

ABSTRACT
Epstein-Barr Virus (EBV) persists for the lifetime of the infected host despite eliciting strong immune responses. This persistence requires a fine balance between the host immune system and EBV immune evasion. Accumulating evidence suggests an important role for natural killer (NK) cells in this balance. NK cells can kill EBV-infected cells undergoing lytic replication in vitro, and studies in both humans and mice with reconstituted human immune systems have shown that NK cells can limit EBV replication and prevent infectious mononucleosis. We now show that NK cells, via NKG2D and DNAM-1 interactions, recognize and kill EBV-infected cells undergoing lytic replication and that expression of a single EBV lytic gene, BZLF1, is sufficient to trigger sensitization to NK cell killing. We also present evidence suggesting the possibility of the existence of an as-yet-unidentified DNAM-1 ligand which may be particularly important for killing lytically infected normal B cells. Furthermore, while cells entering the lytic cycle become sensitized to NK cell killing, we observed that cells in the late lytic cycle are highly resistant. We identified expression of the vBcl-2 protein, BHRF1, as one effective mechanism by which EBV mediates this protection. Thus, contrary to the view expressed in some reports, EBV has evolved the ability to evade NK cell responses.
IMPORTANCE This report extends our understanding of the interaction between EBV and host innate responses. It provides the first evidence that the susceptibility to NK cell lysis of EBV-infected B cells undergoing lytic replication is dependent upon the phase of the lytic cycle. Induction of the lytic cycle is associated with acquired sensitization to NK cell killing, while progress through the late lytic cycle is associated with acquired resistance to killing. We provide mechanistic explanations for this novel observation, indicating important roles for the BZLF1 immediate early transactivator, the BHRF1 vBcl-2 homologue, and a novel ligand for the DNAM-1 NK cell receptor.
INTRODUCTION
Epstein-Barr Virus (EBV), one of eight human herpesviruses, is carried by over 90% of the world's adult population. Primary EBV infection occurs in the oropharynx, leading to infection of B lymphocytes (1, 2). These infected B cells can support the lytic cycle, in which more than 80 viral genes are expressed to generate new infectious virus, but they more frequently host nonproductive infections through expression of a limited number of so-called latent EBV genes (latency III genes) that drive lymphoproliferation as an alternative mechanism of expanding the infected cell pool. In vitro, this growth transformation is demonstrated by the ready establishment of lymphoblastoid cell lines (LCLs) following infection of resting B cells. Following initial infection in vivo, EBV downregulates the expression of all viral proteins and enters a true latent phase (latency 0) in the memory B cell population, where it establishes a lifelong infection (1). Periodically the virus reactivates and undergoes full lytic replication, which both aids the expansion of the virus within the host and enables transmission to new hosts (2).
A major component of the immune control of EBV is considered to be the strong and persistent T cell responses both to the transformation-associated latency III EBV gene products and to several lytic cycle-associated EBV proteins (3). However, an increasing body of evidence suggests that natural killer (NK) cells have an important role to play in the virus-host balance. NK cells expand following primary infection with EBV (4, 5), and patients with genetic defects leading to loss or impairment of NK cell differentiation or function are prone to complications associated with EBV infection (6). Furthermore, mice with reconstituted human immune system components that are experimentally infected with EBV experience enhanced symptoms resembling infectious mononucleosis and EBV-associated lymphomagenesis when depleted of NK cells; these pathogenic outcomes of NK cell depletion were shown to be due to loss of control over EBV lytic replication (7).
Successful persistence of viruses in the infected host requires some degree of evasion of the various potent immune responses. Like other herpesviruses, in addition to establishing antigenically silent latent infections, EBV has multiple mechanisms to evade both CD8+ and CD4+ T cell responses to viral proteins expressed following reactivation of the lytic cycle or growth transformation (8). However, the possible existence of EBV evasion mechanisms against NK cells is unclear.
Other human herpesviruses, most notably human cytomegalovirus (CMV) but also Kaposi's sarcoma-associated herpesvirus (KSHV), herpes simplex virus 1 (HSV-1) and HSV-2, varicella-zoster virus (VZV), and human herpesvirus 7 (HHV-7), all possess some NK cell evasion mechanism, most frequently but not exclusively involving modulation of NKG2D ligands (9–12). In one respect it could be argued that EBV evades NK cell responses through infecting B lymphocytes and, in growth-transformed cells, maintaining high levels of major histocompatibility complex (MHC) class I molecules that ligate inhibitory receptors on NK cells. Certainly, EBV-transformed latently infected LCLs are not killed unless they are experimentally defective for HLA expression (13). With regard to B cells lytically infected with EBV, however, there is evidence only that EBV sensitizes them to NK cell recognition and killing. This evidence was derived entirely from studies with malignant cell lines, principally the AKBM line derived from Burkitt lymphoma cells engineered to express two selection markers, green fluorescent protein (GFP) and truncated CD2, when induced into the lytic cycle through ligation of surface immunoglobulin (14). The switch from latent to lytic infection in AKBM cells triggers an upregulation of NKG2D ligands that is at least partly responsible for the sensitization to NK cell killing. However, the mechanism of NKG2D ligand upregulation in the lytic cycle was not determined, and due to technical limitations of these earlier experiments, the possibility of counteracting evasion mechanisms was not investigated. Importantly, the generality and relevance of the AKBM observations to normal B cell infection has not been demonstrated.
In the present study, we identified the immediate early protein BZLF1 as being able to sensitize cells to NK cell killing through upregulating the ULBP NKG2D ligands. We also identified the vBcl-2 homologue BHRF1 as a potential NK evasion gene that could protect BZLF1-sensitized cells from NK cell killing. Consistent with these findings, we demonstrated that whereas AKBM cells in the early stages of the lytic cycle were killed by NK cells, AKBM cells at the late stages of the lytic cycle were resistant. Importantly, this phenomenon was also observed in lytically infected LCLs, even though these nonmalignant cells were killed primarily through NK cell receptor/ligand combinations that differed from those utilized in NK cell killing of lytic AKBM cells.
MATERIALS AND METHODS
Cell lines.
The NK cell line NKL (15) was maintained in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and 200 IU/ml interleukin-2 (IL-2). The NK cell line NK-92 (16) was maintained in RPMI 1640 supplemented with 10% FCS, 10% horse serum, 5% human serum, and 400 IU/ml IL-2. Both NKL and NK-92 were obtained from the American Tissue Culture Collection, and their activating receptor profiles were determined for this study (Fig. 1). AKBM cells are a derivate of the Akata Burkitt lymphoma cell line engineered to carry a reporter plasmid that expresses GFP when the cells enter the lytic cycle. These cells were maintained in RPMI 1640 supplemented with 8% FCS and were induced into the lytic cycle by cross-linking surface IgG molecules as previously described (14). The EBV-negative Burkitt lymphoma cell line DG75 (17) and EBV-transformed LCLs (18) were maintained in RPMI 1640 supplemented with 8% FCS. DG75-control and DG75-BHRF1 were generated through transduction and nerve growth factor receptor (NGFR) sorting as described above and maintained in RPMI 1640 supplemented with 8% FCS. A doxycycline (DOX)-inducible BZLF1 expression vector, pRTS-CD2-BZLF1, or control vector with the reverse BZLF1 sequence (pRTS-CD2-control) (19) was introduced into DG75 by electroporation and rCD2 selection. BZLF1 expression was induced by addition of DOX, and the induced cells were positively selected by magnetic cell sorting with anti-NGFR microbeads and LS columns (Miltenyi Biotech). Human embryonic kidney (HEK) 293 cells (20) were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FCS.
