

ABSTRACT
Elevated secretion of inflammatory factors is associated with latent Epstein-Barr virus (EBV) infection and the pathology of EBV-associated diseases; however, knowledge of the inflammatory response and its biological significance during the lytic EBV cycle remains elusive. Here, we demonstrate that the immediate early transcriptional activator BZLF1 suppresses the proinflammatory factor tumor necrosis factor alpha (TNF-α) by binding to the promoter of TNF-α and preventing NF-κB activation. A BZLF1Δ207-210 mutant with a deletion of 4 amino acids (aa) in the protein-protein binding domain was not able to inhibit the proinflammatory factors TNF-α and gamma interferon (IFN-γ) and reduced viral DNA replication with complete transcriptional activity during EBV lytic gene expression. TNF-α depletion restored the viral replication mediated by BZLF1Δ207-210. Furthermore, a combination of TNF-α- and IFN-γ-neutralizing antibodies recovered BZLF1Δ207-210-mediated viral replication, indicating that BZLF1 attenuates the antiviral response to aid optimal lytic replication primarily through the inhibition of TNF-α and IFN-γ secretion during the lytic cycle. These results suggest that EBV BZLF1 attenuates the proinflammatory responses to facilitate viral replication.
IMPORTANCE The proinflammatory response is an antiviral and anticancer strategy following the complex inflammatory phenotype. Latent Epstein-Barr virus (EBV) infection strongly correlates with an elevated secretion of inflammatory factors in a variety of severe diseases, while the inflammatory responses during the lytic EBV cycle have not been established. Here, we demonstrate that BZLF1 acts as a transcriptional suppressor of the inflammatory factors TNF-α and IFN-γ and confirm that BZLF1-facilitated escape from the TNF-α and IFN-γ response during the EBV lytic life cycle is required for optimal viral replication. This finding implies that the EBV lytic cycle employs a distinct strategy to evade the antiviral inflammatory response.
INTRODUCTION
Infection by the Epstein-Barr virus (EBV) causes infectious mononucleosis and several malignant cancers, including Burkitt's lymphoma, Hodgkin's lymphoma, nasopharyngeal carcinoma (NPC), and gastric carcinoma, as well as posttransplant lymphomas (1–5). EBV infection is persistent worldwide, but the frequency of EBV-associated NPC is highest in southern China, while Burkitt's lymphoma is most commonly found in equatorial Africa (2, 3). Although the exact mechanism by which EBV causes tumorigenesis remains to be fully defined, two important cofactors are strongly involved in EBV pathogenesis: genetic susceptibility and local diet. Unique polymorphisms of NPC-associated EBV have been identified in Chinese individuals, indicating the existence of EBV variants with higher pathogenic potential for NPC than that seen in the typical Western strains that cause infectious mononucleosis (6–8).
Latent infection with limited gene expression is the default EBV cycle, whereas the lytic cycle is essential for transmission (1, 9). Lytic replication during primary infection or reactivation from the latent cycle is initiated by the expression of the immediate early (IE) viral transactivators BZLF1 and BRLF1. BZLF1, an EBV-encoded transcription factor of the basic-leucine zipper (b-ZIP) family, activates both viral and cellular genes by binding to BZLF1-responsive elements (ZREs), including several transcription factors and inflammatory factors (10).
Inflammatory mediators have complex roles in cancer and infectious diseases, either limiting or promoting these disorders (11–15). Several proinflammatory factors have been fully characterized in experimental and clinical studies, including tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin-1α (IL-1α), and IL-1β. TNF-α serves as an antiviral immune factor operating via two different mechanisms: induction of apoptosis in infected cells and activation of the antiviral response in uninfected cells (16–19). For successful infection and replication, viruses employ multiple strategies to escape or hijack the host defenses, including innate immunity and the inflammatory response (15, 17, 20). The EBV lytic cycle evades the host inflammatory responses through the activity of BZLF1, which inhibits both IFN-γ signaling and tumor necrosis factor receptor 1 (TNFR1) signaling (21–23). BZLF1 suppresses the NF-κB signaling pathway by directly binding the p65 subunit (24, 25), acting as an alternative evasion mechanism for NF-κB-responsive inflammatory responses during EBV lytic replication (26).
