

ABSTRACT
HIV-1 infection leads to the progressive depletion of the CD4 T cell compartment by various known and unknown mechanisms. In vivo, HIV-1 infects both activated and resting CD4 T cells, but in vitro, in the absence of any stimuli, resting CD4 T cells from peripheral blood are resistant to infection. This resistance is generally attributed to an intracellular environment that does not efficiently support processes such as reverse transcription (RT), resulting in abortive infection. Here, we show that in vitro HIV-1 infection of resting CD4 T cells induces substantial cell death, leading to abortive infection. In vivo, however, various microenvironmental stimuli in lymphoid and mucosal tissues provide support for HIV-1 replication. For example, common gamma-chain cytokines (CGCC), such as interleukin-7 (IL-7), render resting CD4 T cells permissible to HIV-1 infection without inducing T cell activation. Here, we find that CGCC primarily allow productive infection by preventing HIV-1 triggering of apoptosis, as evidenced by early release of cytochrome c and caspase 3/7 activation. Cell death is triggered both by products of reverse transcription and by virion-borne Vpr protein, and CGCC block both mechanisms. When HIV-1 RT efficiency was enhanced by SIVmac239 Vpx protein, cell death was still observed, indicating that the speed of reverse transcription and the efficiency of its completion contributed little to HIV-1-induced cell death in this system. These results show that a major restriction on HIV-1 infection in resting CD4 T cells resides in the capacity of these cells to survive the early steps of HIV-1 infection.
IMPORTANCE A major consequence of HIV-1 infection is the destruction of CD4 T cells. Here, we show that delivery of virion-associated Vpr protein and the process of reverse transcription are each sufficient to trigger apoptosis of resting CD4 T cells isolated from peripheral blood. While these 2 mechanisms have been previously described in various cell types, we show for the first time their concerted effect in inducing resting CD4 T cell depletion. Importantly, we found that cytokines such as IL-7 and IL-4, which are particularly active in sites of HIV-1 replication, protect resting CD4 T cells from these cytopathic effects and, primarily through this protection, rather than through enhancement of specific replicative steps, they promote productive infection. This study provides important new insights for the understanding of the early steps of HIV-1 infection and T cell depletion.
INTRODUCTION
Early human immunodeficiency virus type 1 (HIV-1) infection is characterized by rapid and substantial depletion of both activated and resting CD4 T cells (1). The acute phase of both human HIV-1 infection and simian immunodeficiency virus (SIV) infection of macaques is characterized by ≥90% of viral RNA+ cells displaying a resting phenotype (2). It is during this stage that the main HIV-1 reservoir and source of virus rebound in vivo is established in resting memory T cells. Latency can be directly established upon infection of resting CD4 T cells (3–7). In comparison to that in activated T cells, HIV-1 infection is inefficient in resting CD4 T cells in vitro (8–10). Multiple blocks have been described for key steps of HIV-1 infection in resting CD4 T cells, including inefficient reverse transcription (RT), nuclear import, integration, transcription, and virus release (for review, see references 11 and 12).
In vitro study of HIV-1 infection of resting CD4 T cells has relied predominantly on cells drawn from peripheral blood, which is conveniently sampled from both uninfected and infected individuals. However, common culture methods which deprive the cells of survival factors such as hormones and cytokines normally present in circulation create a nonphysiological situation of increased stress (13, 14) which could exacerbate resting CD4 T cell resistance to infection. Common gamma-chain cytokines (CGCC), such as interleukin-7 (IL-7), IL-2, IL-15, and IL-4, are present at steady state (IL-7) to maintain T cell survival (15, 16) and homeostasis (17) or during immune responses to assist cell proliferation, activation, and differentiation (IL-2, IL-4, and IL-15). When treated with these cytokines in vitro, resting peripheral blood CD4 T cells become more permissive to HIV-1 infection (4, 18, 19). HIV-1 replication in vivo occurs predominantly in lymphoid tissues (LT) and mucosa where IL-7 (20) and IL-4 (21) are abundant. Cells drawn from LT are highly infectible ex vivo (22). Interestingly, T cell activation appears low to nonexistent under the influence of these factors (4, 19), and we have utilized IL-4 to assist infection of blood-derived cells to function as an in vitro model of HIV-1 replication and latency in resting CD4 T cells (4). However, it is still unclear which effects of cytokine treatment permit HIV-1 productive infection in resting CD4 T cells (18).
Several years ago, the presence of apoptotic cells that were not productively infected with HIV-1 was observed in lymphoid organs, suggesting HIV-1-mediated bystander killing (23). Several mechanisms for HIV-1-induced bystander killing have been reported, involving various HIV-1 and immune components, including the Vpr protein (24–28). Recently, it has been demonstrated that failure to complete reverse transcription (abortive infection) triggers cell death of resting CD4 T cells within tonsil cell explants (43, 63). In this system, early products of HIV-1 reverse transcription triggered a cascade of proinflammatory events leading to resting, but not activated, T cell death by caspase 1-dependent pyroptosis.
In the present study, we show that the productive infection of resting peripheral blood CD4 T cells is to a large extent limited by HIV-1-induced cell death. We observed that this death was triggered by both RT-dependent and Vpr-dependent pathways. T cell death presented features of apoptosis, with early cytochrome c release and caspase 3/7 activation. This killing could be immediately aborted by submitogenic cytokine treatment added contemporaneously with, or after, infection. Although cytokines very modestly enhanced early HIV-1 replication steps, including reverse transcription, cytokine treatment blocked cell death even in the absence of RT completion. In these cells, artificially aborting HIV-1 reverse transcription did not increase cell death, and increasing HIV-1 RT efficiency with SIV Vpx provided only a modest increase in cell survival. Thus, our data suggest that HIV-1-induced T cell death during the early steps of infection of resting peripheral blood CD4 T cells is a primary cause of abortive infection, rather than the other way around. Strategies aiming to prevent T cell depletion by suppressing programmed cell death might lead to an enhancement of HIV-1 infection and potentially to an increase in the HIV-1 reservoir.
MATERIALS AND METHODS
Viruses and VLPs.
Schematics of reporter viruses and virus-like particle (VLP) genomes are shown in Fig. 1. NLENG1-ESI (29), NLENHSA-ESI (30), NLHGESI (4), and Y1IΔRTΔVpr (RT-N136Y) (4) have been previously described. NLENG1-ESI is referred to as “G1ESI” in figures for space reasons. All green fluorescent protein (GFP) and heat-stable antigen (HSA) reporter viruses were restricted to a single round of infection by mutation of the env gene and were pseudotyped with HIV-1 Envelope provided by cotransfection of 293T producer cells with an NL4-3 envelope expression plasmid as previously described (29). Y1IΔRTΔVpr-p6X was constructed from Y1I-RT-N136Y by ablation of vpr through insertion of the HSA/vpr region from NLHGESI and replacement of the p6gag amino acid 17 to 26 coding region with the corresponding coding sequence from SIVmac239, based on virus SIVp6 17-26 from reference 31. This virus-like particle (VLP) packages Vpx protein following cotransfection with a Vpx expression vector, pCI-Vpx. pCI-Vpx was constructed by inserting the Vpx coding region from SIVmac239 into the expression vector pCI (Promega).
FIG 1.

Schematics of viruses used in this study. Viruses in panels A to C have been described previously (24, 29). Y1IΔRTΔVpr-p6X is described in Materials and Methods. Note that for panels C and D, the ablation of reverse transcriptase activity prevents all de novo virus expression in target cells, including YFP fluorescence. aa, amino acids.
Cells.
