

ABSTRACT
Maternal primary cytomegalovirus (CMV) infection, reactivation, or reinfection with a different viral strain may cause fetal injury and adverse pregnancy outcomes. Increasing evidence indicates that fetal injury results not only from direct viral cytopathic damage to the CMV-infected fetus but also from indirect effects through placental infection and dysfunction. CMV alters Wingless (Wnt) signaling, an essential cellular pathway involved in placentation, as evidenced by reduced transcription of canonical Wnt target genes and decreased Wnt3a-induced trophoblast migration. Whether CMV affects the noncanonical Wnt signaling pathway has been unclear. This study demonstrates for the first time that CMV infection inhibits Wnt5a-stimulated migration of human SGHPL-4 trophoblasts and that inhibition of the pathway restores normal migration of CMV-infected cells. Western blot and real-time PCR analyses show increased expression of noncanonical Wnt receptor ROR2 in CMV-infected trophoblasts. Mimicking the CMV-induced ROR2 protein expression via ectopic expression inhibited Wnt5a-induced trophoblast migration and reduced T cell-specific factor (TCF)/lymphoid enhancer-binding factor (LEF)-mediated transcription as measured using luciferase reporter assays. Gene silencing using small interfering RNA (siRNA) duplexes decreased ROR2 transcript and protein levels. In contrast, proliferation of SGHPL-4 trophoblasts, measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was not affected. The siRNA-mediated downregulation of ROR2 in trophoblasts rescued CMV-induced reduction in trophoblast migration. These data suggest a mechanism where CMV alters the expression of the Wnt receptor ROR2 to alter Wnt5a-mediated signaling and inhibit trophoblast motility. Inhibition of this mechanism may be a target for therapeutic intervention for CMV-induced placental damage and consequent fetal damage in congenital CMV infections.
IMPORTANCE Maternal primary cytomegalovirus (CMV) infection, reactivation, or reinfection with a different viral strain may cause fetal injury and adverse pregnancy outcomes. Increasing evidence indicates that fetal injury results not only from direct viral cytopathic damage to the CMV-infected fetus but also from indirect effects through placental infection and placental dysfunction. No effective therapy is currently proven to prevent or treat congenital CMV infection. Understanding the molecular underpinnings of CMV infection of the placenta is essential for therapeutic innovations and vaccine design. CMV alters canonical Wingless (Wnt) signaling, an essential cellular pathway involved in placental development. This study suggests a mechanism in which CMV alters the expression of noncanonical Wnt receptor ROR2 to alter motility of placental cells, which has important implications in the pathogenesis of CMV-induced placental dysfunction. Inhibition of this mechanism may be a target for therapeutic intervention for CMV-induced placental damage and consequent fetal damage in congenital CMV infection.
INTRODUCTION
Cytomegalovirus (CMV) is now the leading infectious cause of congenital malformation in developed countries, with a mean prevalence of 0.64% (ranging from 0.2 to 2%) of pregnancies (1–4). This translates to an estimated 120,000 infants born each year in developed countries with congenital CMV infection, of whom ∼25% develop symptoms, including sensorineural hearing loss (SNHL), vision loss, seizures, and mental disability (1–3, 5–7). Congenital CMV infection has also been associated with fetal death in utero, neonatal death, intrauterine growth restriction (IUGR), preterm birth, and preeclampsia (5, 8–12). Clinical disease in the neonate may result not only from direct viral cytopathic damage to the CMV-infected fetus (13) but also from indirect effects through placental infection (8, 14–16). No effective therapy is currently proven to prevent or treat congenital infection. Studies of prevention using vaccines are continuing, and recent studies on efficacy of administration of CMV hyperimmunoglobulin or antiviral drugs to pregnant women with primary CMV infection await confirmation in ongoing randomized studies (17). Understanding the molecular underpinnings of CMV infection of the placenta and the fetus is essential for therapeutic innovations and vaccine design.
The pathological changes observed in CMV-infected placentae in vivo include avascular and immature villi, thrombosis of villous capillaries, villitis, and necrosis of villous tissue and trophoblasts (16, 18, 19). We and others have reported that CMV infection mediates alterations in cytokines, cell-cell and cell-matrix adhesion molecules, key integrins, and peroxisome proliferator-activated receptor γ (PPARγ) signaling in trophoblasts (8, 15, 20–31). In vitro, CMV dramatically reduces invasion and migration of trophoblasts (25, 32). The molecular signaling underlying the observed CMV-induced placental damage remains to be elucidated, and cellular mechanisms known to alter trophoblast motility provide logical targets. Intracellular signaling pathways have been identified as regulators of trophoblast invasion and migration, including cascades mediated by phosphatidylinositide 3-kinases (PI3K), extracellular signal-regulated kinase 1 (ERK1), ERK2, focal adhesion kinase (FAK), Akt/protein kinase B, mechanistic target of rapamycin (mTOR), and Smads (33, 34). Additionally, the Wingless (Wnt) signaling pathway plays an important role in the differentiation and migration of trophoblasts (35). Abnormal expression of Wnt signaling has been reported to occur in placental tissue from clinical cases of spontaneous abortion (36) and preeclampsia (37), suggesting that aberrant Wnt signaling contributes to the pathogenesis of these diseases of pregnancy. Wnt ligands are evolutionarily conserved and couple to various receptors to activate different downstream pathways. Wnt3a ligand is generally thought to activate β-catenin-dependent (canonical) Wnt signaling. Activation of the canonical pathway involves stabilization and nuclear translocation of β-catenin, followed by T cell-specific factor (TCF)/lymphoid enhancer-binding factor (LEF)-mediated transcription of Wnt target genes, including c-myc and ccnd1 (38). CMV has been reported to inhibit Wnt3a-induced migration of trophoblasts by affecting canonical Wnt signaling (39). In contrast, Wnt5a ligand mediates intracellular signaling via a β-catenin-independent (noncanonical) Wnt pathway to regulate cell polarity and migration in a variety of tumor types, including metastatic melanoma (40), thyroid carcinoma (41), and colon cancer (42). Wnt5a associates with tyrosine kinase-like orphan receptor 2 (ROR2) (43, 44), which leads to Rho GTPase (RhoA and Rac)-dependent activation of the Wnt-Jun N-terminal kinase (JNK) pathway and activation of the Wnt-Ca2+ pathway through calcium/calmodulin-dependent protein kinase II (CaMKII) (45–47). The role of this noncanonical Wnt pathway in trophoblast migration has not been investigated so far and needs to be elucidated given the importance of Wnt signaling in embryonic development and disease.