FIG 1.

Activating receptor expression profiles of NK cell lines and primary NK cells. NKL, NK92, and two enriched primary NK cell lines were stained for NKG2D, DNAM-1, NKp30, and NKp46 surface expression and analyzed using flow cytometry. Solid black lines represent staining of each activating receptor, and gray-filled histograms represent the isotype control.
Plasmids.
The BZLF1 and BRLF1 genes from the B95.8 prototype EBV (GenBank accession numbers CAA24861.1 and CAA24814.1) were subcloned into the pCDNA3-IRES-nls-GFP plasmid vector (21) and were verified by restriction digest and sequence analysis. BHRF1, also from the B95.8 prototype EBV, was cloned into the pLZRS-IRES-ΔNGFR vector (22) to generate retroviruses expressing BHRF1 and the truncated nerve growth factor receptor (ΔNGFR) for selection of infected cells.
Transfection and electroporation.
Transient transfection of HEK 293 cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Plasmid DNA was transfected into DG75 cells by electroporating cells at 270 V and 950 μF in 4-mm cuvettes. Cells transduced with PLZRS-NGFR vectors were positively selected for the expression of NGFR using the MACSelect NGFR-transfected cell selection kit (Miltenyi Biotec) according to the manufacturer's instructions to establish stably transduced cell lines.
Isolation of NK cells.
Blood was taken from healthy donors with ethical consent according to the human tissue act. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lympholyte cell separation medium (Cedarlane Labs), and untouched NK cells were isolated from PBMCs using the NK cell isolation kit (Miltenyi Biotec) according to the manufacturer's protocol.
Antibodies.
For flow cytometry experiments, fluorescein isothiocyanate (FITC)-conjugated, phycoerythrin (PE)-conjugated, and unconjugated antibodies to CD19 (HIB19), NGFR (ME20.4), and CD155 (TX24) were purchased from BioLegend. The FITC-conjugated anti-DNAM-1 (11A8), allophycocyanin (APC)-conjugated anti-NKp30 (P30-15), and APC-conjugated anti-human IgG Fc (HP6017) were also purchased from BioLegend. The APC-conjugated anti-NKp46 (9E2) was purchased from EBioscience. The APC-conjugated anti-NKG2D antibody (1D11), anti-CD112 antibody (R2.525), and Alx647-conjugated antibody to active caspase-3 (C92-605) were purchased from BD Biosciences. APC-conjugated and PE-conjugated antibodies to ULBP2/5/6 (165903) and MICA/B (159207) were purchased from R&D Biosystems. Recombinant human DNAM-1/CD226 Fc chimera protein (666-DN-050) was also purchased from R&D Biosystems. The BZLF1 (BZ.1) antibody (23) was generated by our investigators, and the BcLF1 (V3) antibody (24) was a kind gift from Gary Pearson, previously of Georgetown University, Washington, DC. To detect unconjugated antibodies, peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated or Alx647-conjugated secondary antibodies against mouse IgG1 (RMG1-1) or IgG2a (RMG2a-62) were purchased from BioLegend. For blocking experiments, antibodies to NKG2D (1D11), DNAM-1 (DX-11), and NKp46 (9E2) were purchased from BD Biosciences. For Western blotting, the anticalregulin antibody was purchased from Santa Cruz Biotechnology, the BZLF1 antibody (BZ.1) is described above, and the BHRF1 antibody was purified from cultures of the 5B11 hybridoma (25) obtained from Elliott Kieff, Harvard.
Flow cytometry analysis.
Stained cell samples were detected on a BD Biosciences Accuri C6 flow cytometer. Data were analyzed using FlowJo software (TreeStar).
Cytotoxicity assays.
NKL and NK92 cells and freshly isolated NK cells were used as effectors in cytotoxicity assays. AKBM cells were used as targets at 24 h postinduction with anti-IgG. DG75 cells were used as targets at 24 h posttransfection with control-GFP, BZLF1-GFP, or BRLF1-GFP expression plasmids. DG75 cells stably expressing control-NGFR or BHRF1-NGFR vectors were used as targets at 24 h posttransfection with control- or BZLF1-GFP expression plasmids. LCLs were screened for levels of spontaneous lytic cycle, and those containing suitable proportions of BZ.1+ cells (≥1%) were selected for use as targets in NK cell assays. Effector and target cells were combined at different ratios and incubated for 4 to 16 h. In 4-h assays, cytotoxicity was determined by caspase-3 staining by flow cytometry. Specific cytotoxicity was calculated as follows: % caspase-3-positive target cells after coincubating with NK cells for 4 h − % caspase-3-positive target cells after 4 h of incubation alone. For blocking experiments, NK cells were incubated with saturating amounts of blocking antibody (30 μg/ml) for 1 h at 37°C and then washed three times before use as effectors in cytotoxicity assays.
In 16-h cytotoxicity assays, killing was measured by determining the decline in numbers of target cells against a control population of target cells not killed by NK cells. Killing was calculated with the following the equation: killing (%) = 100 − [(experimental GFP %/control GFP %) × 100].
In the degranulation assay, DG75 target cells and the NKL cell line were cocultured with FITC-conjugated anti-CD107a antibody for 5 h. The cells were then washed and stained with combinations of APC-conjugated anti-NKG2D with PE-conjugated anti-CD19 to separate the NKL population from the DG75 population. Stained cells were analyzed by flow cytometry.
Western blotting.
Total cell lysates were denatured in reducing sample buffer and then sonicated and heated to 100°C for 5 min. Solubilized proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on 4 to 12% acrylamide gradient bis-Tris NuPage minigels with morpholinepropanesulfonic acid running buffer (Invitrogen). Separated proteins were electroblotted to polyvinylidene difluoride membranes and probed with specific antibodies. Samples were then subjected to chemiluminescent detection using the Millipore ECL detection kit (Millipore).
Quantitative PCR (qPCR) assay.
Total RNA, isolated from cultured cell lines using the Qiagen RNeasy kit, was treated with DNase I (Turbo DNA-free kit; Ambion) and then reverse transcribed using qScript cDNA SuperMix (Quanta Biosciences). Quantitative reverse transcription-PCR (qRT-PCR) assays for MICA, MICB, ULBP2, ULBP5, ULBP6, CD112, and CD155 were performed with TaqMan gene expression assays (Applied Biosystems) duplexed with b2m assays for normalization.
Statistical analysis.
Where statistical analysis was performed, data were analyzed with Student t tests or one-way analysis of variance (ANOVA) as described in the figure legends. Analysis was performed using Prism 5 software (GraphPad Software).
RESULTS
The switch from latent to lytic infection sensitizes B cells to NK cell killing.
We previously reported that the switch from the latent to the lytic cycle in AKBM cells induced sensitivity to NK cell killing (14). Those experiments were conducted by sorting induced AKBM cells for the expression of rCD2/GFP to isolate homogeneous populations of cells in the lytic cycle. While that methodology provided valuable information, it was not suitable for the additional investigations planned in the present study. We therefore designed a novel method of measuring NK cell killing in mixed populations of target cells using flow cytometry.