CD4+ T cells were isolated by negative selection of healthy HIV-negative donor peripheral blood using the Dynabeads Untouched magnetic separation kit (Invitrogen) as previously described (29). Blood was purchased from the New York Blood Center. CD4+ T cells were typically ≥99% CD25− CD69− CD38dim HLA-DR− quiescent cells. CD4+ T cells were cultured at 2 × 106 cells per ml in Advanced RPMI 1640 (Life Technologies) with 10% fetal bovine serum (Atlanta Biologicals) plus 1% penicillin and streptomycin (Life Technologies), 1% l-glutamine (HyClone), and 50 μM β-mercaptoethanol (Sigma). IL-7 (2 ng/ml; BioLegend) or IL-4 (12.5 ng/ml; R&D Systems) was added once on the day of infection or when indicated. Where indicated, cells were activated with Dynabeads human T-activator CD3/CD28 (Invitrogen), per the manufacturer's instructions, for the amount of time indicated. IL-2 (50 U/ml; obtained from NIH) was added 24 h after bead stimulation.
Infections.
Virus stocks were filtered (0.45 μm) and then treated with Benzonase (Novagen; 50 U/ml, 30 min, 37°C) to eliminate contaminating plasmids. Virus titers were determined by TaqMan reverse transcription-quantitative PCR (RT-qPCR) for HIV-1 RNA (target in integrase) and normalized to 125 to 320 ng p24gag equivalents per million cells. Infections were performed by spinoculation (4) in the presence of 5 μg/ml DEAE-dextran (Sigma) for 2 h at 1,200 × g and 37°C. T cells were then washed twice and placed back in culture. Mock infections were performed identically in the absence of the virus inoculum. Antiviral treatments included efavirenz (EFV; 3 μM), zidovudine (AZT; 5 μM or as indicated), and raltegravir (RAL; 1 μM). All antiretrovirals were obtained from the NIH AIDS Research and Reference Reagent Program. In some experiments, cells were treated with staurosporine (BioVision; 1 μM) or deoxynucleosides (50 μM).
Survival.
In most experiments, T cell survival was assessed by flow cytometry using forward and side scatter parameters and 7-aminoactinomycin D (7AAD) staining. 7AAD (BD Pharmingen; 10 μg/ml) was added 15 min prior to flow cytometer acquisition.
Annexin V/SYTOX staining.
Annexin V Pacific Blue (Invitrogen) staining was performed per the manufacturer's instructions and followed by SYTOX AADvanced dead cell stain (Life Technologies) incorporation. SYTOX functions similarly to 7AAD by incorporating into the DNA of permeable cells.
Cytochrome c release assay.
Mitochondrial depolarization was assessed by intracellular staining for cytochrome c as previously described (33). Briefly, cell plasma membranes were permeabilized with digitonin for 3 to 5 min to eliminate cytoplasmic but not mitochondrial cytochrome c. Cells were then immediately fixed with 4% paraformaldehyde (PFA) and subsequently stained in a saponin-containing buffer with a fluorescein isothiocyanate (FITC)-conjugated anti-cytochrome c antibody (eBioscience; 2B5).
Caspase activation detection.
Activated caspase 3/7 was stained using a CellEvent caspase-3/7 green flow cytometry assay kit (Life Technologies) per the manufacturer’s instructions, followed by Sytox staining. Caspase 1 activation was measured using the FLICA 660 caspase 1 assay (ImmunoChemistry Technologies) per the manufacturer's instructions, and measurement was followed by 4′,6-diamidino-2-phenylindole (DAPI) staining (Life Technologies).
Cell cycle analysis.
Cells were stained for DNA and RNA content by 7AAD-pyronin Y (PY) staining as previously described (34, 35). For these experiments, we infected T cells with the HSA reporter HIV-1 instead of GFP reporter HIV-1, as cytosolic GFP fluorescence is lost during the saponin permeabilization step. Surface HSA expression was detected by staining with anti-mCD24 (M1/69; BD Pharmingen) prior to PY-7AAD staining. For PY-7AAD, 1 × 106 cells from each condition were stained. Permeabilization was performed with 0.02% saponin, and cells were successively incubated with 10 μg/ml 7AAD, 10 μg/ml actinomycin D, and 0.1 mg/ml PY.
PCR methods for quantification of HIV-1 DNA and RNA.
All methods, including those using primers and TaqMan probes, have been previously described (4, 30). Briefly, DNA from cells was purified using the DNeasy blood and tissue kit or the AllPrep kit (Qiagen). Cell-associated RNA and viral RNA in culture medium were prepared using the RNeasy kit (Qiagen). DNA was analyzed using the QuantiTect probe PCR kit (Qiagen), and RNA was analyzed by RT-qPCR using the QuantiTect probe RT-PCR kit (Qiagen). Integrated HIV-1 was detect by Alu PCR as described previously (4).
RESULTS
Common gamma-chain cytokine treatment is sufficient for the survival and productive HIV-1 infection of resting CD4 T cells.
Resting peripheral blood CD4 T cells were isolated by negative selection from HIV-1-negative donors (4), infected with the HIV-1 NL4-3-derived GFP reporter virus pseudotyped with HIV-1 Envelope (Fig. 1A) (36), and then monitored for GFP expression and survival over 7 days (Fig. 2). In order to test the ability of IL-7 to influence productive infection, cells were infected, then divided into separate wells, and then treated once with low-dose IL-7 (2 ng/ml) at different times after infection or with nothing. IL-7 increased the number of productively infected cells, as has been previously demonstrated (4, 18, 19). In the absence of IL-7, 2% of the initial population of T cells became GFP positive, whereas 16.9% of the cells became GFP positive when treated with IL-7 on day 0. While the yield of GFP+ cells decreased progressively when IL-7 treatment was delayed, more than 65% of the maximum was still obtained when IL-7 was added 2 days after infection (Fig. 2A to C). Similar results were obtained using another common gamma-chain cytokine, IL-4, albeit with slightly less efficiency (Fig. 2D).
FIG 2.

IL-7 treatment at any time after infection is sufficient to rescue cells from HIV-1-induced T cell death and allow productive infection. Resting peripheral blood CD4 T cells were labeled with eFluor670, infected with the single-round HIV-1 GFP reporter virus NLENG1-ES-IRES, and then placed in culture for up to 7 days. IL-7 (2 ng/ml) was added once at the indicated times. Cells were harvested daily, stained with 7AAD, and analyzed by flow cytometry for GFP expression, proliferation, and survival. (A) Kinetics of GFP expression with IL-7 treatment at various times after infection or with no treatment. The y axis is the percentage of live GFP+ cells normalized to total cells, live or dead. (B) Kinetics of cell death following infection. Forward and side scatter plus 7AAD labeling were used to gauge cell viability in flow cytometry. Data show cells treated with IL-7 or not treated at various times following infection. dpi, day postinfection. ** and * indicate survival on day 7 of mock-infected cells in the presence or absence of IL-7, respectively. (C) Stacked bar graph combining GFP expression and survival data collected on day 7 from results in panels A and B. The x axis indicates the day of IL-7 treatment. Ø indicates no IL-7. The total height of bars (black plus green bars) equals the percentage of cells alive. (D) Effect of IL-4 treatment on HIV-1 infection and CD4 T cell survival. Stacked bar graph combining GFP expression and survival equivalent to data in panel C but using IL-4 (12.5 ng/ml) instead of IL-7.