The proven importance of Wnt signaling in trophoblast motility led us to investigate Wnt5a signaling in trophoblast motility and to analyze the role of the Wnt receptor ROR2 in CMV-induced reduction in trophoblast migration. Wnt5a induced migration of trophoblasts in a concentration-dependent manner. CMV infection inhibited this Wnt5a-mediated trophoblast migration and altered expression of ROR2. Ectopic expression of ROR2 inhibited Wnt5a-mediated trophoblast migration and reduced TCF/LEF-mediated transcription as measured by luciferase reporter assays. Small interfering RNA (siRNA) targeting ROR2 partially rescued CMV-inhibited trophoblast migration. These results suggest that CMV infection controls the expression of tyrosine kinase receptor ROR2 to regulate trophoblast motility, which has important implications in the pathogenesis of CMV-induced placental dysfunction.
MATERIALS AND METHODS
Cells, viruses, and reagents.
The human extravillous trophoblast line SGHPL-4 was derived from first-trimester chorionic villous tissue (32, 48). These trophoblasts were chosen because they share many phenotypic and functional properties with extravillous trophoblasts in vivo (32) and support productive replication of CMV (49). SGHPL-4 trophoblasts were maintained in Ham's F10 nutrient mix (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin–l-glutamine at 37°C in 5% CO2.
CMV AD169 strain (ATCC) was propagated in MRC-5 fibroblasts. Virus stock titers were determined via standard plaque assays using MRC-5 fibroblasts. For CMV infections, cells were plated on 12-well tissue culture plates at a density of 0.5 × 106 cells per well. The next day, cells were infected with CMV at multiplicity of infection (MOI) of 1 PFU/cell. Briefly, viral inoculum was added to the cells and allowed to adsorb for 4 h at 37°C in 5% CO2. The virus inoculum was then removed and replaced with fresh medium.
Nonsilencing RNA duplexes (Ambion; 20 μM), ROR2 siRNA duplexes (Ambion; 20 μM [s9760]), full-length ROR2-overexpressing plasmid (700 ng), and empty plasmid pcDNA3.1 (700 ng) transfections were conducted using Lipofectamine 2000 reagent (Invitrogen) and Opti-MEM I reduced serum medium (Invitrogen) according to the manufacturer's instructions. The efficiency of overexpression or knockdown of ROR2 was determined using real-time PCR and Western blot analyses.
Real-time PCR.
Total RNA was collected from biological triplicates of mock- and CMV-infected cells using the RNAqueous total RNA isolation kit (Life Technologies) according to the manufacturer's instructions. Real-time PCR was performed using KAPA SYBR Fast 2X mastermix (Geneworks) and the Stratagene Mx3000P detection system (Agilent Technologies). Oligonucleotides (Sigma) used to detect expression of the corresponding genes are shown in Table 1. Data were analyzed using the MxPro qPCR software. Cycle threshold (CT) values of each sample were normalized to CTs of the housekeeping gene eukaryotic translation initiation factor 4a2 (eIF4a2) by calculating the difference of the CT values (dCT). Relative expression levels (fold change) were determined using the value of mock-infected cells at each time point examined.
TABLE 1.
Real-time PCR primers
| Gene | Primer sequence |
|
|---|---|---|
| Forward (5′-3′) | Reverse (5′-3') | |
| ror2 | GGATCCGAACGACCCTTTAG | AGTAACCTTTCAGAGTTGGAATCG |
| mapk8 | TCTGGTATGATCCTTCTGAAGCA | TCCTCCAAGTCCATAACTTCCTT |
| rhoA | GGAAAGCAGGTAGAGTTGGCT | GGCTGTCGATGGAAAAACACAT |
| eif4a2 | GTGTGAACTGGACCCTGTTG | TATTTAACATTCAAACTTCATTAAGACATG |
Western blot analyses and antibodies.
Protein extracts were prepared and analyses were performed as described previously (50). The primary antibodies used were mouse anti-ROR2 (QED Bioscience; 1:1,000), rabbit anti-JNK (Cell Signaling; 1:1,000), mouse anti-RhoA (1B12; Abcam; 1:1,000), mouse anti-CMV immediate early/early (IE/E) (Dako clone CCH2+DDG9; 1:1,000), and mouse anti-β-actin (Sigma; 1:2,000). The secondary antibodies used were goat anti-mouse IgG-horseradish peroxidase (HRP) (Thermo Scientific; 1:2,000) and goat anti-rabbit IgG-HRP (Pierce; 1:2,000).
Immunofluorescence.
Immunofluorescence staining was performed as described previously (15). Three days after CMV infection, SGHPL-4 cells were stained with mouse anti-CMV IE/E (Dako clone CCH2+DDG9; 1:1,000) goat anti-mouse Alexa Fluor 594 antibodies and 4′,6-diamidino-2-phenylindole (DAPI). For image analyses, 5 fields of view were captured. The number of CMV-infected cells was determined by measuring the intensity of IE/E and DAPI staining using ImageJ software. The percentage of IE/E-positive cells was calculated as (average IE/E staining/average DAPI staining) × 100.
Migration assay.