Because EBV-harboring tumor cells are latently infected and the induction of the EBV lytic cycle results in cell killing, artificial activation of lytic replication may represent a promising therapeutic strategy for EBV-associated cancers (10, 27). However, a small amount of spontaneous lytic replication was observed in terminally differentiated plasma cells, peripheral blood B lymphocytes, and nasopharyngeal cells infected with a specific EBV strain from a Chinese NPC patient (7, 28, 29); this replication may restore the reservoirs of EBV in epithelial cells and contribute to its pathogenesis during both primary and persistent infection. Notably, the spontaneous replication may drive the EBV lytic life cycle into two distinct fates in vivo, viral clearance and transmission, following dramatically different immune and inflammatory responses. Unfortunately, the dynamic inflammatory responses and the unique inflammatory factors that facilitate or attenuate EBV pathogenesis have not been fully defined.
In the present study, we demonstrated that BZLF1 inhibits the transcription of proinflammatory factors during the early lytic life cycle by binding to their promoters and disrupting NF-κB activation. As a result, BZLF1 facilitates viral replication by reducing the inflammatory antiviral response.
MATERIALS AND METHODS
Cells and antibodies.
EBV-positive Burkitt's lymphoma-derived P3HR-1 cells and the BJAB EBV-negative lymphoma cell line previously described (30, 31) were a gift from Erle Robertson (University of Pennsylvania) and were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS) and antibiotics (penicillin and streptomycin). EBV-negative human nasopharyngeal carcinoma CNE-1 cells were previously described (32) and purchased from the Cell Facility of Sun Yat-sen University. EBV-negative human nasopharyngeal carcinoma HNE-1 cells, which were previously described (33), were a gift from Musheng Zeng (Cancer Center of Sun Yat-sen University). Both of these cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% FBS and antibiotics. The 293T cell line was purchased from ATCC (CRL-3216), and the GP2-293 cell line was purchased from Clontech Laboratories, Inc. (Mountain View, CA), maintained in our laboratory, and cultured in DMEM containing 10% FBS and antibiotics. Anti-green fluorescent protein (anti-GFP) (catalog no. 2955), anti-GST (anti-glutathione S-transferase) (catalog no. 2624), and anti-myc (catalog no. 2272) antibodies (Ab) and an NF-κB pathway sampler kit (catalog no. 9936) were purchased from Cell Signaling Technology Inc. (Beverly, MA). Anti-BZLF1 (sc-53904), anti-Ea-D (BMRF1; sc-58121), and anti-EBNA1 (sc-57719) were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX). The mouse anti-LMP2A antibody, which was previously described (34), was a gift from Musheng Zeng (Cancer Center of Sun Yat-sen University).
Plasmids.
BZLF1 was amplified from viral cDNA of EBV lytic replication from B95.8 cell lines and subcloned into pcDNA3.1-HA, pEBG, pEGFP-C2, and pLXRN vectors (Clontech Laboratories, CA). BZLF1 deletions or single mutants were constructed by QuikChange mutagenesis following the standard procedures. Firefly luciferase promoter reporters TNF-α-luc, IFN-γ-luc, interleukin 1β-luc (IL-1β-luc), and IL-8-luc contain 1 kb of the cellular TNF-α, IFN-γ, IL-1β, and IL-8 promoters, respectively. NF-κB binding activity reporters contain multiple κB elements, respectively. NF-κB binding site-mutated TNF-α-luc promoter reporters were constructed using QuikChange mutagenesis. pBMLF1-luc, pBMRF1-luc, and pEBNA1-luc promoter reporters contain 400 bp of the promoter regions amplified from the EBV DNA genome of B95.8 cells. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) promoter-derived Renilla luciferase expression vector pRL-GAPDH was constructed by replacing the TK promoter of pRL-TK with the cellular GAPDH promoter. The TNF-α small hairpin RNAs (shRNAs) were subcloned into the pLKO.1 lentiviral vector, and the sequences are GACTCAGCGCTGAGATCAATC, GGAGCCAGCTCCCTCTATTTA, and CTGTAGCCCATGTTGTAGCAA.