In the same experiment, we also analyzed the influence of IL-7 on cell survival measured using cell morphology (scatter profile) and 7AAD incorporation (Fig. 2B and C). Uninfected and untreated cells remained 80% viable over 7 days in culture, while IL-7-treated uninfected cells remained >95% viable (Fig. 2B and C). When T cells were infected in the absence of cytokines, a sharp drop in cell survival was observed between days 2 and 4 postinfection. When T cells were treated with IL-7 on the day of infection (day 0), viability was restored to the level of uninfected IL-7-treated cells. Death in infected cultures began prior to the maximum number of GFP+ cells emerging, suggesting that cell killing was triggered by early replication events. IL-7 treatment at later days was able to abort the ongoing cell death, indicating that the killing mechanism was reversible. Similar results were again obtained using IL-4, but also with somewhat lower efficiency (Fig. 2D).
Importantly, IL-7 protection preserved the viability of both the GFP-positive and -negative cells in the infection cultures, suggesting that cell death was happening in cells destined for either productive or nonproductive infection. In fact, most of the HIV-induced cell death in cultures treated with IL-7 after day 3 was in cells which would never have become productively infected (i.e., a reduction in the height of the black portion of the bars in Fig. 2C). When looking at the effect of IL-7 on T cell cycle progression using pyronin Y and 7AAD staining, 78% of the cells were still in G0, versus essentially all untreated cells remaining in G0 (Fig. 3). Importantly, there was no difference in cell cycle status between cells expressing and cells not expressing HIV-1, which held true for both untreated and IL-7-treated cells. Control cells treated with anti-CD3/CD28 beads after infection became activated, and the productively infected activated cells were enriched in the G2/M phase, consistent with cell cycle arrest by Vpr (37, 38).
FIG 3.
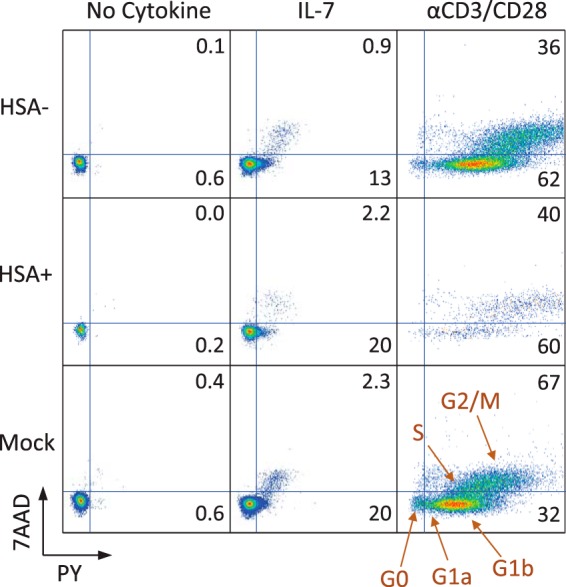
Productive infection does not correlate with cell cycle progression. Resting CD4 T cells were infected with an HIV-1 reporter virus expressing cell surface protein heat-stable antigen (HSA; mCD24) (30) and treated or not with IL-7. At 1 day postinfection, some cells were activated with anti-CD3/CD28 activation beads. At 4 days postinfection, cells were stained with anti-mCD24 and PY plus 7AAD for cell cycle progression.
The percentage of cells productively infected in the absence of cytokine was variable among donors, ranging from 2.1% to 6.6%. Interestingly, the reducing agent β-mercaptoethanol, which we add to culture medium to extend the life of labile factors such as cytokines, had a positive effect on productive infection in both the presence and absence of cytokine treatment (Fig. 4A). We also examined the specific survival of naive and memory T cell subsets after 5 days of infection based on the expression of markers CD45RO, CCR7, and CD62L (Fig. 4B). We observed that all the subsets generated productively infected cells in the absence of cytokine and that IL-7 enhanced both productive infection and cell survival. Memory subsets (CD45RO+) were more infectible in the absence of cytokine than were naive (CD45RO−) cells, especially effector memory cells (CD45RO+ CCR7− and CD45RO+ CD62L−).
FIG 4.
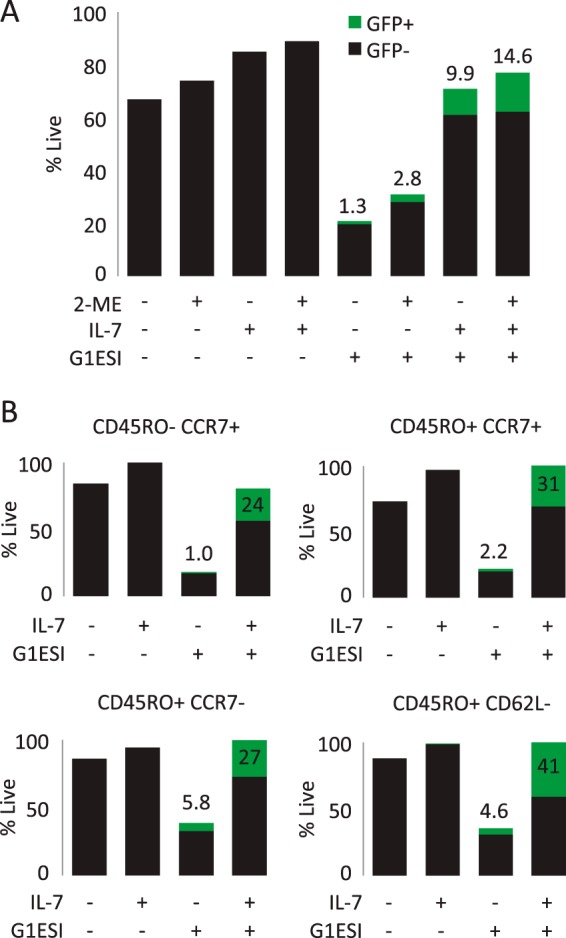
Productive infection and survival of individual T cell subsets. (A) Effect of β-mercaptoethanol (2-ME) on productive infection and T cell survival in presence or absence of IL-7 treatment. T cell preparation and infection were performed in the presence or absence of β-mercaptoethanol in the culture medium. Numbers above bars indicate the percent GFP+ cells. (B) Productive infection and survival of T cell subsets identified at 5 days postinfection based on expression of CD45RO, CCR7, and CD62L. Percentages were normalized to the maximal survival condition. Numbers above bars indicate the percent GFP+.
HIV-1-induced apoptosis of peripheral blood resting CD4 T cells.
We next asked what mechanism was driving resting CD4 T cell death in this system. The rapid decrease in the cell size (Fig. 5A) and early exposure of phosphatidylserine (Fig. 5B) suggested that the cells were undergoing classic apoptosis. We then examined cytochrome c release from mitochondria (Fig. 5C), observing a clear reduction in staining as an indication of intrinsic apoptosis induction. Activitation of caspase 3/7, the common effector caspases that are activated downstream of cytochrome c release, was also induced (Fig. 5D). We observed a clear subset of cells positive for active caspase 3/7 in the absence of cell membrane permeabilization (DNA labeling by SYTOX), demonstrating that caspase activation preceded membrane permeability, a further indication that the infected cells were undergoing classic apoptosis. However, we were not successful in our attempts to block T cell death with inhibitors of caspase 3 and 9 activation (not shown), and the pan-caspase inhibitor Z-VAD-fmk was extremely toxic to the cells in our system (not shown). Caspase 1-dependent pyroptosis has recently been demonstrated to be triggered by abortive reverse transcripts following infection of tonsil cell cultures (43, 63) and gut lamina propria CD4 T cells (39). We tested caspase 1 activation by flow cytometry, observing a consistent activation of caspase 1 in dying cells and importantly also in control cells induced to apoptosis by staurosporine (Fig. 5E). Caspase 1 activation was observed only in cells already positive for phosphatidylserine externalization and cell shrinkage (Fig. 5F); thus, caspase 1 activation was a downstream event, not an initiator of the programmed cell death observed. Consistent with this conclusion, caspase 1 inhibitors (Z-WEHD-fmk and Z-YVAD-fmk) were incapable of blocking HIV-1-induced T cell death (not shown).