To validate this new assay, target AKBM cells were induced into the lytic cycle by treatment for 1 h with anti-IgG. At 24 h postinduction, cells were incubated with NKL effector cells at various effector-to-target ratios. After 4 h of coincubation, cells were harvested and stained for cell surface CD19 to differentiate effector and target cells, and for intracellular activated caspase-3 as a marker of NK cell-induced killing. Figure 2A shows CD19 staining to differentiate NK cells from the target population, AKBM cells. Within the target population, cells undergoing the latent or lytic cycle were differentiated by GFP expression (latent infection, GFP negative; lytic infection, GFP positive), and activated caspase-3 was measured in each target population to determine levels of cytotoxicity.
FIG 2.

EBV-infected cells undergoing lytic infection are sensitive to NK cell killing. AKBM cells were induced into the lytic cycle and used as targets in 4-h cytotoxicity assays. (A) Cells were stained for CD19 to differentiate effector and target cells, and AKBM cells undergoing lytic infection were identified by GFP expression. Cells were stained for caspase-3 as a marker of NK cell-induced killing. (B to D) NK cell killing was measured in latent and lytic populations at increasing effector/target cell (E:T) ratios. Effector cells used were NKL cells (B), NK-92 cells (C), and freshly isolated NK cells (D). (E) NKL cells were incubated with blocking antibodies prior to use in cytotoxicity assays, and NK cell killing was measured in the lytic population of AKBM cells at an effector/target cell ratio of 4:1. Data shown are mean values from three separate experiments, error bars represent standard errors, and significance was determined using t tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
In healthy cells, caspase-3 exists as an inactive proenzyme; cleavage of this protein produces the active form of the enzyme, activated caspase-3 (here referred to simply as caspase-3), which plays a central role in the execution phase of apoptosis (26). Cytotoxic lymphocytes such as NK cells and CD8+ T cells are able to kill target cells through two main mechanisms, Fas/FasL interaction and the release of cytotoxic granules containing perforin and granzyme. Killing mediated through either mechanism will initiate a caspase cascade in target cells, resulting in conversion of pre-caspase-3 to activated caspase-3 in a target cell; immunostaining and flow cytometry for activated caspase-3 can therefore be used as an early marker of target cell killing by effector cells.
As shown in Fig. 2B, with increasing effector/target cell ratios, the levels of caspase-3 increased in lytic cells but not in the latent cells; this reflects the increased cytotoxicity to lytic cells. At the highest effector-to-target ratio (4:1), levels of caspase-3-positive cells in the lytic population reached 23%, compared to just 3% in latent cells. This confirms the previous finding of our lab that AKBM cells in the lytic cycle are susceptible to killing by NK cells and shows that caspase-3 induction can be used as a marker for NK cell killing in this setting.
NK cells are a highly polymorphic population of cells controlled by different activating and inhibitory receptor ligand combinations. To show that the previous result is not unique to the NKL effectors, the experiment was repeated with two alternative sources of NK cells: the NK cell line NK-92 and polyclonal NK cells freshly isolated from peripheral blood. Figure 2C shows that NK-92 cells activated caspase-3 in 55% of lytic AKBM cells, compared to fewer than 1% of latent cells, at an effector/target cell ratio of 4:1. Similarly, Fig. 2D shows that freshly isolated blood NK cells activated caspase-3 in 50% of lytic cells and just 2% of latent cells. Thus, the same observation was made with the three different sources of NK cells.
NK cell killing of lytically infected AKBM cells was shown previously to be mediated through the activating receptor NKG2D, expressed on NK cells. This observation was confirmed in the present study by performing caspase-3 cytotoxicity assays in the presence of blocking antibodies directed against activating receptors expressed on NK cells (Fig. 2E). The inclusion of either a control antibody or a blocking antibody against the NKp46 natural cytotoxicity receptor (NCR) did not decrease the level of caspase-3 induced in target cells. A DNAM-1-blocking antibody showed a small decrease in caspase-3 induction, though this result did not reach significance. When a blocking antibody directed against NKG2D was added to cytotoxicity assay mixtures, a significant decrease in caspase-3 induction was observed. These results exactly match those previously reported (14) with conventional 52Cr release assays on purified lytic AKBM populations.
EBV-infected cells in the late-stage lytic cycle are protected from NK cell killing.
A major advantage of the flow cytometry-based cytotoxicity assay is that it allows simultaneous in situ analysis of different target cell populations that might be refractory to physical separation methods. We therefore repeated the NKL cytotoxicity assays on AKBM target cells, which were then immunostained intracellularly for BZLF1 and BcLF1 expression as markers of the early and late lytic cycle. Figure 3A shows that this staining protocol allowed us to differentiate three populations of cells: latently infected cells expressing neither BZLF1 nor BcLF1, early lytic cells expressing BZLF1 but not BcLF1, and late lytic cells expressing BZLF1 and BcLF1. Caspase-3 was measured in all three populations of cells and cytotoxicity calculated. The results in Fig. 3B show that, as expected, latently infected AKBM cells were resistant to NK cell killing. However, the analysis of different lytic populations revealed a remarkable result: whereas cells in early lytic cycle were highly sensitive to NK cell killing, with activation of caspase-3 observed in around 40% of the BZLF1+ BcLF1− population at an effector-to-target cell ratio of 4:1, the BZLF1+/BcLF1+ cells in the late-stage lytic cycle were completely protected from NK cell killing.
FIG 3.

EBV-infected cells in the late-stage lytic cycle are protected from NK cell killing. AKBM cells were induced into the lytic cycle and used as targets in 4-h cytotoxicity assays using NKL cells. (A) Cells were stained for BZLF1 and BcLF1 to differentiate cells in the latent (BZLF1− BcLF1−), early lytic (BZLF1+ BcLF1−), and late lytic (BZLF1+ BcLF1+) cycles. (B) Caspase-3 positivity was assayed in each of the three populations as a measure of NK cell killing. Data shown are mean values from three separate experiments, and error bars represent standard errors.
This novel observation suggested to us that sensitization of AKBM cells to NK cells was a very early event following activation of the lytic cycle and that EBV may have active mechanisms for evading the NK cell response.
BZLF1 can induce expression of NKG2D ligands and sensitize B cells to NK cell killing.
We hypothesized that the EBV immediate early gene BZLF1 or BRLF1 might cause the sensitization seen in previous experiments, as sensitization appears to be an early event and because the HCMV counterpart of EBV BZLF1, IE-1, has been shown to activate transcription of NKG2D ligands (27). We therefore investigated the two immediate early genes of EBV for their effect on the expression of NKG2D ligands in EBV-negative cells. In the first instance, BZLF1 and BRLF1 were transiently expressed in HEK 293 cells using bicistronic plasmid vectors that coexpress the gene of interest along with GFP, which allows identification of transfected cells using flow cytometry. Using an antibody that detects ULBP2, -5, and -6, the levels of the ULBP ligands of the NKG2D receptor were measured on GFP-positive cells by flow cytometry at 24 h posttransfection. While cells transfected with BRLF1-GFP showed no significant change in ULBP expression compared to cells transfected with control-GFP (Fig. 4A), increased ULBP expression was detected in those cells transfected with BZLF1-GFP (Fig. 4B).
FIG 4.
