion yielded a score of 6, qualifying this assay as extremely strong for the bacterial entry assay (30, 31). This combinatorial assay can therefore be applied to the screening of siRNA libraries with the aim of obtaining information on either bacterial entry or vacuolar rupture (33).
FIG 4.

Differential immunofluorescence staining for quantification of extracellular versus total L. monocytogenes numbers. HeLa cells were previously transfected with control (A) or c-Met-specific (B) siRNAs in 96-well plates and infected with L. monocytogenes EGDe PrfA*β-lact. Indirect immunofluorescence was performed 1 day after CCF4 cleavage acquisition, and plates were kept at 4°C without probenecid for 4 days in order to decrease the CCF4 cell load. Extracellular bacteria were labeled with a secondary goat anti-rabbit antibody–Alexa Fluor 647 stain (red), and total bacteria were labeled with a secondary goat anti-rabbit antibody–Alexa Fluor 488 stain (green) after cell permeabilization. Nuclei were stained with Hoechst 33342 (blue). Intracellular and extracellular bacteria are seen in green and red, respectively. Inactivation of c-Met reduces the number of intracellular bacteria displaying a very distinct green signal. Bars = 100 μm; inset bars = 8 μm. (C) Measurement of the number of intracellular bacteria per cell from images displayed in panels A and B. The experiment was repeated three times (4 wells per condition in each experiment). The results from one representative experiment are shown and were statistically analyzed using a t test. *, P < 0.05.
DISCUSSION
Internalization of L. monocytogenes within host cells leads to the rapid rupture of its vacuolar compartment and to bacterial escape into the cytosol (18). The pore-forming toxin LLO and two bacterial phospholipases play a major role in this intracellular step, but it is increasingly recognized that host factors also positively or negatively modulate L. monocytogenes vacuolar escape (11–13, 15). Here, we present a microscopy-based method that detects L. monocytogenes escape from its internalization compartment and that can be applied to the identification of host factors affecting vacuolar rupture. Since cytoplasmic access also depends on cellular invasion, we designed a supplementary microscopy readout to discriminate the number of extracellular versus total bacterial numbers, allowing investigation of the cell invasion step and the vacuolar rupture step in a single experiment.
Previous methods have been described to investigate the different steps of the L. monocytogenes intracellular cycle. Genomewide siRNA screens performed in Drosophila S2 cells relied extensively on manual visual inspection of the images (17, 34) and were not specifically designed to measure vacuolar escape. A more specific method to identify host factors involved in vacuolar escape relied on the detection of bacterial proliferation in the cytoplasm of host cells (12), which required extensive infection periods; this method used phalloidin labeling to distinguish vacuolar from cytosolic L. monocytogenes, since free bacteria in the cytoplasm recruit F-actin after vacuolar rupture. Finally, a method was developed by Henry et al. (18), who designed a new indicator of L. monocytogenes vacuolar escape by using a fluorescent cell wall-binding domain (CBD) of the L. monocytogenes phage endolysin Ply118. This last method allowed identification of cytosolic bacteria shortly after escape and unravelling of the characteristics of the markers of the newly lysed vacuoles but was restricted to the population of cells effectively transfected with a plasmid encoding a CBD-YFP (yellow fluorescent protein) fusion protein.
Our method may be used in conjunction with time-lapse studies (19), fluorescence-activated cell sorter analysis (35), and high-throughput screening (26), allowing deciphering of which factors are associated with L. monocytogenes entry into host cells and identification of factors driving vacuolar rupture. We have generated a reporter system that can be adapted to studies in other Gram-positive bacteria for which internalization in host cells has been reported (36).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Institut Pasteur (PTR460 grant to J.P.-C. and J.E.), Institut National de la Santé et de la Recherche Médicale (Unité 604), Institut National de la Recherche Agronomique (Unité Sous contract 2020), Fondation Les Mousquetaires, and European Research Council Advanced Grant (233348 MODELIST and 670823 BacCellEpi). The Imagopole is part of the France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INSB-04-01, Investments for the Future) and received additional support from the Conseil de la Region Ile-de-France (program Sesame 2007, project Imagopole [S.S.]) and from the Fondation Française pour la Recherche Médicale (FRM) Programme Grands Equipements [N.A.]). P.C. is a Howard Hughes Medical Institute Senior International Research Scholar. J.E. is supported by an ERC starting grant (Rupteffects, 261166) and by the Institut Pasteur Carnot-MIE program. P.C. and J.E. acknowledge support from LabEx IBEID.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02302-15.