FIG 5.

HIV-1 infection-induced apoptosis of resting CD4 T cells. CD4 T cells were infected or mock infected with the single-round GFP reporter virus in the presence or absence of IL-7 and analyzed at 3 days postinfection. (A) Morphology of CD4 T cells and DNA staining by SYTOX (cell death-dependent DNA stain) incorporation. The box indicates small forward-scatter (FSC)-low cells. (B) Annexin V and SYTOX staining. (C) Cytochrome c staining. The dimmer peak corresponds to cytochrome c release from mitochondria. (D) Caspase 3/7 activation and SYTOX incorporation. (E) Caspase 1 activation and DAPI incorporation. (F) Phosphatidylserine externalization and caspase 1 activation in CD4 T cells after infection in the absence of cytokine. Caspase 1 activation was measured with the FLICA 660 caspase 1 assay 3 days after infection. Annexin V labeling was performed immediately after FLICA staining. The histogram on the right shows size (forward scatter [FSC]) of cells infected in the absence of cytokine. FSC was reduced by the time of phosphatidylserine externalization and prior to caspase 1 activation. Panels A to E show results from one experiment representative of at least 3 experiments for each condition.
IL-7 treatment at the time of infection substantially reduced the HIV-induced increase of all indicators of apoptosis. Infected and mock-infected cells treated with IL-7 were also very similar, with only an HIV-induced increase in DNA staining observed between these two conditions. CGCC are known inducers of the antiapoptotic factor Bcl-2 (14, 16, 40, 41), which blocks the mitochondrial pathway of apoptosis, and as expected, IL-7 and IL-4 upregulated Bcl-2 expression, with IL-7 increasing Bcl-2 more than 3-fold and IL-4 increasing expression by 1.9-fold as measured by flow cytometry (data not shown). Cytokine-induced modifications of the balance of pro- and antiapoptotic Bcl family molecules are likely effectors for the survival effect observed here (14, 40). The inhibition of Stat5 and phosphatidylinositol 3-kinase (PI3K), two important signaling pathways downstream of the IL-7 receptor, did not interfere with the protective effect of IL-7 (not shown). Further investigation will be required to understand the precise survival mechanisms induced by these cytokines.
Identification of RT-dependent and Vpr-dependent apoptosis.
We next investigated which components of HIV-1 infection lead to resting CD4 T cell death. Recently, it has been shown that the process of integration into activated T cells can trigger apoptosis and that inhibiting integration with raltegravir blocks this cause of cell death (42). We tested raltegravir in our system, finding that it did not increase cell survival (Fig. 6A). Addition of the nonnucleoside RT inhibitor efavirenz (EFV) at the time of infection (hour 0) prevented most but not all HIV-1-induced cell death (Fig. 6B), suggesting that reverse transcripts were also a major trigger of HIV-1-induced T cell death, though not the only inducers. Interestingly, addition of the nucleoside RT inhibitor zidovudine (AZT) at a conventional dose (5 μM) was very inefficient in preventing HIV infection of resting CD4 T cells (Fig. 6C) and failed to prevent HIV RT-induced cell death. Intensification of AZT treatment by 50-fold (250 μM) to near-toxic concentrations could reduce the productive infection and partially protect the cells.
FIG 6.

Early HIV-1-induced death of resting peripheral blood T cells results from the additive effects of reverse transcription and virion-borne Vpr. (A) Effect of raltegravir on cell death. CD4 T cells were infected with GFP reporter virus in the presence or absence of raltegravir (1 μM) and with or without IL-4 or IL-7 treatment. Data were collected 6 days postinfection. Ø, no interleukin. (B) Stacked bar graph combines GFP expression (green) and cell viability (total bar height) assessed 6 days after infection. CD4 T cells were infected with the single-round GFP reporter virus or mock infected (centrifuged with no virus). At the indicated times relative to infection, the nonnucleoside RT inhibitor efavirenz (EFV) was added. Where indicated, IL-7 was added on the day of infection. The asterisk indicates the addition of EFV 24 h after infection. (C) Effects of AZT (indicated micromolar concentrations) and EFV (3 μM) on survival and GFP expression. Numbers above bars indicate the percent GFP+. (D) Independent and additive contributions of virion Vpr and reverse transcription to cell death. Cells were infected with a virus that expresses all HIV-1 proteins except Envelope or with mutant viruses that do not express either Vpr or an active reverse transcriptase or both. In each case, cells were treated or not with EFV or IL-7. Stacked bar graph combines GFP expression and cell viability assessed 6 days after infection. Panels A and B show results from one representative experiment of 3. Data are averages from 3 independent infections performed in the same experiment. The arrow indicates that the addition of EFV completely prevented all cell death induced by this virus. (E) Vpr-independent killing is protected by EFV at a high multiplicity of infection. Survival experiment equivalent to that shown in Fig. 3B using 3 times the dose of the NLHGESI ΔVpr virus.
In order to further characterize the cell death induced by HIV-1 infection, we performed a time course of EFV addition after infection (Fig. 6B). Addition of EFV 24 h after infection failed to block most of the RT-dependent cell death but did block all GFP expression. Since GFP expression depends upon completion of reverse transcription, this is a strong indication that very few to no genomes had completed reverse transcription by 24 h. From this, we infer that incomplete RT products were sufficient to trigger this component of cell death, consistent with the death of abortively infected resting tonsil CD4 T cells (43). After 72 h of infection, EFV could not rescue cell death but could decrease GFP expression compared with no EFV treatment (0.4% versus 1.4% GFP+ cells), indicating that even at this late time point most reverse transcription was still incomplete.
Next, we investigated the cause of the component of cell death that was not dependent on reverse transcription. A likely candidate was HIV-1 Vpr (for a review, see reference 44). Cell death induced by Vpr is usually associated with its effect on cell cycle arrest in proliferating cells, but this has also been described in nonproliferating cells (45, 46). We infected cells with a panel of 4 viruses that delivered virion-associated Vpr, functional RT, both together, or neither (Fig. 6D). The Vpr mutant virus that contains no virion-associated Vpr induced cell death, but less than the virus which delivered both Vpr and RT. Addition of EFV completely prevented all cell death induced by this virus (Fig. 6D, arrow). In an additional experiment, we increased the virus dose, resulting in a stronger infection and more cell death caused by the ΔVpr virus; nevertheless, all HIV-1-induced cell death was still prevented by EFV (Fig. 6E). A ΔRT ΔVpr double mutant induced no cell death compared with mock-infected T cells, confirming that Vpr and reverse transcription were the key components for HIV-1-induced cell death in resting peripheral blood CD4 T cells. Importantly, treatment with IL-7 rescued cells from both RT- and Vpr-dependent cell death (Fig. 6D).
Cytokine rescue does not require completion of reverse transcription.