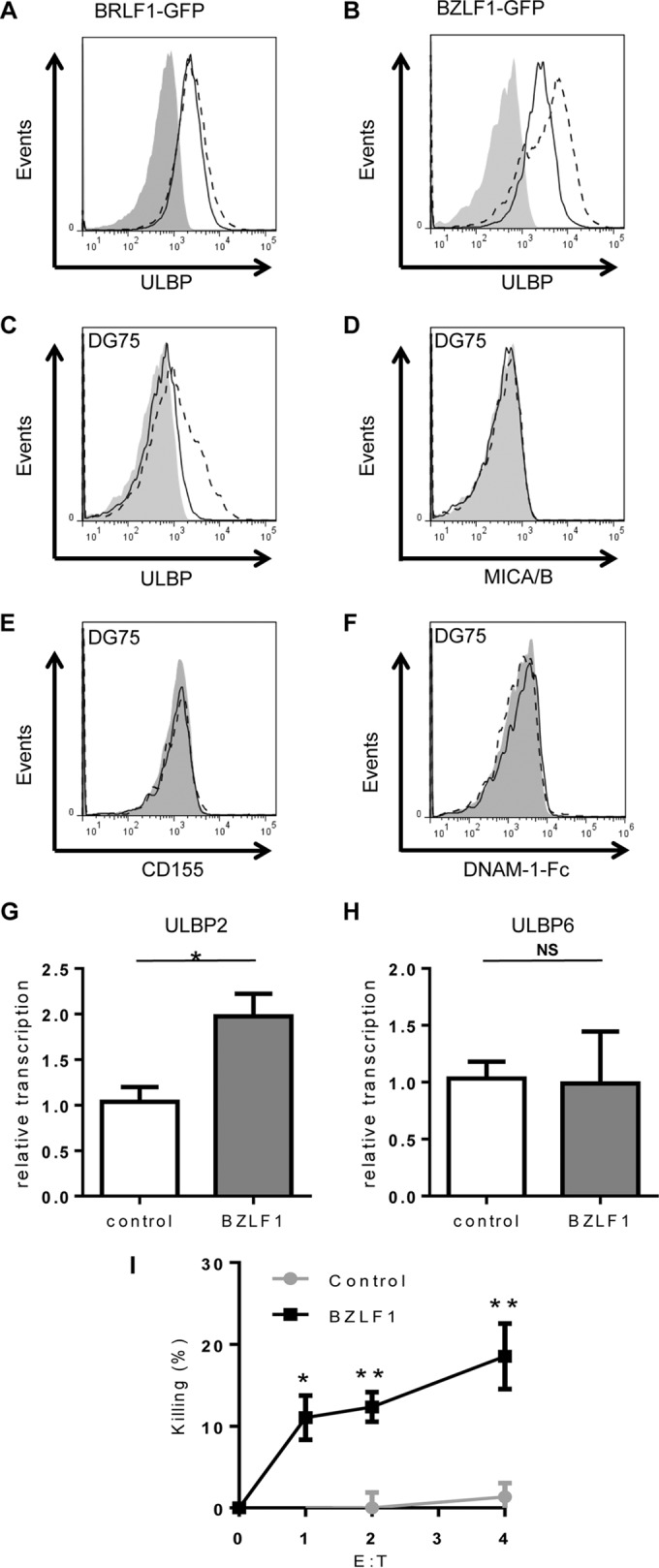
BZLF1 induces expression of NKG2D ligands and sensitizes B cells to NK cell killing. (A to F) HEK 293 cells (A and B) or DG75 cells (C to F) transiently expressing control-GFP (solid black line) and BRLF1-GFP (dashed black line) (A) or BZLF1-GFP (dashed black line) (B to F) were investigated for surface expression of NK cell-activating receptor ligands using flow cytometry. Gray-filled histograms represent isotype control staining. Results shown are representative of three separate experiments. (G and H) Total RNA was iso lated from control DG75 and BZLF1-expressing DG75 cells and then reverse transcribed to cDNA. Relative transcription levels of ULBP2 (G) and ULBP6 (H) were measured by qPCR assay, normalized to measured B2m transcripts. Data shown are mean values from three separate experiments, error bars represent standard errors, and significance was determined using t tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (I) DG75 cells transfected with control or BZLF1 expression plasmids were used as targets in NK cell killing assays using NKL cells, and specific cytotoxicity was calculated.
As B cells are the natural reservoir for EBV and the original NK cell sensitivity data were obtained in the Burkitt lymphoma cell line AKBM, we next investigated the effect of BZLF1 on NK cell ligand expression in an EBV-negative Burkitt lymphoma cell line, DG75. Following electroporation to introduce BZLF1-GFP or control-GFP vectors into DG75, the levels of NK cell ligands were measured by flow cytometry. Expression of BZLF1 in DG75 B cells, at levels comparable to but not exceeding BZLF1 levels in the lytic cycle (19), had effects similar to those seen in 293 cells, in that ULBP expression significantly increased (Fig. 4C). Expression of two additional NKG2D ligands, the MHC class I chain-related proteins MICA and MICB, was unaffected by expression of BZLF1 (Fig. 4D). As discussed above, NK cells may be activated by many different receptors. With this in mind, the effect of BLZF1 on the two known DNAM-1 ligands was also tested, but BZLF1 caused no increase in the expression of either CD155 (Fig. 4E) or CD112 (data not shown) or in binding of DNAM-1-Fc fusion protein (Fig. 4F).
To confirm the previous result and further investigate the effect of BZLF1 on the expression of NK cell-activating ligands, mRNA expression levels were measured in the absence and presence of BZLF1 protein. As the antibody used in the previous experiment recognizes ULBP2, -5, and -6, the transcription levels of the genes for these three proteins was measured. DG75 cells expressing inducible BZLF1 (19) were enriched, and total RNA was then isolated and reverse transcribed to generate cDNA. The relative transcription level of each ULBP gene was then measured using qPCR. The level of ULBP2 transcript was increased 2-fold in BZLF1-expressing DG75 cells compared to control cells (P < 0.05) (Fig. 4G). No upregulation of ULBP6 transcription level was observed (Fig. 4H), and no ULBP5 transcription was detected in either control DG75 or BZLF1-expressing DG75 cells (data not shown). Transcription levels of DNAM-1 ligand were also measured in the same assay, but no CD112 or CD155 transcripts were detected in either DG75 or BZLF1-expressing DG75 cells (data not shown).
As BZLF1 clearly increases the expression of ULBPs in these cells, we next investigated whether BZLF1 expression alone is able to sensitize B cells to killing by NK cells. In order to test this, DG75 cells were again transfected with BZFL1 expression vector and used as targets in cytotoxicity assays. A high baseline expression of caspase-3 in viable electroporated DG75 cells precluded the use of the cytotoxicity assay used for Fig. 2 and 3, so an alternative method of measuring NK cell killing by flow cytometry was used. Cells were incubated with NK cells for 16 h, and the percentage of GFP-tagged target B cells remaining after this time was measured at different effector/target cell ratios. Specific cytotoxicity was calculated by comparing the percentage of GFP-positive cells after 16 h of incubation with NK cells with that in cultures of transfected cells alone. Figure 4I shows that cells expressing the control-GFP vector were not depleted by NK cells, while expression of BZLF1 sensitized cells to NK cell killing, as there was a significant depletion of BZLF1-GFP target cells at all effector/target cell ratios.
BHRF1 protects B cells from BZLF1-induced NK killing.