Two days postinfection (p.i.), SGHPL-4 trophoblasts were trypsinized and collected in Ham's F10 nutrient mix (Invitrogen) supplemented with 1% FBS and 1% penicillin-streptomycin–l-glutamine, and 5 × 104 cells were seeded into Transwells with 8.0-μm-pore-size membranes (Corning) in medium containing phosphate-buffered saline (PBS) control or human recombinant Wnt5a protein (200 ng/ml or 400 ng/ml) (R&D Systems). The mock- or CMV-infected cells were allowed to migrate for 16 h at 37°C in 5% CO2. The 16-h time point was empirically determined to be ideal for SGHPL-4 cell migration. Three days after CMV infection, the nonmigrated cells were removed from the upper side of the membranes, migrated trophoblasts were stained with 1% crystal violet solution (Sigma), and five nonoverlapping images were taken of each membrane. The average color intensity was determined using ImageJ analysis software.
Proliferation assay.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays were performed as described previously (51). Briefly, MTT reagent diluted in culture medium was incubated with cells for 2 h at 37°C in 5% CO2. Medium was removed and isopropanol added to each well. After 10 min of incubation at room temperature with gentle shaking, absorbance was measured at 565 nm.
Luciferase assay.
SGHPL-4 trophoblasts were cotransfected with 700 ng of full-length ROR2-expressing plasmid or empty plasmid pcDNA3.1, 1 μg of either TopFlash (which contains multimeric TCF/LEF sequences upstream of a firefly luciferase reporter gene) or FopFlash (which contains mutated TCF/LEF binding sites, used as a specificity control for TopFlash activity) expression plasmids (Millipore, Temecula, CA), and 100 ng of pRL-TK (Renilla-thymidine kinase [TK]-luciferase vector [Promega], as a transfection control), using Lipofectamine 2000. Seven hours later, the cells were treated with 150 ng/ml of human recombinant Wnt3a (R&D Systems) for 16 h prior to luciferase activities being measured using a Glomax 96 microplate luminometer (Turner Biosystems, Sunnyvale, CA). Firefly luciferase activity was normalized for transfection efficiency by dividing it by the Renilla luciferase activity.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism version 6. Comparison of two groups was carried out using a t test. Statistical significance was accepted at a P value of <0.05. Data are expressed as means + standard errors of the means (SEMs).
RESULTS
CMV infection inhibits Wnt5a-induced migration of human trophoblasts.
Immunofluorescence staining of CMV-infected human SGHPL-4 extravillous trophoblasts at 3 days postinfection (p.i.) showed that IE/E protein was present (Fig. 1A), indicating that viral entry into the cell and induction of productive viral replication had occurred. Image analyses revealed that ∼15% of trophoblasts were infected with CMV. As no study to date has addressed whether Wnt5a-mediated signaling affects trophoblast motility, transwell assays were performed. Wnt5a increased migration of SGHPL-4 trophoblasts in a dose-dependent manner (Fig. 1B). Compared with controls, treatment with 200 ng/ml and 400 ng/ml of Wnt5a increased migration 2-fold and ∼3.5-fold, respectively. Analyses of the effect of CMV infection on this Wnt5a-induced migration in SGHPL-4 trophoblasts showed that CMV infection caused a decrease in migration compared with uninfected trophoblasts (Fig. 1C) (P < 0.05).
FIG 1.
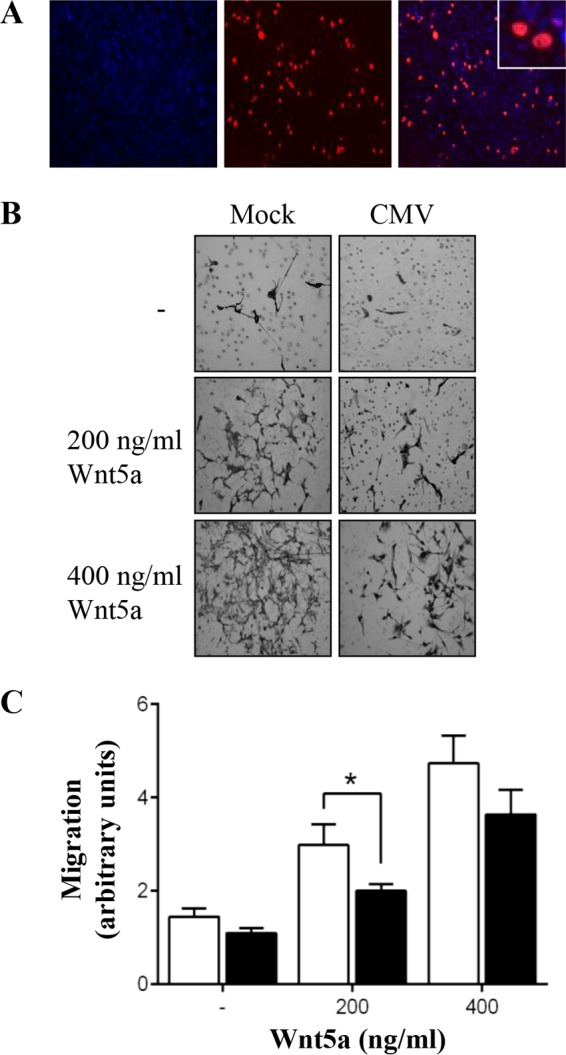
CMV infection inhibits Wnt5a-induced migration of SGHPL-4 trophoblasts. Human trophoblasts were infected with CMV (■) at an MOI of 1 PFU/cell or mock infected (□). (A) CMV-infected cells were analyzed by immunofluorescence at 3 days p.i. Representative images of CMV IE/E protein (red) and DAPI (blue) are shown. (B) Cell migration was analyzed using Transwell assays, with or without addition of recombinant Wnt5a protein. Images are representative of four independent experiments. (C) Average migration, combined from four independent experiments, was determined by capturing five images (20 fields per treatment) from each well and measuring the intensity using ImageJ software (means + SEMs; *, P < 0.05).
CMV infection alters expression of noncanonical Wnt signaling receptor ROR2.
The effect of CMV infection on the mRNA expression of β-catenin-independent Wnt signaling mediators was studied in human SGHPL-4 trophoblasts using real-time PCR analyses and Western blotting. CMV infection increased ROR2 mRNA levels ∼2-fold, although this change did not reach statistical significance. At the protein level, ROR2 receptor expression increased 4.5-fold in CMV-infected trophoblasts compared to values for uninfected cells (Fig. 2B and C) (P < 0.05). CMV infection also increased mRNA expression of the GTPase RhoA (∼1.5-fold; P < 0.01) (Fig. 2A) compared with that in uninfected SGHPL-4 trophoblasts. However, this increase in RhoA was not observed at the protein level (Fig. 2B). The level of expression of the kinase JNK did not significantly change during CMV infection in trophoblasts (Fig. 2).