As stated above, abortive reverse transcription can trigger cell death in resting tonsil (43) and lamina propria (39) CD4 T cells, and our results demonstrate that reverse transcription is also toxic to peripheral blood CD4 T cells. However, it remains to be tested whether agents which facilitate reverse transcription reduce cell death (tested in Fig. 8), and whether agents such as cytokines which inhibit cell death depend upon facilitating completion of reverse transcription, or if they can protect cells in the presence of abortive reverse transcripts. To test the latter idea, we investigated the influence of IL-7 on cell survival in the presence of abortive reverse transcripts. As we inferred from Fig. 6B, EFV at 24 h postinfection resulted in abortive reverse transcription, seen here both as a reduction in early reverse transcripts and most profoundly as a reduction in complete reverse transcripts (Fig. 7A), resulting in cell death as observed in Fig. 6B (Fig. 7B). (The effects of IL-7 on RT kinetics are further investigated in Fig. 8.) IL-7 treatment resulted in a modest increase in completed reverse transcripts; however, a direct effect on the process of reverse transcription is difficult to infer because of the confounding influence of IL-7 on infected cell survival. Unsurprisingly, IL-7 did not overcome the block to reverse transcription imposed by EFV; however, IL-7 completely rescued cells from death whether or not reverse transcription was aborted with EFV (Fig. 7B). Thus, the protection from cell death provided by IL-7 was independent of the efficiency or completion of reverse transcription.
FIG 8.

Influence of increased RT efficiency on infection and cell death. Vpx from SIVmac239 was delivered by VLP in trans at the time of infection. Where indicated, IL-7 or efavirenz (EFV) was added at the time of infection. (A) Kinetics of GFP expression. (B) Kinetics of HIV-1 2-LTR DNA circle formation quantified by qPCR. (C) Kinetics of late HIV-1 cDNA formation quantified by quantitative PCR. (D) Kinetics of cell death. (E) GFP expression and survival on day 6. (F) Effect of deoxynucleosides (dNs) on infection and survival. T cells were infected with the indicated dose of virus (nanograms of p24gag per 106 cells) and treated the same day with dNs (50 μM). Data were collected on day 5 postinfection. Numbers above bars indicate the percent GFP+. Results in panels A, B, and C are from one experiment representative of 3. Results in panels D and E are averages and standard deviations from 3 independent experiments. Results in panel F are representative of 3 experiments. dpi, day postinfection.
FIG 7.

Rescue of resting CD4 T cells by IL-7 does not require completion of HIV-1 reverse transcription. CD4 T cells were infected with a single-round GFP reporter HIV-1 virus and incubated overnight without cytokine. Where indicated, the cells were treated with EFV 24 h after infection. Where indicated, IL-7 was added 27 h postinfection. (A) Quantity of early and late products of RT. At 6 days after infection, cells were extracted for DNA content and RT efficiency was assessed by measuring the levels of incomplete “early” and near-full-length “late” HIV-1 cDNA by qPCR. (B) Stacked bar graph combining GFP expression and survival assessed 6 days after infection. Results in panels A and B are from one representative experiment of two.
Influence of enhancing reverse transcription on infection and cell death.
It has been recently demonstrated that the cellular protein SAMHD1, which is constitutively expressed in several nondividing cell types such as monocytes/macrophages (47), dendritic cells (48, 49), and resting CD4 T cells (50), functions as a restriction factor against retroviral infection by limiting the efficiency of reverse transcription. This effect was initially associated with the capacity of SAMHD1 to deplete the intracellular deoxynucleoside triphosphate (dNTP) pool, although, or in addition, it has recently been found that SAMHD1 RNase activity directly degrades retroviral cDNAs (51). Vpx, an accessory protein present in HIV-2 and most simian immunodeficiency viruses but absent from HIV-1, is capable of inducing SAMHD1 degradation and enhancing reverse transcription (52). Delivery of Vpx in trans via coinfection of cells with HIV-1 and virus-like particles (VLPs) containing SIVmac239 Vpx protein greatly enhances productive HIV-1 infection of myeloid cells (53) as well as resting CD4 T cells (50, 54).
Here, we were interested in whether the facilitation of HIV-1 RT efficiency by Vpx would influence HIV-1-induced cell death in resting peripheral blood CD4 T cells. We constructed an HIV-1-based virus-like particle (VLP) lacking both reverse transcriptase activity and Vpr expression and which also contained a modified p6Gag protein (31) to incorporate SIVmac239 Vpx in the virions (Fig. 1D). This VLP can bind and fuse to CD4+ CXCR4+ cells and deliver Vpx protein but will not undergo reverse transcription or generate HIV proteins or yellow fluorescent protein (YFP) in target cells. We observed that coinfection of resting CD4 T cells with the Vpx VLP accelerated the appearance of GFP+ cells by 1 day and increased the total production of GFP+ cells severalfold (Fig. 8A). As in Fig. 2, IL-7 also increased the production of GFP+ cells but not the kinetics of their first appearance. IL-7 further increased the production of GFP+ cells in the Vpx+ infections. Importantly, the production of late reverse transcripts (Fig. 8B) and 2-long-terminal-repeat (LTR) circles, which are generated in the nucleus and function as a measure of nuclear import of the preintegration complex (Fig. 8C), were greatly accelerated by Vpx, while IL-7 had no further influence on reverse transcription in the Vpx infection. Although IL-7 alone induced an increase in the number of late reverse transcripts and 2-LTR circles, this may be due to its survival promotion rather than an influence on the process of reverse transcription. This would be consistent with the initial appearance of these DNA species being unaffected by IL-7. One clear inference to be made here is that the mechanisms of infection enhancement by IL-7 and Vpx are therefore complementary rather than overlapping. Prior studies of the effects of Vpx on HIV-1 infection have typically employed endpoint analysis to reveal increased yields of productively infected cells, while here we employed kinetic analysis to reveal an acceleration in the completion of reverse transcription in the presence of Vpx.
Despite this strong improvement of HIV-1 reverse transcription, HIV-1-induced cell death was also observed in the presence of Vpx (no IL-7), including both RT-dependent (reduced by 31% compared to that in the absence of Vpx, P = 0.031) and Vpr-dependent (statistically identical, P = 0.21) components (Fig. 8D and E). The reduction in RT-dependent cell death and increase in productively infected T cells observed in the presence of Vpx treatment seem likely to be a result of increased RT completion; however, IL-7 was able to further increase the productive infection of Vpx-treated cells, indicating that many cells destined for productive infection were still dying. Similar results were obtained by applying deoxynucleosides (dNs) at the time of infection to enhance reverse transcription (Fig. 8F) (55).
DISCUSSION
Despite being considered to harbor a significant viral reservoir contributing to HIV-1 rebound after cessation of antiretroviral therapy (56), resting peripheral blood CD4 T cells have been historically observed to be resistant to direct HIV-1 infection in vitro (8, 9, 57, 58). This resistance occurs at several levels, including the presence of restriction factors, which lowers reverse transcription efficiency; a low dNTP concentration, which also reduces reverse transcription kinetics; cytoskeletal actin/cofilin inhibition; and other downstream events which occur less efficiently in resting CD4 T cells. Progression of T cells to the G1b cell cycle phase was initially considered necessary for completion of reverse transcription and productive infection (18, 57). However, it has subsequently been demonstrated that productive infection of quiescent peripheral blood CD4 T cells in G0-G1a can proceed, though at considerably lower speed and efficiency than those of activated T cells (5, 59, 60). We confirm this and observe that completion of HIV-1 reverse transcription in resting CD4 T cells peaked around 2 to 3 days postinfection in cells in G0-G1a and that productive infection, measured by GFP expression, peaked after day 3. This is very slow in comparison to the infection of activated T cells or T cell lines, where completion of reverse transcription, nuclear import, and integration can all be detected within a few hours of infection (58). Addition of a low dose of IL-7 after infection had very little effect on RT kinetics, but it did increase the yield of RT products and GFP+ cells, which we attribute primarily to a reduction in cell death (discussed below). Thus, we did not observe any correlation between HIV-1 early expression and cell cycle progression, such that cells clearly still in G0 or G1a were infected and became GFP+. The enhancement of reverse transcription and overall infection efficiency by Vpx without IL-7 further argues that cell cycle progression per se does not determine permissiveness to infection.