As BZLF1 is the master transactivator of the EBV lytic cycle, the data in Fig. 4 provide at least one explanation for why AKBM cells in the early lytic cycle are susceptible to NK cell killing. We next sought to explain why AKBM cells in the late lytic cycle became resistant to NK cell killing despite the levels of BZLF1 protein being maintained during the late lytic cycle (Fig. 5A). BHRF1 is an early lytic cycle protein whose maximal levels are not achieved until about 12 h postinduction, coincident with the appearance of late lytic cycle antigens (Fig. 5A and B). As BHRF1 is a vBcl-2 homologue with powerful antiapoptotic functions (28, 29), we hypothesized that it might be a contributor to the protection against NK cells.
FIG 5.
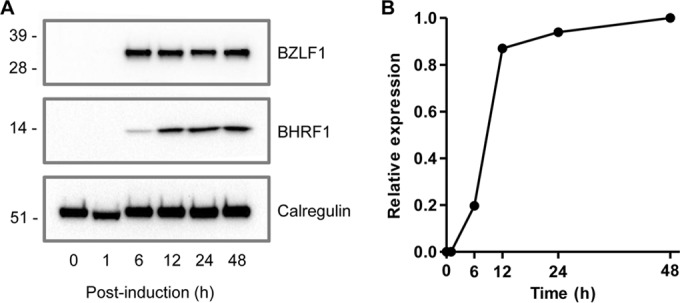
Maximum expression levels of BHRF1 protein occur beyond 12 h postinduction of the lytic cycle. AKBM cells were induced into the lytic cycle by cross-linking of surface immunoglobulin. (A) Levels of BHRF1 (middle panel) and BZLF1 (upper panel) proteins were measured at the indicated time points postinduction using Western blot analysis. The level of calregulin (lower panel) was detected as a loading control. (B) Relative expression of BHRF1 protein was calculated using Bio-Rad Image Lab densitometry software and compared to that of the calregulin control at each time point.
To test this possibility, BHRF1 was coexpressed with BZLF1 in DG75 cells to determine if BHRF1 could counteract the sensitization caused by BZLF1. DG75 cells were transduced with either control or BHRF1-expressing retroviral vectors coexpressing a truncated NGFR as a selectable marker. Following magnetic selection, these cell lines were shown to be 100% NGFR positive (Fig. 6A). The two cell lines were then electroporated with either control or BZLF1-GFP expression vectors, as for Fig. 4, and levels of BHRF1 and BZLF1 proteins in these DG75 lines were monitored by immunoblotting (Fig. 6B). Finally the four cell lines were used as targets in cytotoxicity assays to measure sensitivity to NK cell killing (Fig. 6C). As expected, there was no significant NK cell killing of DG75-control and DG75-BHRF1 cells. As seen before, expression of BZLF1 in control DG75 cells caused the cells to become sensitive to NK cell killing, but expression of BZLF1 in DG75 cells stably expressing BHRF1 resulted in no sensitization. Therefore, BHRF1 is able to completely antagonize BZLF1 and protect B cells from NK cell killing.
FIG 6.

BHRF1 protects B cells from BZLF1-induced NK cell killing. DG75 cells were transduced with control- or BHRF1-NGFR-expressing retroviral vectors. (A) Following magnetic enrichment, cells were stained for expression of NGFR. Cells were then transfected with control- or BZLF1-GFP expression vectors. (B) Expression of BHRF1 (top panel) and BZLF1 (middle panel) proteins in the four different cell lines was determined by Western blot analysis. Calregulin expression (bottom panel) was measured as a loading control. (C) The four cell lines were then used as targets in killing assays using NKL cells at increasing effector/target cell ratios. Data shown are mean values from three separate experiments, and error bars represent standard errors. (D) Surface expression of ULBP was measured on DG75-control cells (gray-filled histograms), DG75-control cells expressing BZLF1 (solid black line), and DG75-BHRF1 cells expressing BZLF1 (dashed black line). Data shown are representative of three separate experiments. (E) The four DG75 cell lines mentioned above were cocultured with NKL cells and FITC-conjugated anti-CD107a antibody for 5 h. The surface CD107a expression of NKL cells from four cultures was analyzed by flow cytometry. Data shown are mean values from three separate experiments, and error bars represent standard errors. The significance was determined using one-way ANOVA tests (*, P < 0.05; **, P < 0.01).
From what is known about BHRF1, we anticipated that this vBcl-2 protects B cells from NK cell killing through its antiapoptotic function rather than by directly reversing the effects of BZLF1 through downregulation of ULBPs. To rule out the latter possibility, we assayed the surface expression of ULBP (Fig. 6D). As before, BZLF1-transfected DG75 cells revealed elevated expression of ULBP relative to that in control-transfected DG75 cells. BZLF1-expressing DG75-BHRF1 cells showed a similar elevated ULBP expression, showing that BHRF1 has no effect on ULBP expression.
Despite being resistant to NK cell killing, we hypothesized that due to increased ULBP expression, BZLF1-expressing DG75-BHRF1 cells will still be recognized by NK cells, causing the NK cells to become activated and degranulate. To confirm this hypothesis, degranulation of NK cells was studied following coculture with DG75 cells expressing BZLF1 and BHRF1. Figure 6E shows, as expected, an increased degranulation in NKL cells stimulated with BZLF1-expressing DG75 cells compared to control DG75 cells. This increased degranulation was unchanged in NKL cells stimulated with BZLF1-expressing DG75-BHRF1 cells, despite BHRF1 protecting these cells from NKL cytotoxicity. This suggests that BHRF1 is able to protect cells from NK cell killing through its intrinsic antiapoptotic function despite NK cells still recognizing and degranulating in response to such cells.
LCLs in the late-stage lytic cycle are also protected from NK cell killing.
While the AKBM and DG75 cell lines were useful tools for establishing and characterizing the phenomena of lytic cycle sensitization and protection from NK cell killing, respectively, in the early and late phases of the lytic cycle, it could be argued that they are malignant cell models and that the relevance to normal B cell infection is unclear. Indeed, due to technical difficulties, it has not previously been shown that lytically infected normal B cells can be killed by NK cells. Our new flow cytometry-based cytotoxicity assay (Fig. 2 and 3) provided an opportunity to address this question in the present study.
EBV naturally infects and transforms B cells in vitro, establishing a continuously growing but nonmalignant LCL. EBV infection in LCLs is predominantly nonproductive, expressing only a limited number of growth-transforming latent viral genes and showing resistance to NK cell killing. However, viral gene expression can be quite heterogeneous, and in many LCL cultures a small proportion of cells can spontaneously enter the lytic cycle. We assayed a panel of different LCL cultures for the presence of cells undergoing a spontaneous lytic cycle and selected suitable lines (i.e., those with >1% BZ.1+ cells) as targets in NK cell cytotoxicity assays. Cell were cocultured with NKL cells for 4 h, harvested, and stained for the expression of CD19, BZLF1, and BcLF1 to distinguish CD19+ target cells in latent infection (expressing neither BZLF1 nor BcLF1), early lytic infection (expressing BZLF1 but not BcLF1), and late lytic infection (expressing both BZLF1 and BcLF1). Caspase-3 was measured in all three populations of cells and cytotoxicity calculated. The results obtained using multiple LCL cultures (Fig. 7A) were remarkably similar to the earlier results using the AKBM model. Latently infected LCLs were resistant to killing by NK cells, cells in the early stages of the lytic cycle were highly sensitive to NK cell killing, and cells in the late lytic cycle were completely resistant to NK cell killing.