FIG 2.
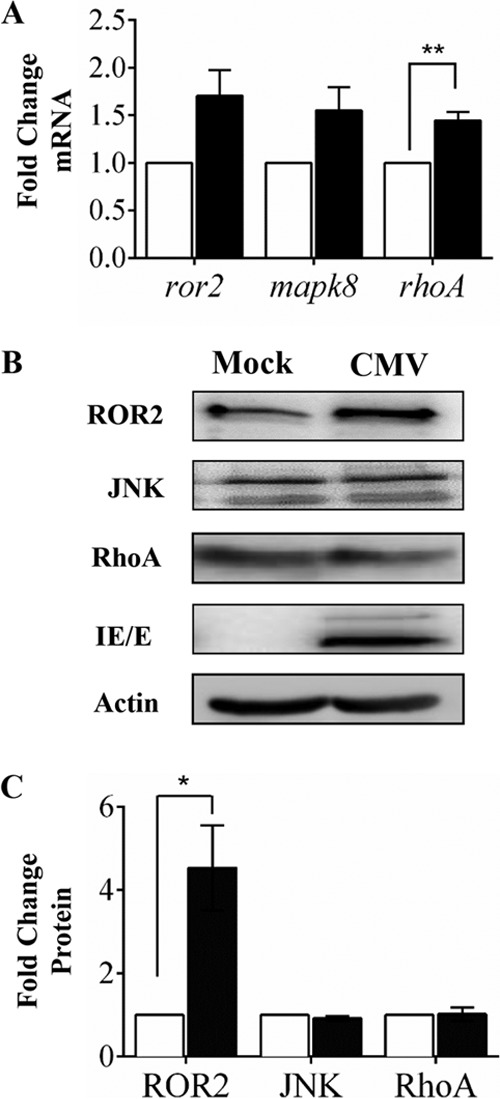
CMV infection alters expression noncanonical Wnt receptor ROR2 in SGHPL-4 trophoblasts. Human trophoblasts were infected with CMV (■) at an MOI of 1 PFU/cell or mock infected (□). (A) Three days postinfection, mRNA expression was analyzed using real-time PCR. Data, displayed as relative to mock infected, are combined from three independent experiments (means + SEMs; ** P < 0.01). (B) Protein expression was analyzed using Western blotting. Data are representative of three independent experiments. (C) Densitometry analysis of data presented in panel B. Data are combined from three independent experiments (means + SEMs; *, P < 0.05).
Increased ROR2 expression inhibits Wnt5a-induced migration and TCF/LEF-mediated transcription in SGHPL-4 trophoblasts.
The influence of Wnt5a treatment and/or increased ROR2 expression on cell proliferation was first studied by transfecting SGHPL-4 trophoblasts with ROR2-expressing plasmid. Western blotting and MTT assays showed that although ROR2 expression was significantly increased in these ROR2 plasmid-transfected cells (Fig. 3A), ectopic ROR2 expression, with or without Wnt5a treatment, did not alter the amount of proliferating trophoblasts (Fig. 3B).
FIG 3.

ROR2 overexpression inhibits Wnt5a-induced migration and TCF/LEF-mediated transcription in SGHPL-4 trophoblasts. Human trophoblasts were transfected with control or ROR2 expressing plasmid. Twenty-four hours posttransfection, the cells were stimulated with Wnt5a (200 ng/ml) or PBS control for 16 h. (A) ROR2 protein expression was analyzed using Western blotting (*, longer exposure). (B) Proliferation was analyzed using MTT assays (means + SEMs). (C) Cell migration was measured using Transwell assays. Images are representative of three independent experiments. (D) Average migration was determined by capturing five images (15 fields per treatment) from each well and measuring the intensity using ImageJ software (means + SEMs; **, P < 0.01). (E and F) Cells were transfected with control or ROR2 plasmid with either TopFlash (E) or FopFlash (F) and pRL-TK plasmids and 7 h later stimulated with Wnt3a (150 ng/ml) or PBS control for 16 h. Data are representative of two independent experiments (means + SEMs; *, P < 0.05).
The effect of increased ROR2 expression on trophoblast migration was studied in these trophoblasts. Ectopic ROR2 expression alone did not alter trophoblast migration (Fig. 3C, top images, and D), suggesting that ROR2 does not play a role in basal trophoblast migration. However, increased ROR2 protein caused a statistical significant reduction in Wnt5a-induced migration of these trophoblasts (Fig. 3C, lower images, and D) (P < 0.01).
The consequence of increased expression of ROR2 on TCF/LEF-mediated transcription was examined using a luciferase Wnt reporter assay. While FopFlash activity remained unchanged (Fig. 3F), increased ROR2 protein impaired Wnt3a-induced activity of TopFlash (Fig. 3E) (P < 0.05), suggesting that ROR2 signaling antagonizes canonical Wnt signaling.
Gene silencing of ROR2 partially restores CMV-induced reduction in trophoblast migration.
Functional studies on the effect of ROR2 knockdown were performed in order to further clarify the role of ROR2 in CMV-induced reduction in trophoblast migration. Trophoblasts were transfected with nonsilencing (NS) control RNA duplexes or siRNA duplexes that specifically target ROR2, followed by infection with CMV (Fig. 4). Real-time PCR analyses showed that ROR2 siRNA reduced ROR2 mRNA levels to ∼40% of that in NS RNA control-treated cells (100%) (Fig. 4A) (P < 0.001). Western blotting demonstrated that ROR2 translation was also reduced by the ROR2-specific siRNA treatment (Fig. 4B), as expected.
FIG 4.