In a study of IL-7, resting CD4 T cells, and HIV infection, Ducrey-Rundquist et al. observed a correlation between T cell cycle progression to G1b induced by mitogenic levels of IL-7 and productive infection. However, this particular study employed Env− viruses pseudotyped with the vesicular stomatitis virus envelope G protein (VSV-G) (18). It has since been demonstrated that VSV-G is not suitable for infection of cells lacking the low-density lipoprotein (LDL) receptor for VSV-G, including resting CD4 T cells (61, 62), so it is likely that VSV-G pseudotyping favored the infection of cells that were stimulated beyond G0. In our experiments, we employed only viruses bearing HIV-1 Envelope (CXCR4-tropic) and we treated the cells only after infection and with a submitogenic dose of IL-7, thus limiting its effect on cell cycle progression, especially during the first days, when the fate of the cells and of infection is decided (4). It has been observed that IL-7 promotes generation of infectious virions in resting CD4 T cells, enhancing env expression (5). In our present work, we were not concerned about the overall replicative capacity of HIV-1 within resting CD4 T cells but rather the events leading to productive infection. It is possible that IL-7 enhancement of cellular resistance to viral toxicity contributes to the reported increase in infected cells and viruses expressing HIV-1 envelope (5).
The major effect of IL-7 was to prevent cell death induced by HIV-1 infection, both the Vpr-induced and reverse transcription-induced components, which we showed to result in apoptosis. Importantly, the survival effect of the cytokine treatment increased both the fraction of productively infected cells (GFP+ cells) and the survival of GFP-negative cells bearing genomes that did not express viral proteins. This enhancement of the viability of all cells is potentially important, as it suggests that factors such as IL-7 and IL-4, which promote infection through viability enhancement, may contribute to the establishment of the latent reservoir. Further, care should be taken if attempts are made to suppress HIV-1-induced “bystander” killing of nonproductively infected cells using cytokines (63), as enhancement of latent reservoirs might be an unintended consequence (3–7). The protection conferred by cytokines in vitro coincides with the observation that lymphoid organs and mucosa provide a privileged environment for HIV-1 infection in resting T cells. It remains to be determined what the capacity of peripheral blood resting T cells is to support HIV-1 infection in vivo. Indeed, the first 2 to 3 days of in vitro culture imposed on isolated peripheral blood T cells in the absence of cytokine may prime them for an increased sensitivity to HIV-1 infection-induced cell death.
T cell death is a major hallmark of both acute/primary HIV-1 infection and the progression to AIDS, and productively infected T cells have a short half-life in vivo as a result of multiple mechanisms of elimination, including cytotoxic T-lymphocyte (CTL) killing and the expression of toxic proteins (64). The cell death that we examined did not result from productive infection but was still observed when reverse transcription was interrupted 1 day after infection, prior to its completion. The death that we observed rather resembles bystander HIV-1-induced cell killing, a phenomenon first reported by Finkel et al., who showed, in lymph node sections from both human and macaque, that the majority of the dying cells were not productively infected (negative for viral mRNA) (23). One major mechanism for bystander T cell killing by HIV-1 results from expression of Env glycoprotein on the surface of infected cells, which can induce T cell death through its interaction with CD4 and coreceptors on uninfected cells (reviewed in references 27 and 65). This type of T cell death is likely to be particularly relevant in late stages of HIV-1 infection and is most highly associated with late-stage CXCR4-tropic viruses (66). In contrast, in our model, infection with cell-free viral particles bearing gp120 Env but lacking RT activity and virion-associated Vpr proteins did not induce cell death, as far as we could measure. More recently, it was also shown that cytotoxicity can be associated with integration-induced DNA damage response in activated T cells (42), a process inhibited by the integrase inhibitors such as raltegravir. Again, in our model we did not see any protection by raltegravir. It is possible that the quiescent state of resting T cells maintains a limited DNA damage response (67–69) which is sufficient to protect them from HIV-1 integration-induced cell death.
We observed cell death caused by two factors: reverse transcription and virion-associated Vpr. Elimination of these two factors removed all measurable resting peripheral blood CD4 T cell killing by HIV in the first few days after infection. This is not to say that HIV infection and the associated late-stage gene expression were not cytopathic but that up to and including integration, reverse transcription and virion Vpr were the drivers of cell death. Vpr is a multifunctional HIV-1 protein and has been associated with various effects on HIV-1 infection processes as well as the induction of cell cycle arrest (70) in the G2/M phase and cell death by apoptosis (46, 71; reviewed in reference 44). The cytotoxic effect of Vpr is often correlated with its antiproliferative capacity (71, 72; reviewed in reference 73), although a recent study segregated these functions (74). We have recently demonstrated that after infection of resting CD4 T cells, Vpr blocks activation-induced proliferation (4), which we attribute to de novo Vpr synthesis (unpublished data). Virion Vpr can induce apoptosis in primary activated T cells (32, 75), and apoptosis can also be induced by extracellular Vpr on unstimulated T cells (76). Here, we demonstrate a direct cytopathic effect of virion-associated Vpr in resting, nonproliferating CD4 T cells. One identified pathway for Vpr cytopathicity is direct disruption of mitochondrial membrane potential, release of cytochrome c and apoptosis-inducing factor (AIF), followed by caspase 9 activation (32, 76). These effects can be relieved by Bcl-2 (76). This effect is compatible with our observations (cytochrome c release) and with the idea that IL-7 can rescue Vpr-induced cell death. In the present study, we observed that common gamma-chain cytokines IL-7 and IL-4 protect resting CD4 T cells from this Vpr-induced cell killing.
The Greene group has reported a novel form of HIV-1-induced cell death in tonsil CD4 T cells, implicating the detection of HIV-1 cDNA by the cytosolic receptor IFI16 (77) and the subsequent activation of a caspase 1-dependent inflammatory response triggering death by pyroptosis (43, 63). Steele et al. have reported HIV-1-induced pyroptosis of gut lamina propria cells (39). The former study contains several similarities to ours, including protection from cell death by a nonnucleoside RT inhibitor such as efavirenz. Doitsh et al. also observed that the nucleoside RT inhibitor AZT favored the production of partial RT products and did not protect resting CD4 T cells from cell death. In our hands, AZT was inefficient at blocking primary HIV-1 infection of resting peripheral blood CD4 T cells. As AZT is poorly effective in some nonproliferating cell types such as monocytes (78), due to inefficient phosphorylation, this could be occurring in resting CD4 T cells as well. While Doitsh et al. observed pyroptosis in tonsil cells, we observed only features of conventional apoptosis in peripheral blood T cells, and we were unable to protect cells with caspase 1 inhibitors. Rather, we observed cytochrome c release and caspase 3/7 activation. However, inhibition of caspase 3 or caspase 9 alone did not protect the cells, suggesting redundant signaling in the cell death program induced by HIV-1. The fact that IL-7 (and IL-4) prevented this cell death suggests that the cell death program triggered by HIV-1 relies primarily on a mitochondrial apoptotic pathway. Indeed, CGCC can promote survival by influencing the levels of the Bcl-2 family members and prevent mitochondrial depolarization and all subsequent death programs. We see at least two possible explanations why resting peripheral blood CD4 T cells do not undergo cell death by pyroptosis. First, peripheral blood CD4 T cells do not constitutively express IFI16 (79). Second, it has been reported that cytosolic DNA detection in monocytes can lead to both apoptosis and pyroptosis (80). Pyroptosis was triggered only with relatively larger amounts of DNA (within the context of their system). In fact, recent work indicates that induction of pyroptosis in tonsillar cells requires delivery of multiple viral particles via cell-cell transmission, indicating that strong detection is required for triggering (81).