FIG 7.

LCLs are also protected from NK cell killing in the late-stage lytic cycle, but killing of cells in the early lytic cycle is mediated by DNAM-1. LCLs were screened for the presence of cells undergoing spontaneous lytic cycle and used as targets in 4-h cytotoxicity assays using NKL cells. Cells were stained for BZLF1 and BcLF1 to differentiate latent, early lytic, and late lytic cells and stained for caspase-3 as a marker of NK cell-induced killing. (A) NK cell killing was measured in the three populations at increasing effector/target cell ratios. (B) NKL cells were incubated with blocking antibodies prior to use in cytotoxicity assays and NK cell killing measured in the early lytic population of LCLs at an effector/target cell ratio of 4:1. Data shown are mean values from three separate experiments using four different LCLs, error bars represent standard errors, and significance was determined using t tests (**, P < 0.01). (C to F) LCLs were stained for BZLF1 to detect cells undergoing spontaneous lytic cycle, and levels of MICA/B (C), ULBP (D), CD155 (E), and CD112 (F) were measured by flow cytometry. Solid black lines represent BZLF− (latent) cells, dashed black lines represent BZLF1+ (lytic) cells, and gray-filled histograms represent isotype control staining of bulk LCLs. HeLa cells were used as a positive control for CD155 expression (E), and K562 cells were used as a positive control for MICA/B, ULBP, and CD112 expression (C, D, and F). Results shown are representative of multiple separate experiments using multiple antibodies to CD155 and CD112. (G) Total RNA was isolated from LCLs and then reverse transcribed to cDNA. Relative transcription levels of CD112 and CD155 were measured by qPCR assay, normalized to measured B2m transcripts. The error bars represent standard errors for three different LCLs. HeLa cells served as a standard for relative transcription in this assay. (H) LCLs were stained for BZLF1 to detect cells undergoing spontaneous lytic cycle, and levels of DNAM-1 ligands were measured using DNAM-1-Fc fusion protein by flow cytometry. Solid black lines represent BZLF− (latent) cells, dashed black lines represent BZLF1+ (lytic) cells, and gray-filled histograms represent isotype control staining of bulk LCLs. K562 cells were used as a positive control.
Although NK cell recognition and killing of AKBM cells have been shown to be mediated by NKG2D/ULBP interactions, differing reports exist in the literature as to the expression of NKG2D ligands on LCLs (30–32). We therefore examined whether NK cell killing of LCLs undergoing the lytic cycle is mediated through NKG2D, by performing cytotoxicity assays in the presence of blocking antibodies directed against different activating receptors (Fig. 7B). In contrast to what we observed previously in experiments with the AKBM cells, blocking NKG2D or NKp46 had no effect on NK cell killing of LCLs expressing BZLF1, but including a blocking antibody against DNAM-1 substantially ablated NK cell killing of target cells. Furthermore, staining of LCLs with antibodies to NKG2D ligands failed to detect expression of either MICA/B or ULBP (Fig. 7C and D). These data suggest that NK cell killing of LCLs is mediated predominantly through DNAM-1 and that the precise mechanism(s) of sensitization of lytically infected B cells to NK cell killing may depend on the cellular origin or phenotype.
DNAM-1 has two known cellular ligands, CD155 and CD112 (33). As with NKG2D ligands, there is some disagreement in the literature as to the expression of DNAM-1 ligands on LCLs. To ascertain if the sensitization of LCLs undergoing early lytic cycle was due to increased expression of known DNAM-1 ligands, we stained LCLs from different donors with antibodies against CD155 and CD112. The results showed that neither latently nor lytically infected cells in LCL cultures expressed CD155 (Fig. 7E) or CD112 (Fig. 7F) despite clear staining on control cells (HeLa for CD155 and K562 for CD112). This experiment was repeated using multiple antibodies to both ligands and multiple LCLs from different donors, and in all cases neither CD155 nor CD112 expression was detected. Interestingly, when CD155- or CD112-blocking antibodies were included in cytotoxicity assays, they had no effect on NK cell killing of lytic LCLs (data not shown). These data indicate that while NK cell killing of lytically infected LCLs is mediated through the DNAM-1 receptor on NK cells, the LCLs do not express detectable amounts of either of the two known DNAM-1 ligand proteins.
DISCUSSION
In this study, we have demonstrated that the acquisition of sensitivity to NK cell killing of-EBV infected B cells upon entry into the lytic cycle is not an artifact of the unusual malignant cell line model in which the observation was first made. This phenomenon of sensitization to NK cell killing is also observed in independently established, normal LCLs, in which a small subpopulation of cells spontaneously enters the lytic cycle. The cytotoxicity assay that we developed to be able to investigate NK killing of the minor population of lytically infected cells within LCL cultures has also allowed another important finding: that during the late stages of the EBV lytic cycle, EBV-infected B cells acquire a profound resistance to NK cell killing.
In the AKBM cell model, sensitization of lytically infected cells appears to be mediated predominantly by upregulation of ULBPs, which are ligands for the NKG2D activating receptor on NK cells. Furthermore, we showed that expression of a single EBV gene, that for the lytic transactivator BZLF1, causes a significant upregulation of these NKG2D ligands in an EBV-negative B cell line and coincidentally sensitizes the cells to killing by NK cells. This upregulation of surface ULBP expression correlates with increased transcript levels of ULBP2 in BZLF1-transfected DG75 cells (Fig. 4G). BZLF1 is a powerful transcription factor that not only initiates a cascade of EBV lytic cycle gene expression but also regulates more than 270 cellular genes in AKBM cells (34). The BZLF1-regulated cellular genes identified by chromatin immunoprecipitation (ChIP) analysis do not include those for known NK cell receptor ligands. However, our present analysis indicates that BZLF1 expression leads to a 2-fold increase in ULBP2 transcripts (Fig. 4G). It is therefore likely that BZLF1 indirectly targets ULBP2 gene transcription and/or that BZLF1 indirectly targets ULBP2 posttranscriptionally. It is known that BZLF1 binds to DNA damage response proteins, causing their mislocalization and, consequently, increased DNA damage in cells expressing BZLF1 (35). NKG2D ligands are known to be upregulated in response to a number of stress signals, including DNA damage (36), raising the possibility that upregulation of NKG2D ligands by BZLF1 may be an indirect result of induced DNA damage.
As mentioned above, BZLF1 is the master regulator of EBV lytic virus replication and thus is critical for the virus life cycle. The sensitization to NK cell killing initiated by BZLF1 expression and/or by other early lytic genes is therefore a price that the virus must pay. Though seemingly counterintuitive, EBV's ability to initiate an NK cell response to control viral infection is an evolutionary advantage to the virus, since NK cell control of EBV is an important factor in establishing a stable relationship between host and virus, thus allowing asymptomatic EBV persistence. An absence of effective NK cell responses in immunodeficiencies such as X-linked lymphoproliferative (XLP) disease and X-MEN syndrome is associated with EBV-related pathogenic complications (6, 32). In addition, two reports have described patients with CD16 mutation who experienced prolonged EBV infections and complications such as EBV-associated Castleman's disease (37, 38). As well as NK cell deficiencies, the NK cell phenotype has been shown to be correlated with the outcome of EBV infection. Two reports have shown that certain polymorphisms in killer immunoglobulin-like receptors (KIRs) can predispose people to infectious mononucleosis or hemophagocytic lymphohistiocytosis (39, 40). Equally, an alternative KIR polymorphism can actually protect against infectious mononucleosis (40).