Dual-luciferase assays.
Fifty nanograms of firefly luciferase reporter plasmids and 10 ng of Renilla luciferase internal control plasmid (pRL-TK or pRL-GAPDH) were cotransfected with 100 ng of wild-type or mutant BZLF1 constructs into 293T cells seeded in a 48-well plate. The cells were harvested 36 h posttransfection and lysed in 1× passive lysis buffer (Promega, WI). Firefly and Renilla luciferase activities were determined in a TriStar multimode reader using a dual-luciferase assay reagent (Promega, WI). For each condition, three independent experiments were performed in triplicate.
Real-time PCR.
Total RNA was extracted using TRIzol reagent (Life Technologies, CA) and subjected to reverse transcription (RT) with reverse transcriptase of Moloney murine leukemia virus (M-MLV) (TaKaRa, Japan). Genomic DNA was extracted with an Allpure cell kit (Magen, China). cDNA and genomic DNA levels were determined by real-time PCR using a SYBR green Real-Time PCR master mix kit (Roche). The primers for real-time PCR were designed using the Primerbank program (35), and the primer pairs used in this study are listed in Table 1. The sequences of the primer pairs for the whole panel of the inflammatory factor real-time PCR array are available upon request.
TABLE 1.
Primer pairs used in this study
| Target | Primer or probe sequencea |
|---|---|
| Real-time PCR primer pairs | |
| TNF-α mRNA | F: AGCCTGTAGCCCATGTTGTAG |
| R: CCGCTCGAGTCACAGGGCAATGAT | |
| IFN-γ mRNA | F: TCGGTAACTGACTTGAATGTCCA |
| R: TCGCTTCCCTGTTTTAGCTGC | |
| GAPDH mRNA | F: ACATCATCCCTGCCTCTAC |
| R: TCAAAGGTGGAGGAGTGG | |
| GAPDH genomic DNA | F: CATCATCCCTGCCTCTACTG |
| R: GCCTGCTTCACCACCTTC | |
| EBNA1 | F: CATTGAGTCGTCTCCCCTTTGGAAT |
| R: TCATAACAAGGTCCTTAATCGCATC | |
| LMP1 | F: CCACTTGGAGCCCTTTGTMTACTC |
| R: TGCCTGTCCGTGCAAATTC | |
| BALF5 | F: AACCTTTGACTCGACCATCG |
| R: ACCTGCTCTTCGATGCACTT | |
| BcLF1 | F: CATCCATGTTCATTGGGACC |
| R: CATTAGTCATACCTGCCAGG | |
| BLLF1 | F: GTCAGTACACCATCCAGAGCC |
| R: TTGGTAGACAGCCTTCGTATG | |
| BMRF1 | F: GCCGTTGAGGCCCACGTTGT |
| R: TGGGAATGGCAGGCGAGGGT | |
| EMSA probe | |
| BMLF1 | Cys5.5-CCATCACGGTCACCTTCATGAGTCAGTGCTTCGCCGGTCGCTGTGGGGCCAATCAACCGA |
| ChIP primer pairs | |
| TNF-α-ChIP | F: CCACTACCGCTTCCTCCAGATG |
| R: GGTGTGCCAACAACTGCCTTTA | |
| BMLF1-ChIP | F: CCAAACTTAGTTCAGGTGTGCCA |
| R: TAGAGTACCAGAAACACCCTCACAT | |
| EBNA1-ChIP | F: TAACGAGCAGAGGGAATGAAAG |
| R: AGGTTTCCTCAGGCTTGGCTA | |
| BSRF1-ChIP | F: GTGAGGTCAGCCGCTTCTTGG |
| R: CGTTCTGTTTAGCCCGCCGTT | |
| GAPDH-ChIP | F: CGAGGAGAAGTTCCCCAACTT |
| R: GACCCTTACACGCTTGGATGA |
The boldface, italic, underlined sequence indicates the ZRE site in the BMLF1 probe.
Chromatin immunoprecipitation (ChIP).