Finally, Vpx, which enhanced the kinetics of reverse transcription, the generation of completed genomes in resting CD4 T cells, and overall productive infection, did so with only a minor reduction in RT-dependent cell death. If specifically abortive reverse transcripts are driving RT-dependent cell death, then it might be expected that the enhancement by Vpx would reduce cell death to a greater extent than what we observed. On the other hand, cells are likely to be penetrated by more than one virus undergoing reverse transcription. Thus, it is possible that both abortive and completed reverse transcripts coexist in cells and that the abortive transcripts drive apoptosis induction even when more reverse transcripts can be completed. Importantly, then, cytokine treatment and Vpx have distinct and additive effects contributing to productive infection, and cell death and RT efficiency are 2 separate factors restricting HIV-1 infection in resting blood CD4 T cells. After the present study was submitted, Muñoz-Arias et al. published a study (79) showing that blood-derived resting CD4 T cells are resistant to pyroptosis and other forms of cell death owing to poor reverse transcription and lower expression of IFI16. The differences in findings between the present study and that by Muñoz-Arias et al. are likely to reflect important differences in the systems employed. Whereas we infected cells using cell-free virions, Muñoz-Arias et al. employed coculture of virus-producing activated peripheral blood lymphocytes (PBLs) or HEK293 cells with resting target cells. It is not clear how these cocultures affect target cell resting status or if factors produced by the activated PBLs or HEK293 cells could protect the cells similarly to cytokines in our system.
REFERENCES
- 1.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, Veazey RS, Notermans D, Little S, Danner SA, Richman DD, Havlir D, Wong J, Jordan HL, Schacker TW, Racz P, Tenner-Racz K, Letvin NL, Wolinsky S, Haase AT. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 2.Haase AT. 2011. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu Rev Med 62:127–139. doi: 10.1146/annurev-med-080709-124959. [DOI] [PubMed] [Google Scholar]
- 3.Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. 2014. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512:74–77. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinité B, Ohlson EC, Voznesensky I, Rana SP, Chan CN, Mahajan S, Alster J, Burke SA, Wodarz D, Levy DN. 2013. An HIV-1 replication pathway utilizing reverse transcription products that fail to integrate. J Virol 87:12701–12720. doi: 10.1128/JVI.01939-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pace MJ, Graf EH, Agosto LM, Mexas AM, Male F, Brady T, Bushman FD, O'Doherty U. 2012. Directly infected resting CD4+ T cells can produce HIV Gag without spreading infection in a model of HIV latency. PLoS Pathog 8:e1002818. doi: 10.1371/journal.ppat.1002818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, O'Doherty U. 2009. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol 83:4528–4537. doi: 10.1128/JVI.01910-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavez L, Calvanese V, Verdin E. 2015. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog 11:e1004955. doi: 10.1371/journal.ppat.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9:1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222. doi: 10.1016/0092-8674(90)90802-L. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson M. 2003. HIV-1 pathogenesis. Nat Med 9:853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- 11.Zack JA, Kim SG, Vatakis DN. 2013. HIV restriction in quiescent CD4(+) T cells. Retrovirology 10:37. doi: 10.1186/1742-4690-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan X, Baldauf HM, Keppler OT, Fackler OT. 2013. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell Res 23:876–885. doi: 10.1038/cr.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. 2000. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell 6:683–692. doi: 10.1016/S1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 14.Rathmell JC, Farkash EA, Gao W, Thompson CB. 2001. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol 167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 15.Hassan J, Reen DJ. 1998. IL-7 promotes the survival and maturation but not differentiation of human post-thymic CD4+ T cells. Eur J Immunol 28:3057–3065. [DOI] [PubMed] [Google Scholar]
- 16.Vella A, Teague TK, Ihle J, Kappler J, Marrack P. 1997. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL-4. J Exp Med 186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. 2000. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol 1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 18.Ducrey-Rundquist O, Guyader M, Trono D. 2002. Modalities of interleukin-7-induced human immunodeficiency virus permissiveness in quiescent T lymphocytes. J Virol 76:9103–9111. doi: 10.1128/JVI.76.18.9103-9111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med 189:1735–1746. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Introini A, Vanpouille C, Lisco A, Grivel JC, Margolis L. 2013. Interleukin-7 facilitates HIV-1 transmission to cervico-vaginal tissue ex vivo. PLoS Pathog 9:e1003148. doi: 10.1371/journal.ppat.1003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moutsopoulos NM, Vazquez N, Greenwell-Wild T, Ecevit I, Horn J, Orenstein J, Wahl SM. 2006. Regulation of the tonsil cytokine milieu favors HIV susceptibility. J Leukoc Biol 80:1145–1155. doi: 10.1189/jlb.0306142. [DOI] [PubMed] [Google Scholar]
- 22.Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, Roederer M, Sherman MP, Chin PS, Goldsmith MA. 2001. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 15:671–682. doi: 10.1016/S1074-7613(01)00217-5. [DOI] [PubMed] [Google Scholar]
- 23.Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba TW, Ruprecht RM, Kupfer A. 1995. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med 1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 24.Bangs SC, McMichael AJ, Xu XN. 2006. Bystander T cell activation—implications for HIV infection and other diseases. Trends Immunol 27:518–524. doi: 10.1016/j.it.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Moon HS, Yang JS. 2006. Role of HIV Vpr as a regulator of apoptosis and an effector on bystander cells. Mol Cells 21:7–20. [PubMed] [Google Scholar]
- 26.Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM. 2010. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 11:110–122. doi: 10.1111/j.1600-0854.2009.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg H, Mohl J, Joshi A. 2012. HIV-1 induced bystander apoptosis. Viruses 4:3020–3043. doi: 10.3390/v4113020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox AL, Siliciano RF. 2014. HIV: not-so-innocent bystanders. Nature 505:492–493. doi: 10.1038/505492a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelderblom HC, Vatakis DN, Burke SA, Lawrie SD, Bristol GC, Levy DN. 2008. Viral complementation allows HIV-1 replication without integration. Retrovirology 5:60. doi: 10.1186/1742-4690-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trinité B, Chan CN, Lee CS, Mahajan S, Luo Y, Muesing MA, Folkvord JM, Pham M, Connick E, Levy DN. 2014. Suppression of Foxo1 activity and down-modulation of CD62L (L-selectin) in HIV-1 infected resting CD4 T cells. PLoS One 9:e110719. doi: 10.1371/journal.pone.0110719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunseri N, O'Brien M, Bhardwaj N, Landau NR. 2011. Human immunodeficiency virus type 1 modified to package simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol 85:6263–6274. doi: 10.1128/JVI.00346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muthumani K, Hwang DS, Desai BM, Zhang D, Dayes N, Green DR, Weiner DB. 2002. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J Biol Chem 277:37820–37831. doi: 10.1074/jbc.M205313200. [DOI] [PubMed] [Google Scholar]
- 33.Christensen ME, Jansen ES, Sanchez W, Waterhouse NJ. 2013. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarisation and cytochrome c release. Methods 61:138–145. doi: 10.1016/j.ymeth.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 34.Toba K, Winton EF, Koike T, Shibata A. 1995. Simultaneous three-color analysis of the surface phenotype and DNA-RNA quantitation using 7-amino-actinomycin D and pyronin Y. J Immunol Methods 182:193–207. doi: 10.1016/0022-1759(95)00050-K. [DOI] [PubMed] [Google Scholar]