Gene silencing of ROR2 in SGHPL-4 trophoblasts. Human trophoblasts were transfected with nonsilencing RNA duplexes or siRNA duplexes that specifically target ROR2. At 24 h posttransfection, trophoblasts were infected with CMV at MOI of 1 PFU/cell or mock infected and analyzed at 3 days p.i. (A) mRNA expression was analyzed using real-time PCR analyses. Data are displayed relative to NS-treated cells (means + SEMs; ***, P < 0.001) B) Protein expression was analyzed using Western blotting. All data are representative of two independent experiments.
The effect of reduced ROR2 expression on trophoblast migration was then analyzed using this ROR2 siRNA-mediated gene silencing (Fig. 5A and B). Downregulation of ROR2 did not significantly alter basal or Wnt5a-stimulated trophoblast migration (Fig. 5A, compare upper images 1 and 3 and upper images 2 and 4). However, downregulation of ROR2 increased migration of CMV-infected cells nearly to that of the wild type—that is, uninfected trophoblasts (86%) (Fig. 5A, compare lower images 2 and 4) (P < 0.01). These data indicate that inhibition of CMV-induced altered expression of ROR2 protein partially restores CMV-reduced trophoblast migration.
FIG 5.

Gene silencing ROR2 partially restores CMV-reduced migration of SGHPL-4 trophoblasts. Human trophoblasts were transfected with nonsilencing RNA duplexes or siRNA duplexes that specifically target ROR2. At 24 h posttransfection, trophoblasts were infected with CMV (■) at an MOI of 1 PFU/cell or mock infected (□). (A) Two days p.i., uninfected or infected cells were seeded in Transwells, stimulated with Wnt5a (200 ng/ml) or PBS control for 16 h, and analyzed 3 days p.i. Images are representative of three independent experiments. (B) Average migration was determined by capturing five images (15 fields per treatment) from each well and measuring the intensity using ImageJ software (means + SEMs; **, P < 0.01). (C) Proliferation was analyzed using MTT assays. Data are presented as means + SEMs and are representative of three independent experiments.
The influence of decreased ROR2 expression on cell proliferation was studied in SGHPL-4 trophoblasts. Knockdown of ROR2 did not alter the amount of proliferating trophoblasts (Fig. 5C), consistent with ROR2 not being involved in trophoblast growth (Fig. 3B).
DISCUSSION
Inadequate trophoblast invasion and migration result in abnormal placentation, which is associated with early pregnancy loss, preeclampsia, IUGR, and stillbirth. All of these pregnancy complications are also associated complications of maternal CMV infection during pregnancy (8–10), consistent with a role for CMV infection in impaired placental establishment and abnormal placental development. Therefore, understanding the molecular mechanisms underlying CMV-induced placental damage is essential in identifying new therapeutic strategies to prevent diseases of pregnancy related to CMV infection.
Previous studies demonstrated that treatment of trophoblasts with recombinant Wnt3a induced elevated expression of β-catenin/TCF target genes, including cyclin D1, TCF4, and β-catenin (52), and increased trophoblast invasion and migration (52, 53). Despite these comprehensive investigations, the involvement of Wnt5a and the noncanonical Wnt pathway in trophoblast differentiation and migration has not been elucidated. Here, we demonstrate for the first time that Wnt5a increased migration of human SGHPL-4 trophoblasts in a dose-dependent manner (Fig. 1). Wnt5a treatment did not result in altered proliferation of trophoblasts (Fig. 3), suggesting that this Wnt ligand does not directly alter trophoblast growth.
Since Wnt5a-mediated β-catenin-independent signaling regulates epithelial-to-mesenchymal transition (EMT) of cancerous cells (54), which is partly a disorder of cell movement, we hypothesized that Wnt5a signaling could be involved in the molecular mechanisms underlying CMV-induced reduction of trophoblast motility. Indeed, CMV infection reduced Wnt5a-induced trophoblast migration (Fig. 1) (P < 0.05) compared to that of uninfected trophoblasts.
CMV has been shown to directly modulate chemokine CCL2 expression within infected cells during viral replication (24), although previously, Hirsch and Schenk (55) reported that the initial upregulation of CCL2 in CMV-infected cells may be due to indirect (paracrine) effects, possibly due to the use of different experimental systems. Likewise, a previous study suggests that CMV reduced trophoblast invasion through paracrine effects that increase interleukin 10 (IL-10) (25). In this and our previous studies, we found no evidence that CMV infection acts on trophoblast migration in a paracrine manner, although further experiments using different experimental approaches are needed to confirm these preliminary observations.
Interestingly, tyrosine kinase receptor ROR2 expression was significantly increased in CMV-infected trophoblasts, while the protein expression of noncanonical Wnt pathway mediators JNK and RhoA (Fig. 2) or EMT markers N-cadherin and Snai1 (data not shown) was not significantly altered. CMV infection increased expression of Wnt5a-mediating receptor ROR2 4.5-fold in CMV-infected trophoblasts (Fig. 2) (P < 0.05), suggesting that ROR2 plays a critical role in the effects of CMV on trophoblast migration. Supporting this proposition is the fact that mimicking this CMV-induced ROR2 protein expression via ectopic expression inhibited Wnt5a-induced trophoblast migration (Fig. 3). In addition, the use of siRNA duplexes targeting ROR2 restored CMV-induced reduction in trophoblast migration (Fig. 5). ROR2 siRNA treatment, however, did not fully recover CMV-reduced trophoblast migration (Fig. 5), indicating that additional components of Wnt signaling (39) or other pathways that affect trophoblast migration are likely also affected by CMV infection
A previous study reported that CMV infection decreases Wnt3a-induced TCF/LEF activity in trophoblasts (39). The current study demonstrated that increased ROR2 protein expression impaired Wnt3a-induced luciferase transcription of the TCF/LEF reporter in human trophoblasts (Fig. 3E) (P < 0.05), as previously observed for the ROR2-ligand Wnt5a (56), suggesting that ROR2 may be involved in antagonizing canonical Wnt signaling in these cells. Therefore, increased ROR2 by CMV may be responsible for inhibition of canonical Wnt signaling downstream of β-catenin stabilization, at the level of TCF/LEF mediated transcription, and consequently CMV-induced reduction in trophoblast motility (Fig. 6).