Tetradecanoyl phorbol acetate (TPA)-induced or uninduced P3HR-1 cells or transfected CNE-1 cells were fixed with 1.42% formaldehyde for 10 min and quenched with 125 mM glycine. After the cells were collected and washed twice with cold phosphate-buffered saline (PBS), the cells (>107) were suspended in 1 ml of lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 0.5% NP-40, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitor cocktail) for 10 min on ice. After they were subjected to sonication eight times for 10 s each time, the cells were centrifuged at 12,000 × g at 4°C for 10 min. Precleared cell lysates were mixed with specific antibodies for 2 h at 4°C; then, 5% pretreated protein A beads were added and the reaction mixture was incubated at 4°C overnight. The immunoprecipitated complexes were washed five times and eluted with 150 μl of Tris-EDTA buffer containing 1% SDS. Cross-linked complexes were reversed and digested with protease K (25 μg/ml) at 65°C overnight. DNA was extracted and analyzed by real-time PCR. The primer pairs that amplify the promoter regions are listed in Table 1.
EMSA.
Cys5.5-labeled electrophoretic mobility shift assay (EMSA) probes were amplified and labeled by PCR. The final sequence of the probe is listed in Table 1, and the ZRE site in the BMLF1 probe is underlined. EMSA was performed as previously described (36). Briefly, nuclear proteins were extracted as follows: the cells were harvested and washed with cold PBS; 1 × 106 cells were resuspended in 50 μl of buffer (10 mM Tris-HCl [pH 7.5], 0.15 M NaCl, 1.5 mM MgCl2, 0.65% NP-40, 1 mM PMSF, 10 mM dithiothreitol [DTT]) and incubated for 10 min at 4°C and then centrifuged at 12,000 rpm and 4°C for 2 min. The supernatant was discarded, and the pellet was resuspended in 15 μl of lysis buffer (20 mM HEPES [pH 7.9], 25% glycerol, 0.4 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA [pH 8.0], 1 mM PMSF, 10 mM DTT) and incubated on ice for 2 h with shaking. After centrifugation at 12,000 rpm and 4°C for 10 min, the supernatants were transferred to a new tube containing 35 μl of buffer (20 mM HEPES [pH 7.9], 20% glycerol, 50 mM KCl, 0.2 M EDTA [pH 8.0], 1 mM PMSF, 10 mM DTT). Nuclear protein extracts were divided into aliquots and stored at −80°C. The DNA-protein binding reaction was performed by mixing 15 μl of nuclear protein extract, 1 μl of probe, and 4 μl of 5× EMSA binding buffer [20% glycerol, 5 mM MgCl2, 2.5 mM EDTA (pH 8.0), 25 U poly(dI:dC), 250 mM NaCl, 50 mM Tris-HCl (pH 7.4), 10 mM DTT]. After incubation for 15 min at 4°C, 5 μl of EMSA loading buffer was added, and the samples were electrophoresed on a 9% PAGE gel at a constant 120 V for 1.5 h. The gels were scanned and imaged with an Odyssey infrared (IR) scanner.
Lentivirus packaging and infection.
For BZLF1 overexpression, GP2-293 cells in 15-cm-diameter dishes were cotransfected with 15 μg of individual pLXRN lentiviral expression vector and 5 μg of pCMV-VSV-G. The supernatants were harvested after 72 h and then concentrated by ultracentrifugation at 100,000 × g for 1 h, and the stocks were dissolved in 1/100 phosphate buffer. For shRNA knockdown, 5 μg of pLKO.1 shRNA plasmids was cotransfected into 293T cells with 5 μg of the miRΔ8.2 packaging plasmid and 5 μg of pCMV-VSV-G. The supernatants were harvested after 72 h. After titration and preliminary testing, lentiviral infection was performed following standard procedures.
Antibody neutralization.
Anti-human TNF-α-neutralizing antibody (rabbit monoclonal Ab [MAb]) (catalog no. 7321S; Cell Signaling Technology, MA) and anti-human IFN-γ-neutralizing antibody (mouse IgG2A) (MAB286; R&D Systems, MN) were purchased and stored in stock solutions. For P3HR-1 and BJAB cell incubation, 10 μg/ml of neutralizing antibodies or the isotype-matched control IgG antibodies (eBioscience, CA) were added directly into the fresh cell culture medium at 6 h after lentiviral infection. The medium and additional antibodies were refreshed every 2 days.