REFERENCES
- 1.Allerberger F, Wagner M. 2010. Listeriosis: a resurgent foodborne infection. Clin Microbiol Infect 16:16–23. doi: 10.1111/j.1469-0691.2009.03109.x. [DOI] [PubMed] [Google Scholar]
- 2.Cossart P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes.. Proc Natl Acad Sci U S A 108:19484–19491. doi: 10.1073/pnas.1112371108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pizarro-Cerda J, Bonazzi M, Cossart P. 2010. Clathrin-mediated endocytosis: what works for small, also works for big. Bioessays 32:496–504. doi: 10.1002/bies.200900172. [DOI] [PubMed] [Google Scholar]
- 4.Pizarro-Cerda J, Cossart P. 2009. Listeria monocytogenes membrane trafficking and lifestyle: the exception or the rule? Annu Rev Cell Dev Biol 25:649–670. doi: 10.1146/annurev.cellbio.042308.113331. [DOI] [PubMed] [Google Scholar]
- 5.Kuhbacher A, Dambournet D, Echard A, Cossart P, Pizarro-Cerda J. 2012. Phosphatidylinositol 5-phosphatase oculocerebrorenal syndrome of Lowe protein (OCRL) controls actin dynamics during early steps of Listeria monocytogenes infection. J Biol Chem 287:13128–13136. doi: 10.1074/jbc.M111.315788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostowy S, Danckaert A, Tham TN, Machu C, Guadagnini S, Pizarro-Cerda J, Cossart P. 2009. Septin 11 restricts InlB-mediated invasion by Listeria.. J Biol Chem 284:11613–11621. doi: 10.1074/jbc.M900231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mostowy S, Janel S, Forestier C, Roduit C, Kasas S, Pizarro-Cerda J, Cossart P, Lafont F. 2011. A role for septins in the interaction between the Listeria monocytogenes invasion protein InlB and the Met receptor. Biophys J 100:1949–1959. doi: 10.1016/j.bpj.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pizarro-Cerda J, Kuhbacher A, Cossart P. 2015. Phosphoinositides and host-pathogen interactions. Biochim Biophys Acta 1851:911–918. doi: 10.1016/j.bbalip.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Pizarro-Cerda J, Kuhbacher A, Cossart P. 2012. Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb Perspect Med 2:pii=a010009. doi: 10.1101/cshperspect.a010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhbacher A, Emmenlauer M, Ramo P, Kafai N, Dehio C, Cossart P, Pizarro-Cerda J. 2015. Genome-wide siRNA screen identifies complementary signaling pathways involved in Listeria infection and reveals different actin nucleation mechanisms during Listeria cell invasion and actin comet tail formation. mBio 6:e00598-15. doi: 10.1128/mBio.00598-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez-Dominguez C, Madrazo-Toca F, Fernandez-Prieto L, Vandekerckhove J, Pareja E, Tobes R, Gomez-Lopez MT, Del Cerro-Vadillo E, Fresno M, Leyva-Cobian F, Carrasco-Marin E. 2008. Characterization of a Listeria monocytogenes protein interfering with Rab5a. Traffic 9:325–337. doi: 10.1111/j.1600-0854.2007.00683.x. [DOI] [PubMed] [Google Scholar]
- 12.Burrack LS, Harper JW, Higgins DE. 2009. Perturbation of vacuolar maturation promotes listeriolysin O-independent vacuolar escape during Listeria monocytogenes infection of human cells. Cell Microbiol 11:1382–1398. doi: 10.1111/j.1462-5822.2009.01338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis MJ, Gregorka B, Gestwicki JE, Swanson JA. 2012. Inducible renitence limits Listeria monocytogenes escape from vacuoles in macrophages. J Immunol 189:4488–4495. doi: 10.4049/jimmunol.1103158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh R, Jamieson A, Cresswell P. 2008. GILT is a critical host factor for Listeria monocytogenes infection. Nature 455:1244–1247. doi: 10.1038/nature07344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radtke AL, Anderson KL, Davis MJ, DiMagno MJ, Swanson JA, O'Riordan MX. 2011. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc Natl Acad Sci U S A 108:1633–1638. doi: 10.1073/pnas.1013262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH. 2008. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 451:350–354. doi: 10.1038/nature06479. [DOI] [PubMed] [Google Scholar]
- 17.Agaisse H, Burrack LS, Philips JA, Rubin EJ, Perrimon N, Higgins DE. 2005. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 309:1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- 18.Henry R, Shaughnessy L, Loessner MJ, Alberti-Segui C, Higgins DE, Swanson JA. 2006. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes.. Cell Microbiol 8:107–119. doi: 10.1111/j.1462-5822.2005.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray K, Bobard A, Danckaert A, Paz-Haftel I, Clair C, Ehsani S, Tang C, Sansonetti P, Tran GV, Enninga J. 2010. Tracking the dynamic interplay between bacterial and host factors during pathogen-induced vacuole rupture in real time. Cell Microbiol 12:545–556. doi: 10.1111/j.1462-5822.2010.01428.x. [DOI] [PubMed] [Google Scholar]