FIG 6.
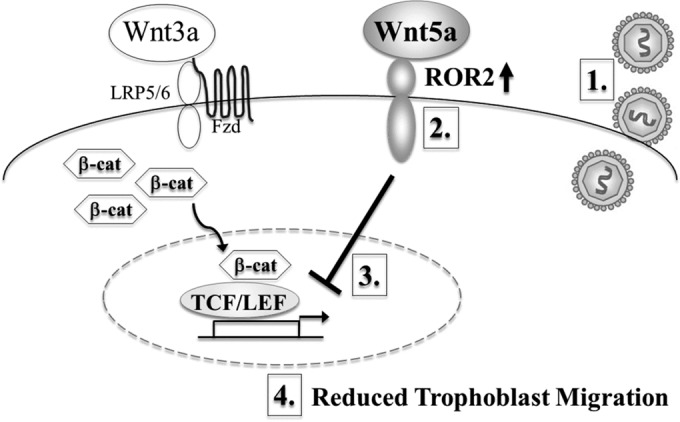
Proposed model of the underlying molecular mechanism of CMV-reduced trophoblast migration. CMV infection of trophoblasts (1) increases the expression of Wnt5a-mediating receptor ROR2 (2). We propose that this increase in tyrosine kinase receptor ROR2 alters Wnt5a-mediated signaling, which leads to inhibition of canonical Wnt signaling downstream of β-catenin stabilization, at the level of TCF/LEF mediated transcription (3), resulting in reduced trophoblast migration (4).
A previous study identified a link between canonical Wnt signaling and CMV-reduced trophoblast motility (39), but did not address the noncanonical Wnt signaling pathway in trophoblasts. We demonstrate that Wnt5a-ROR2 signaling is also critically involved in CMV-reduced trophoblast motility. These findings suggest a mechanism where CMV modulates expression of Wnt receptor ROR2 to alter Wnt5a-mediated signaling, antagonize canonical Wnt signaling, and inhibit trophoblast motility (Fig. 6). This provides the important possibility that inhibiting this mechanism pharmacologically using small molecules provides a target for therapeutic interventions to reduce CMV-induced placental damage and disease consequences of congenital CMV infection.
ACKNOWLEDGMENTS
We thank Guy Whitley at St. George's, University of London in London, United Kingdom, for kindly providing the SGHPL-4 extravillous trophoblast line. We also thank Claire Henry for her technical assistance with the luciferase assays.
REFERENCES
- 1.Dollard SC, Grosse SD, Ross DS. 2007. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol 17:355–363. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 2.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 3.McMullan BJ, Palasanthiran P, Jones CA, Hall BM, Robertson PW, Howard J, Rawlinson WD. 2011. Congenital cytomegalovirus—time to diagnosis, management and clinical sequelae in Australia: opportunities for earlier identification. Med J Aust 194:625–629. [DOI] [PubMed] [Google Scholar]
- 4.Munro SC, Hall B, Whybin LR, Leader L, Robertson P, Maine GT, Rawlinson WD. 2005. Diagnosis of and screening for cytomegalovirus infection in pregnant women. J Clin Microbiol 43:4713–4718. doi: 10.1128/JCM.43.9.4713-4718.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. 1992. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J 11:93–99. doi: 10.1097/00006454-199202000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Munro SC, Trincado D, Hall B, Rawlinson WD. 2005. Symptomatic infant characteristics of congenital cytomegalovirus disease in Australia. J Paediatr Child Health 41:449–452. doi: 10.1111/j.1440-1754.2005.00665.x. [DOI] [PubMed] [Google Scholar]
- 7.van Zuylen W, Hamilton S, Naing Z, Hall B, Shand A, Rawlinson W. 2014. Congenital cytomegalovirus infection: clinical presentation, epidemiology, diagnosis and prevention. Obstet Med 7:140–146. doi: 10.1177/1753495X14552719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasenko JM, Howard J, Arbuckle S, Graf N, Hall B, Craig ME, Rawlinson WD. 2011. Human cytomegalovirus infection is detected frequently in stillbirths and is associated with fetal thrombotic vasculopathy. J Infect Dis 203:1526–1533. doi: 10.1093/infdis/jir121. [DOI] [PubMed] [Google Scholar]
- 9.Pereira L, Petitt M, Fong A, Tsuge M, Tabata T, Fang-Hoover J, Maidji E, Zydek M, Zhou Y, Inoue N, Loghavi S, Pepkowitz S, Kauvar LM, Ogunyemi D. 2014. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J Infect Dis 209:1573–1584. doi: 10.1093/infdis/jiu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams EJ, Embleton ND, Clark JE, Bythell M, Ward Platt MP, Berrington JE. 2013. Viral infections: contributions to late fetal death, stillbirth, and infant death. J Pediatr 163:424–428. doi: 10.1016/j.jpeds.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzoni F, Lunardi S, Liumbruno A, Ferri G, Madrigali V, Fiorentini E, Forli F, Berrettini S, Boldrini A, Ghirri P. 2014. Neonatal screening for congenital cytomegalovirus infection in preterm and small for gestational age infants. J Matern Fetal Neonatal Med 27:1589–1593. doi: 10.3109/14767058.2013.871253. [DOI] [PubMed] [Google Scholar]
- 12.Xie F, Hu Y, Magee LA, Money DM, Patrick DM, Krajden M, Thomas E, von Dadelszen P. 2010. An association between cytomegalovirus infection and pre-eclampsia: a case-control study and data synthesis. Acta Obstet Gynecol Scand 89:1162–1167. doi: 10.3109/00016349.2010.499449. [DOI] [PubMed] [Google Scholar]
- 13.Gabrielli L, Bonasoni MP, Lazzarotto T, Lega S, Santini D, Foschini MP, Guerra B, Baccolini F, Piccirilli G, Chiereghin A, Petrisli E, Gardini G, Lanari M, Landini MP. 2009. Histological findings in foetuses congenitally infected by cytomegalovirus. J Clin Virol 46(Suppl 4):S16–S21. doi: 10.1016/j.jcv.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 14.Scott GM, Chow SS, Craig ME, Pang CN, Hall B, Wilkins MR, Jones CA, Lloyd AR, Rawlinson WD. 2012. Cytomegalovirus infection during pregnancy with maternofetal transmission induces a proinflammatory cytokine bias in placenta and amniotic fluid. J Infect Dis 205:1305–1310. doi: 10.1093/infdis/jis186. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton ST, Scott G, Naing Z, Iwasenko J, Hall B, Graf N, Arbuckle S, Craig ME, Rawlinson WD. 2012. Human cytomegalovirus-induces cytokine changes in the placenta with implications for adverse pregnancy outcomes. PLoS One 7:e52899. doi: 10.1371/journal.pone.0052899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maidji E, Nigro G, Tabata T, McDonagh S, Nozawa N, Shiboski S, Muci S, Anceschi MM, Aziz N, Adler SP, Pereira L. 2010. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am J Pathol 177:1298–1310. doi: 10.2353/ajpath.2010.091210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamilton ST, van Zuylen W, Shand A, Scott GM, Naing Z, Hall B, Craig ME, Rawlinson WD. 2014. Prevention of congenital cytomegalovirus complications by maternal and neonatal treatments: a systematic review. Rev Med Virol 24:420–433. doi: 10.1002/rmv.1814. [DOI] [PubMed] [Google Scholar]