While NK cell control, along with CD4+ and CD8+ immune T cell responses, is clearly important for limiting the pathogenic potential of EBV, the successful persistence of the virus for the life of the infected host implies the existence of some viral immune evasion mechanisms to evade elimination. For CD4+ and CD8+ responses, active mechanisms for evasion during the lytic cycle are well documented (3, 8, 41). However, evasion of NK cell responses in the lytic cycle is poorly understood. It has been suggested that EBV microRNAs, notably miR-BART2, may transcriptionally regulate NK cell ligands (42). However, expression of miR-BART2 is only weakly upregulated, by less than 2-fold, in AKBM cells upon induction of the lytic cycle, which argues against a significant evasion function accounting for our observed resistance of cells in the late lytic cycle to NK cell killing. A more recent study of a relatively complex experimental model of primary infection of PBMCs indicated a clear role for the vIL-10 (BCRF1) in modulating NK cell activity (43). This effect appears to be due to vIL-10 and huIL-10 acting on the NK cells rather than affecting the sensitivity of the EBV-infected cells. While not devaluing the importance of the published data, it is unlikely that BCRF1 contributes to our observed resistance of late lytic cells to NK cells, since early lytic cells in the same culture are highly sensitive to the same NK cells.
Against this background, our novel finding that BHRF1 can afford substantial protection against NK cell lysis is important, as it offers a plausible mechanism for the resistance of cells in the late lytic cycle. However, the lessons from other herpesviruses would suggest that BHRF1 is unlikely to be the only mechanism that EBV has evolved to counteract NK cell responses and enable some virus replication to occur in vivo. Human cytomegalovirus (HCMV) is the most well studied in the context of NK cell evasion, and it has multiple different mechanisms that act in synergy (44). CMV is able to reduce expression of multiple NKG2D ligands: UL16 reduces expression of ULBP1, ULBP2, and MICB, while US142, US18, and US20 reduce expression of MICA (45–48). UL141 blocks surface expression of DNAM-1 ligands CD112 and CD155 (49, 50). CMV also ligates NK cell inhibitory receptors through expression of HLA homologues such as UL18, which binds LIR-1, or stabilizing HLA-C through the action of UL40 (11, 51).
The value of extending our work beyond the AKBM model to nonmalignant LCLs goes beyond showing the generality of the basic observations that cells in the early lytic cycle are sensitized to NK cell killing while cells in the late lytic cycle acquire resistance. The results revealed another interesting point, i.e., that the same result might be achieved through slightly different mechanisms in different cells. Whereas NK cell recognition of lytic AKBM cells is predominantly through upregulation of NKG2D ligands, recognition of LCLs is mediated not through NKG2D but through DNAM-1. Paradoxically, in all the LCLs we tested, neither of the two known DNAM-1 ligands was detected, whether on latent or lytic infected cells. Interestingly, a small but significant increase in CD155 transcripts was observed in lytic LCLs (Fig. 7G), but the magnitude of the elevated transcripts was such that the biological significance is questionable. Preliminary attempts to identify the DNAM-1 ligand responsible for sensitization to NK cell killing were hampered by the inability to obtain significant binding of DNAM-1-Fc fusion protein to LCLs (Fig. 7H), a result that we attribute to the insensitivity of the fusion protein reagent. We hypothesize that LCLs in the lytic cycle express a third, as-yet-undiscovered DNAM-1 ligand. This ligand may be cellular, as is the case with CD155 and CD112. Alternatively, this ligand may be of viral origin; a number of NK receptors recognize pathogenic proteins, so it is possible that EBV expresses an uncharacterized DNAM-1 ligand in the lytic cycle.
This study makes a significant contribution to the knowledge of the basic immunology of EBV infection by greatly extending our knowledge of the interaction of innate responses to virus-infected cells. The discovery of BHRF1 as a bona fide immune evasion gene product capable of protecting cells from NK cell killing may also have wider implications. Although not examined, the mechanism of action implies that BHRF1 might also afford significant protection against EBV-specific cytotoxic CD8+ and CD4+ T cells.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Longnecker R, Kieff E, Cohen J. 2013. Epstein-Barr Virus, p 1898–1959. In Knipe D, Howley P (ed), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rickinson AB, Long HM, Palendira U, Munz C, Hislop AD. 2014. Cellular immune controls over Epstein-Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol 35:159–169. doi: 10.1016/j.it.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Balfour HH Jr, Odumade OA, Schmeling DO, Mullan BD, J Aed Knight JA, Vezina HE, Thomas W, Hogquist KA. 2013. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein-Barr virus infection in university students. J Infect Dis 207:80–88. doi: 10.1093/infdis/jis646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, Harrison N, Swerdlow AJ, Crawford DH. 2005. The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol 129:266–274. doi: 10.1111/j.1365-2141.2005.05452.x. [DOI] [PubMed] [Google Scholar]
- 6.Parvaneh N, Filipovich AH, Borkhardt A. 2013. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol 162:573–586. doi: 10.1111/bjh.12422. [DOI] [PubMed] [Google Scholar]
- 7.Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, Emmel V, Marcenaro E, Leung CS, Antsiferova O, Landtwing V, Bossart W, Moretta A, Hassan R, Boyman O, Niedobitek G, Delecluse HJ, Capaul R, Munz C. 2013. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep 5:1489–1498. doi: 10.1016/j.celrep.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ressing ME, Horst D, Griffin BD, Tellam J, Zuo J, Khanna R, Rowe M, Wiertz EJ. 2008. Epstein-Barr virus evasion of CD8(+) and CD4(+) T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol 18:397–408. doi: 10.1016/j.semcancer.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Campbell TM, McSharry BP, Steain M, Slobedman B, Abendroth A. 2015. Varicella-zoster virus and herpes simplex virus 1 differentially modulate NKG2D ligand expression during productive infection. J Virol 89:7932–7943. doi: 10.1128/JVI.00292-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pende D, Moretta L, Vitale M, Favoreel HW. 2014. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci U S A 111:16118–16123. doi: 10.1073/pnas.1409485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prod'homme V, Tomasec P, Cunningham C, Lemberg MK, Stanton RJ, McSharry BP, Wang EC, Cuff S, Martoglio B, Davison AJ, Braud VM, Wilkinson GW. 2012. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J Immunol 188:2794–2804. doi: 10.4049/jimmunol.1102068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, Wills M, Lehner PJ. 2008. Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc Natl Acad Sci U S A 105:1656–1661. doi: 10.1073/pnas.0707883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Trivedi PP, Ge B, Krzewski K, Strominger JL. 2007. Many NK cell receptors activate ERK2 and JNK1 to trigger microtubule organizing center and granule polarization and cytotoxicity. Proc Natl Acad Sci U S A 104:6329–6334. doi: 10.1073/pnas.0611655104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pappworth IY, Wang EC, Rowe M. 2007. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol 81:474–482. doi: 10.1128/JVI.01777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. 1996. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol 24:406–415. [PubMed] [Google Scholar]