- 20.Kuhbacher A, Cossart P, Pizarro-Cerda J. 2014. Internalization assays for Listeria monocytogenes.. Methods Mol Biol 1157:167–178. doi: 10.1007/978-1-4939-0703-8_14. [DOI] [PubMed] [Google Scholar]
- 21.Klarsfeld AD, Goossens PL, Cossart P. 1994. Five Listeria monocytogenes genes preferentially expressed in infected mammalian cells: plcA, purH, purD, pyrE and an arginine ABC transporter gene, arpJ. Mol Microbiol 13:585–597. doi: 10.1111/j.1365-2958.1994.tb00453.x. [DOI] [PubMed] [Google Scholar]
- 22.Shen A, Higgins DE. 2005. The 5′ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol 57:1460–1473. doi: 10.1111/j.1365-2958.2005.04780.x. [DOI] [PubMed] [Google Scholar]
- 23.Balestrino D, Hamon MA, Dortet L, Nahori MA, Pizarro-Cerda J, Alignani D, Dussurget O, Cossart P, Toledo-Arana A. 2010. Single-cell techniques using chromosomally tagged fluorescent bacteria to study Listeria monocytogenes infection processes. Appl Environ Microbiol 76:3625–3636. doi: 10.1128/AEM.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller C, Mellouk N, Danckaert A, Simeone R, Brosch R, Enninga J, Bobard A. 2013. Single cell measurements of vacuolar rupture caused by intracellular pathogens. J Vis Exp Jun 12:e50116. doi: 10.3791/50116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dramsi S, Levi S, Triller A, Cossart P. 1998. Entry of Listeria monocytogenes into neurons occurs by cell-to-cell spread: an in vitro study. Infect Immun 66:4461–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellouk N, Weiner A, Aulner N, Schmitt C, Elbaum M, Shorte SL, Danckaert A, Enninga J. 2014. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16:517–530. doi: 10.1016/j.chom.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, Jansch L, Garcia-del Portillo F, Schneewind O, Cossart P, European Listeria Genome C . 2002. Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol Microbiol 43:869–881. doi: 10.1046/j.1365-2958.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 28.Ripio MT, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland JA. 1997. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes.. J Bacteriol 179:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braun L, Nato F, Payrastre B, Mazie JC, Cossart P. 1999. The 213-amino-acid leucine-rich repeat region of the Listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol Microbiol 34:10–23. doi: 10.1046/j.1365-2958.1999.01560.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang XD. 2008. Novel analytic criteria and effective plate designs for quality control in genome-scale RNAi screens. J Biomol Screen 13:363–377. doi: 10.1177/1087057108317062. [DOI] [PubMed] [Google Scholar]
- 31.Zhang XD. 2011. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J Biomol Screen 16:775–785. doi: 10.1177/1087057111405851. [DOI] [PubMed] [Google Scholar]
- 32.Pizarro-Cerda J, Lecuit M, Cossart P. 2002. Measuring and analysing invasion of mammalian cells by bacterial pathogens: the Listeria monocytogenes system. Methods Microbiol 31:161–177. doi: 10.1016/S0580-9517(02)31009-2. [DOI] [Google Scholar]
- 33.Kuhbacher A, Gouin E, Mercer J, Emmenlauer M, Dehio C, Cossart P, Pizarro-Cerda J. 2013. Imaging InlC secretion to investigate cellular infection by the bacterial pathogen Listeria monocytogenes.. J Vis Exp Sep 19:e51043. doi: 10.3791/51043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng LW, Viala JP, Stuurman N, Wiedemann U, Vale RD, Portnoy DA. 2005. Use of RNA interference in Drosophila S2 cells to identify host pathways controlling compartmentalization of an intracellular pathogen. Proc Natl Acad Sci U S A 102:13646–13651. doi: 10.1073/pnas.0506461102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nothelfer K, Dias Rodrigues C, Bobard A, Phalipon A, Enninga J. 2011. Monitoring Shigella flexneri vacuolar escape by flow cytometry. Virulence 2:54–57. doi: 10.4161/viru.2.1.14666. [DOI] [PubMed] [Google Scholar]
- 36.Horsley H, Malone-Lee J, Holland D, Tuz M, Hibbert A, Kelsey M, Kupelian A, Rohn JL. 2013. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One 8:e83637. doi: 10.1371/journal.pone.0083637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.