- 18.Garcia AG, Fonseca EF, Marques RL, Lobato YY. 1989. Placental morphology in cytomegalovirus infection. Placenta 10:1–18. doi: 10.1016/0143-4004(89)90002-7. [DOI] [PubMed] [Google Scholar]
- 19.Benirschke K, Kaufmann P. 2013. Pathology of the human placenta. Springer, New York, NY. [Google Scholar]
- 20.Rauwel B, Mariame B, Martin H, Nielsen R, Allart S, Pipy B, Mandrup S, Devignes MD, Evain-Brion D, Fournier T, Davrinche C. 2010. Activation of peroxisome proliferator-activated receptor gamma by human cytomegalovirus for de novo replication impairs migration and invasiveness of cytotrophoblasts from early placentas. J Virol 84:2946–2954. doi: 10.1128/JVI.01779-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabata T, McDonagh S, Kawakatsu H, Pereira L. 2007. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: a quantitative analysis. Placenta 28:527–537. doi: 10.1016/j.placenta.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Warner JA, Zwezdaryk KJ, Day B, Sullivan DE, Pridjian G, Morris CA. 2012. Human cytomegalovirus infection inhibits CXCL12-mediated migration and invasion of human extravillous cytotrophoblasts. Virol J 9:255. doi: 10.1186/1743-422X-9-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tabata T, Petitt M, Zydek M, Fang-Hoover J, Larocque N, Tsuge M, Gormley M, Kauvar LM, Pereira L. 2015. Human cytomegalovirus infection interferes with the maintenance and differentiation of trophoblast progenitor cells of the human placenta. J Virol 89:5134–5147. doi: 10.1128/JVI.03674-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton ST, Scott GM, Naing Z, Rawlinson WD. 2013. Human cytomegalovirus directly modulates expression of chemokine CCL2 (MCP-1) during viral replication. J Gen Virol 94:2495–2503. doi: 10.1099/vir.0.052878-0. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto-Tabata T, McDonagh S, Chang HT, Fisher S, Pereira L. 2004. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol 78:2831–2840. doi: 10.1128/JVI.78.6.2831-2840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher S, Genbacev O, Maidji E, Pereira L. 2000. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol 74:6808–6820. doi: 10.1128/JVI.74.15.6808-6820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou D, Ma Y, Zhang J, McGrath C, Parry S. 2006. Cytomegalovirus infection of trophoblast cells elicits an inflammatory response: a possible mechanism of placental dysfunction. Am J Obstet Gynecol 194:535–541. doi: 10.1016/j.ajog.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 28.Kovács IJ, Hegedus K, Pal A, Pusztai R. 2007. Production of proinflammatory cytokines by syncytiotrophoblasts infected with human cytomegalovirus isolates. Placenta 28:620–623. doi: 10.1016/j.placenta.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Schleiss MR, Aronow BJ, Handwerger S. 2007. Cytomegalovirus infection of human syncytiotrophoblast cells strongly interferes with expression of genes involved in placental differentiation and tissue integrity. Pediatr Res 61:565–571. doi: 10.1203/pdr.0b013e318045be6d. [DOI] [PubMed] [Google Scholar]
- 30.Tao L, Suhua C, Juanjuan C, Zongzhi Y, Juan X, Dandan Z. 2011. In vitro study on human cytomegalovirus affecting early pregnancy villous EVT's invasion function. Virol J 8:114. doi: 10.1186/1743-422X-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabata T, Petitt M, Fang-Hoover J, Rivera J, Nozawa N, Shiboski S, Inoue N, Pereira L. 2012. Cytomegalovirus impairs cytotrophoblast-induced lymphangiogenesis and vascular remodeling in an in vivo human placentation model. Am J Pathol 181:1540–1559. doi: 10.1016/j.ajpath.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaMarca HL, Nelson AB, Scandurro AB, Whitley GS, Morris CA. 2006. Human cytomegalovirus-induced inhibition of cytotrophoblast invasion in a first trimester extravillous cytotrophoblast cell line. Placenta 27:137–147. doi: 10.1016/j.placenta.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Knöfler M. 2010. Critical growth factors and signalling pathways controlling human trophoblast invasion. Int J Dev Biol 54:269–280. doi: 10.1387/ijdb.082769mk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu D, Luo S, Wang Y, Zhuang L, Chen Y, Peng C. 2001. Smads in human trophoblast cells: expression, regulation and role in TGF-beta-induced transcriptional activity. Mol Cell Endocrinol 175:111–121. doi: 10.1016/S0303-7207(01)00397-5. [DOI] [PubMed] [Google Scholar]