- 35.Schmid I, Cole SW, Korin YD, Zack JA, Giorgi JV. 2000. Detection of cell cycle subcompartments by flow cytometric estimation of DNA-RNA content in combination with dual-color immunofluorescence. Cytometry 39:108–116. doi:. [DOI] [PubMed] [Google Scholar]
- 36.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy DN, Refaeli Y, Weiner DB. 1995. The vpr regulatory gene of HIV. Curr Top Microbiol Immunol 193:209–236. [DOI] [PubMed] [Google Scholar]
- 38.Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J Virol 69:6304–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steele AK, Lee EJ, Manuzak JA, Dillon SM, Beckham JD, McCarter MD, Santiago ML, Wilson CC. 2014. Microbial exposure alters HIV-1-induced mucosal CD4+ T cell death pathways ex vivo. Retrovirology 11:14. doi: 10.1186/1742-4690-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rautajoki KJ, Marttila EM, Nyman TA, Lahesmaa R. 2007. Interleukin-4 inhibits caspase-3 by regulating several proteins in the Fas pathway during initial stages of human T helper 2 cell differentiation. Mol Cell Proteomics 6:238–251. [DOI] [PubMed] [Google Scholar]
- 41.Graninger WB, Steiner CW, Graninger MT, Aringer M, Smolen JS. 2000. Cytokine regulation of apoptosis and Bcl-2 expression in lymphocytes of patients with systemic lupus erythematosus. Cell Death Differ 7:966–972. doi: 10.1038/sj.cdd.4400724. [DOI] [PubMed] [Google Scholar]
- 42.Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. 2013. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature 498:376–379. doi: 10.1038/nature12274. [DOI] [PubMed] [Google Scholar]
- 43.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789-801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guenzel CA, Herate C, Benichou S. 2014. HIV-1 Vpr—a still “enigmatic multitasker”. Front Microbiol 5:127. doi: 10.3389/fmicb.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stewart SA, Poon B, Jowett JB, Xie Y, Chen IS. 1999. Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc Natl Acad Sci U S A 96:12039–12043. doi: 10.1073/pnas.96.21.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arunagiri C, Macreadie I, Hewish D, Azad A. 1997. A C-terminal domain of HIV-1 accessory protein Vpr is involved in penetration, mitochondrial dysfunction and apoptosis of human CD4+ lymphocytes. Apoptosis 2:69–76. doi: 10.1023/A:1026487609215. [DOI] [PubMed] [Google Scholar]
- 47.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.St Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, Wu L. 2012. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9:105. doi: 10.1186/1742-4690-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med 18:1682–1687. doi: 10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, Seo D, Kim J, White TE, Brandariz-Nunez A, Diaz-Griffero F, Yun CH, Hollenbaugh JA, Kim B, Baek D, Ahn K. 2014. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med 20:936–941. doi: 10.1038/nm.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, Margottin-Goguet F. 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bobadilla S, Sunseri N, Landau NR. 2013. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther 20:514–520. doi: 10.1038/gt.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9:87. doi: 10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plesa G, Dai J, Baytop C, Riley JL, June CH, O'Doherty U. 2007. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J Virol 81:13938–13942. doi: 10.1128/JVI.01745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. 1998. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A 95:8869–8873. doi: 10.1073/pnas.95.15.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korin YD, Zack JA. 1998. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J Virol 72:3161–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vatakis DN, Bristol G, Wilkinson TA, Chow SA, Zack JA. 2007. Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4+ T cells. J Virol 81:3574–3582. doi: 10.1128/JVI.02569-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swiggard WJ, O'Doherty U, McGain D, Jeyakumar D, Malim MH. 2004. Long HIV type 1 reverse transcripts can accumulate stably within resting CD4+ T cells while short ones are degraded. AIDS Res Hum Retroviruses 20:285–295. doi: 10.1089/088922204322996527. [DOI] [PubMed] [Google Scholar]
- 60.Swiggard WJ, Baytop C, Yu JJ, Dai J, Li C, Schretzenmair R, Theodosopoulos T, O'Doherty U. 2005. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol 79:14179–14188. doi: 10.1128/JVI.79.22.14179-14188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu D, Wang W, Yoder A, Spear M, Wu Y. 2009. The HIV envelope but not VSV glycoprotein is capable of mediating HIV latent infection of resting CD4 T cells. PLoS Pathog 5:e1000633. doi: 10.1371/journal.ppat.1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amirache F, Levy C, Costa C, Mangeot PE, Torbett BE, Wang CX, Negre D, Cosset FL, Verhoeyen E. 2014. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 123:1422–1424. doi: 10.1182/blood-2013-11-540641. [DOI] [PubMed] [Google Scholar]
- 63.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509-514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ventoso I, Navarro J, Munoz MA, Carrasco L. 2005. Involvement of HIV-1 protease in virus-induced cell killing. Antiviral Res 66:47–55. doi: 10.1016/j.antiviral.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 65.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. 2006. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jekle A, Keppler OT, De Clercq E, Schols D, Weinstein M, Goldsmith MA. 2003. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J Virol 77:5846–5854. doi: 10.1128/JVI.77.10.5846-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korwek Z, Sewastianik T, Bielak-Zmijewska A, Mosieniak G, Alster O, Moreno-Villanueva M, Burkle A, Sikora E. 2012. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair (Amst) 11:864–873. doi: 10.1016/j.dnarep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Li D, Wang D, Liu X, Yin N, Song Y, Lu SH, Ju Z, Zhan Q. 2012. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J Cell Biochem 113:3643–3652. doi: 10.1002/jcb.24228. [DOI] [PubMed] [Google Scholar]
- 69.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. 2014. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15:37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levy DN, Fernandes LS, Williams WV, Weiner DB. 1993. Induction of cell differentiation by human immunodeficiency virus 1 vpr. Cell 72:541–550. doi: 10.1016/0092-8674(93)90073-Y. [DOI] [PubMed] [Google Scholar]
- 71.Stewart SA, Poon B, Jowett JB, Chen IS. 1997. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J Virol 71:5579–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu Y, Gelbard HA, Roshal M, Pursell S, Jamieson BD, Planelles V. 2001. Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J Virol 75:3791–3801. doi: 10.1128/JVI.75.8.3791-3801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muthumani K, Choo AY, Premkumar A, Hwang DS, Thieu KP, Desai BM, Weiner DB. 2005. Human immunodeficiency virus type 1 (HIV-1) Vpr-regulated cell death: insights into mechanism. Cell Death Differ 12(Suppl 1):S962–S970. [DOI] [PubMed] [Google Scholar]
- 74.Nishizawa M, Kamata M, Mojin T, Nakai Y, Aida Y. 2000. Induction of apoptosis by the Vpr protein of human immunodeficiency virus type 1 occurs independently of G(2) arrest of the cell cycle. Virology 276:16–26. doi: 10.1006/viro.2000.0534. [DOI] [PubMed] [Google Scholar]
- 75.Arokium H, Kamata M, Chen I. 2009. Virion-associated Vpr of human immunodeficiency virus type 1 triggers activation of apoptotic events and enhances fas-induced apoptosis in human T cells. J Virol 83:11283–11297. doi: 10.1128/JVI.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. 2000. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med 191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428-432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balzarini J, Van Herrewege Y, Vanham G. 2002. Metabolic activation of nucleoside and nucleotide reverse transcriptase inhibitors in dendritic and Langerhans cells. AIDS 16:2159–2163. doi: 10.1097/00002030-200211080-00008. [DOI] [PubMed] [Google Scholar]
- 79.Muñoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. 2015. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe 18:463-470. doi: 10.1016/j.chom.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ. 2013. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galloway NL, Doitsh G, Monroe KM, Yang Z, Munoz-Arias I, Levy DN, Greene WC. 2015. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep 12:1555-1563. doi: 10.1016/j.celrep.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]