- 16.Gong JH, Maki G, Klingemann HG. 1994. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8:652–658. [PubMed] [Google Scholar]
- 17.Ben-Bassat H, Goldblum N, Mitrani S, Goldblum T, Yoffey JM, Cohen MM, Bentwich Z, Ramot B, Klein E, Klein G. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int J Cancer 19:27–33. doi: 10.1002/ijc.2910190105. [DOI] [PubMed] [Google Scholar]
- 18.Leibold W, Flanagan TD, Menezes J, Klein G. 1975. Induction of Epstein-Barr virus-associated nuclear antigen during in vitro transformation of human lymphoid cells. J Natl Cancer Inst 54:65–68. [DOI] [PubMed] [Google Scholar]
- 19.Zuo J, Thomas WA, Haigh TA, Fitzsimmons L, Long HM, Hislop AD, Taylor GS, Rowe M. 2011. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog 7:e1002455. doi: 10.1371/journal.ppat.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 21.Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, Wiertz EJ, Rowe M. 2009. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog 5:e1000255. doi: 10.1371/journal.ppat.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heemskerk MH, Hoogeboom M, Hagedoorn R, Kester MG, Willemze R, Falkenburg JH. 2004. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med 199:885–894. doi: 10.1084/jem.20031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young LS, Lau R, Rowe M, Niedobitek G, Packham G, Shanahan F, Rowe DT, Greenspan D, Greenspan JS, Rickinson AB, et al. . 1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J Virol 65:2868–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vroman B, Luka J, Rodriguez M, Pearson GR. 1985. Characterization of a major protein with a molecular weight of 160,000 associated with the viral capsid of Epstein-Barr virus. J Virol 53:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson GR, Luka J, Petti L, Sample J, Birkenbach M, Braun D, Kieff E. 1987. Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 160:151–161. doi: 10.1016/0042-6822(87)90055-9. [DOI] [PubMed] [Google Scholar]
- 26.Slee EA, Adrain C, Martin SJ. 2001. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 27.Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. 2007. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol 178:961–969. doi: 10.4049/jimmunol.178.2.961. [DOI] [PubMed] [Google Scholar]
- 28.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. 1993. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A 90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. 2009. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, Staubli G, Gysin C, Berger C, Munz C, Chijioke O, Nadal D. 2014. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 124:2533–2543. doi: 10.1182/blood-2014-01-553024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rancan C, Schirrmann L, Huls C, Zeidler R, Moosmann A. 2015. Latent membrane protein LMP2A impairs recognition of EBV-infected cells by CD8+ T cells. PLoS Pathog 11:e1004906. doi: 10.1371/journal.ppat.1004906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaigne-Delalande B, Li FY, O'Connor GM, Lukacs MJ, Jiang P, Zheng L, Shatzer A, Biancalana M, Pittaluga S, Matthews HF, Jancel TJ, Bleesing JJ, Marsh RA, Kuijpers TW, Nichols KE, Lucas CL, Nagpal S, Mehmet H, Su HC, Cohen JI, Uzel G, Lenardo MJ. 2013. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 341:186–191. doi: 10.1126/science.1240094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, Vitale M, Moretta L, Lopez M, Moretta A. 2003. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med 198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramasubramanyan S, Osborn K, Al-Mohammad R, Naranjo Perez-Fernandez IB, Zuo J, Balan N, Godfrey A, Patel H, Peters G, Rowe M, Jenner RG, Sinclair AJ. 2015. Epstein-Barr virus transcription factor Zta acts through distal regulatory elements to directly control cellular gene expression. Nucleic Acids Res 43:3563–3577. doi: 10.1093/nar/gkv212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Deng W, Hau PM, Liu J, Lau VM, Cheung AL, Huen MS, Tsao SW. 2015. Epstein-Barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab Invest 95:937–950. doi: 10.1038/labinvest.2015.69. [DOI] [PubMed] [Google Scholar]
- 36.Gasser S, Orsulic S, Brown EJ, Raulet DH. 2005. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Vries E, Koene HR, Vossen JM, Gratama JW, von dem Borne AE, Waaijer JL, Haraldsson A, de Haas M, van Tol MJ. 1996. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood 88:3022–3027. [PubMed] [Google Scholar]
- 38.Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, Pandey R, Campbell KS, Orange JS. 2012. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest 122:3769–3780. doi: 10.1172/JCI64837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huo L, Jiang MY, Li Q, Zhu YP. 2015. Novel association of killer cell immunoglobulin-like receptor genes with EBV-infectious diseases in children. Biomed Environ Sci 28:303–307. doi: 10.3967/bes2015.042. [DOI] [PubMed] [Google Scholar]
- 40.Qiang Q, Zhengde X, Chunyan L, Zhizhuo H, Junmei X, Junhong A, Zheng C, Henter JI, Kunling S. 2012. Killer cell immunoglobulin-like receptor gene polymorphisms predispose susceptibility to Epstein-Barr virus associated hemophagocytic lymphohistiocytosis in Chinese children. Microbiol Immunol 56:378–384. doi: 10.1111/j.1348-0421.2012.00443.x. [DOI] [PubMed] [Google Scholar]
- 41.Quinn LL, Zuo J, Abbott RJ, Shannon-Lowe C, Tierney RJ, Hislop AD, Rowe M. 2014. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog 10:e1004322. doi: 10.1371/journal.ppat.1004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385. doi: 10.1016/j.chom.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. 2012. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog 8:e1002704. doi: 10.1371/journal.ppat.1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod'homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, Wang EC, Griffin CA, Davison AJ. 2008. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 41:206–212. doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR. 2009. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 83:12345–12354. doi: 10.1128/JVI.01175-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S, Davies J, McSharry BP, Weekes MP, Antrobus PR, Prod'homme V, Blanchet FP, Sugrue D, Cuff S, Roberts D, Davison AJ, Lehner PJ, Wilkinson GW, Tomasec P. 2014. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 10:e1004058. doi: 10.1371/journal.ppat.1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rolle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Soderberg-Naucler C, Cosman D, Karre K, Cerboni C. 2003. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J Immunol 171:902–908. doi: 10.4049/jimmunol.171.2.902. [DOI] [PubMed] [Google Scholar]
- 48.Spreu J, Stehle T, Steinle A. 2006. Human cytomegalovirus-encoded UL16 discriminates MIC molecules by their alpha2 domains. J Immunol 177:3143–3149. doi: 10.4049/jimmunol.177.5.3143. [DOI] [PubMed] [Google Scholar]
- 49.Prod'homme V, Sugrue DM, Stanton RJ, Nomoto A, Davies J, Rickards CR, Cochrane D, Moore M, Wilkinson GW, Tomasec P. 2010. Human cytomegalovirus UL141 promotes efficient downregulation of the natural killer cell activating ligand CD112. J Gen Virol 91:2034–2039. doi: 10.1099/vir.0.021931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomasec P, Wang EC, Davison AJ, Vojtesek B, Armstrong M, Griffin C, McSharry BP, Morris RJ, Llewellyn-Lacey S, Rickards C, Nomoto A, Sinzger C, Wilkinson GW. 2005. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol 6:181–188. doi: 10.1038/ni1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prod'homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR, Stanton RJ, Borysiewicz LK, Lopez-Botet M, Wilkinson GW, Tomasec P. 2007. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1− NK cells. J Immunol 178:4473–4481. doi: 10.4049/jimmunol.178.7.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]