- 35.Knöfler M, Pollheimer J. 2013. Human placental trophoblast invasion and differentiation: a particular focus on Wnt signaling. Front Genet 4:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao SH, Shuai W, Tong J, Wang L, Chen P, Duan T. 2013. Increased Dickkopf-1 expression in patients with unexplained recurrent spontaneous miscarriage. Clin Exp Immunol 172:437–443. doi: 10.1111/cei.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Zhang L, Jia L, Wang P, Gao Y. 2013. Association of Wnt2 and sFRP4 expression in the third trimester placenta in women with severe preeclampsia. Reprod Sci 20:981–989. doi: 10.1177/1933719112472740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chien AJ, Conrad WH, Moon RT. 2009. A Wnt survival guide: from flies to human disease. J Invest Dermatol 129:1614–1627. doi: 10.1038/jid.2008.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angelova M, Zwezdaryk K, Ferris M, Shan B, Morris CA, Sullivan DE. 2012. Human cytomegalovirus infection dysregulates the canonical Wnt/beta-catenin signaling pathway. PLoS Pathog 8:e1002959. doi: 10.1371/journal.ppat.1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connell MP, Fiori JL, Xu M, Carter AD, Frank BP, Camilli TC, French AD, Dissanayake SK, Indig FE, Bernier M, Taub DD, Hewitt SM, Weeraratna AT. 2010. The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene 29:34–44. doi: 10.1038/onc.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kremenevskaja N, von Wasielewski R, Rao AS, Schofl C, Andersson T, Brabant G. 2005. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 24:2144–2154. doi: 10.1038/sj.onc.1208370. [DOI] [PubMed] [Google Scholar]
- 42.Dejmek J, Dejmek A, Safholm A, Sjolander A, Andersson T. 2005. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res 65:9142–9146. doi: 10.1158/0008-5472.CAN-05-1710. [DOI] [PubMed] [Google Scholar]
- 43.Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, Vijayakumar S, Economides AN, Aaronson SA. 2010. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev 24:2517–2530. doi: 10.1101/gad.1957710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kani S, Oishi I, Yamamoto H, Yoda A, Suzuki H, Nomachi A, Iozumi K, Nishita M, Kikuchi A, Takumi T, Minami Y. 2004. The receptor tyrosine kinase Ror2 associates with and is activated by casein kinase Iepsilon. J Biol Chem 279:50102–50109. doi: 10.1074/jbc.M409039200. [DOI] [PubMed] [Google Scholar]
- 45.Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, Mundlos S, Shibuya H, Takada S, Minami Y. 2003. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto H, Yoo SK, Nishita M, Kikuchi A, Minami Y. 2007. Wnt5a modulates glycogen synthase kinase 3 to induce phosphorylation of receptor tyrosine kinase Ror2. Genes Cells 12:1215–1223. doi: 10.1111/j.1365-2443.2007.01128.x. [DOI] [PubMed] [Google Scholar]
- 47.Ford CE, Qian Ma SS, Quadir A, Ward RL. 2013. The dual role of the novel Wnt receptor tyrosine kinase, ROR2, in human carcinogenesis. Int J Cancer 133:779–787. doi: 10.1002/ijc.27984. [DOI] [PubMed] [Google Scholar]
- 48.Shiverick KT, King A, Frank H, Whitley GS, Cartwright JE, Schneider H. 2001. Cell culture models of human trophoblast II: trophoblast cell lines—a workshop report. Placenta 22(Suppl A):S104–S106. [DOI] [PubMed] [Google Scholar]
- 49.LaMarca HL, Sainz B Jr, Morris CA. 2004. Permissive human cytomegalovirus infection of a first trimester extravillous cytotrophoblast cell line. Virol J 1:8. doi: 10.1186/1743-422X-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doyon P, van Zuylen WJ, Servant MJ. 2013. Role of IkappaB kinase-beta in the growth-promoting effects of angiotensin II in vitro and in vivo. Arterioscler Thromb Vasc Biol 33:2850–2857. doi: 10.1161/ATVBAHA.113.302487. [DOI] [PubMed] [Google Scholar]
- 51.van Zuylen WJ, Garceau V, Idris A, Schroder K, Irvine KM, Lattin JE, Ovchinnikov DA, Perkins AC, Cook AD, Hamilton JA, Hertzog PJ, Stacey KJ, Kellie S, Hume DA, Sweet MJ. 2011. Macrophage activation and differentiation signals regulate schlafen-4 gene expression: evidence for Schlafen-4 as a modulator of myelopoiesis. PLoS One 6:e15723. doi: 10.1371/journal.pone.0015723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pollheimer J, Loregger T, Sonderegger S, Saleh L, Bauer S, Bilban M, Czerwenka K, Husslein P, Knofler M. 2006. Activation of the canonical wingless/T-cell factor signaling pathway promotes invasive differentiation of human trophoblast. Am J Pathol 168:1134–1147. doi: 10.2353/ajpath.2006.050686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sonderegger S, Haslinger P, Sabri A, Leisser C, Otten JV, Fiala C, Knofler M. 2010. Wingless (Wnt)-3A induces trophoblast migration and matrix metalloproteinase-2 secretion through canonical Wnt signaling and protein kinase B/AKT activation. Endocrinology 151:211–220. doi: 10.1210/en.2009-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ford CE, Punnia-Moorthy G, Henry CE, Llamosas E, Nixdorf S, Olivier J, Caduff R, Ward RL, Heinzelmann-Schwarz V. 2014. The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecol Oncol 134:338–345. doi: 10.1016/j.ygyno.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Hirsch AJ, Shenk T. 1999. Human cytomegalovirus inhibits transcription of the CC chemokine MCP-1 gene. J Virol 73:404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sonderegger S, Husslein H, Leisser C, Knofler M. 2007. Complex expression pattern of Wnt ligands and frizzled receptors in human placenta and its trophoblast subtypes. Placenta 28(Suppl A):S97–S102. [DOI] [PMC free article] [PubMed] [Google Scholar]