Wild-type and BZLF1-KO EBV BAC-harboring BJAB cells and HNE-1 cells.
Wild-type and BZLF1 knockout (BZLF1-KO) EBV bacterial artificial chromosome (BAC) genomic DNAs have been previously described (37, 38). To establish the EBV BAC-harboring BJAB cell line, both EBV BAC genomic DNAs were transfected into BJAB cells by electroporation using Amaxa cell line Nucleofector kit V and Amaxa Nucleofector II. After 48 h, the dead cells were removed, and the cells were sorted by the use of a BD FACSAria II cell sorting system. GFP-positive cells were collected and cultured in fresh RPMI 1640 containing 10% FBS and 25 μg/ml hygromycin for 2 weeks. To establish the EBV BAC-harboring epithelial cell line, both EBV BAC DNAs were transfected into HNE-1 cells using Lipofectamine 2000. After 48 h, the cells were digested and separated, followed by selection with 50 μg/ml hygromycin for 2 weeks. Then, EBV BAC-harboring cells were induced by TPA plus sodium butyrate and validated by Western blotting with anti-Ea-D, anti-BZLF1, and anti-EBNA1 antibodies.
Induction of EBV lytic replication.
To detect lytic replication and virion production, P3HR-1 cells or EBV BAC-harboring cells in logarithmic growth were cultured in fresh medium containing 10% FBS and 40 ng/ml TPA or 40 ng/ml TPA plus 3 mM sodium butyrate and incubated for different times as needed. For performance of a ChIP assay under conditions of lytic replication, P3HR-1 cells in logarithmic growth were induced by 40 ng/ml TPA and 3 mM sodium butyrate for 12 h. After induction, the media were refreshed with RPMI 1640 with 10% FBS and incubated for different times, as needed.
Detection of virion production.
P3HR1 cells or EBV BAC-harboring BJAB cells were induced by TPA or transduced with lentivirus-based BZLF1 for 3 or 5 days. The supernatants were collected and centrifuged twice at 12,000 × g at 4°C for 10 min to remove cell debris. After 500 μl of the supernatant was digested with 100 U DNase I for 20 min at 37°C, 20 μg/ml proteinase K and 1% SDS were added, followed by additional incubation for 30 min at 56°C. Then, viral DNA was extracted and analyzed using real-time PCR.
Statistical analysis.
The data were analyzed using SPSS (Statistical Package for the Social Sciences) 13.0 software, and one-way analyses of variance (ANOVA), the Bonferroni method, and chi-square tests were performed.
RESULTS
BZLF1 suppresses expression of TNF-α and proinflammatory factors.
To investigate the evasion of the proinflammatory response during EBV lytic infection, we established an EBV open reading frame (ORF)-expressing library by amplifying 82 EBV ORF-encoding regions from lytic EBV cDNA of B95.8 cells and subcloning into the pEGFP vector; we then performed a systematic screening using a firefly luciferase reporter driven by the TNF-α promoter. The TNF-α reporter was cotransfected into 293T cells with EBV ORF-expressing plasmids and Renilla luciferase-expressing plasmid pRL-TK as an internal control. After transfection for 24 h, the cells were not infected or were infected with herpes simplex virus 1 (HSV-1) (multiplicity of infection [MOI] = 10) to induce TNF-α transcription. Five ORFs (IE and early genes BZLF1, BMLF1, and BMRF1 and late genes BGLF4 and BGLF5) showed dramatically reduced TNF-α transcription compared with the vector control, and the well-known LMP1 target activated TNF-α transcription, which validates our screen. Among these genes, BZLF1 was the key IE gene during EBV lytic replication and exhibited the strongest inhibition, and therefore, we focused on its potential to mediate the proinflammatory response during EBV lytic replication. We confirmed that BZLF1 inhibited TNF-α transcriptional activity to approximately 10% of the control level during both TPA and HSV-1 induction (Fig. 1A). The inhibition of TNF-α secretion by BZLF1 was further validated using an enzyme-linked immunosorbent assay (ELISA; Fig. 1B).
FIG 1.
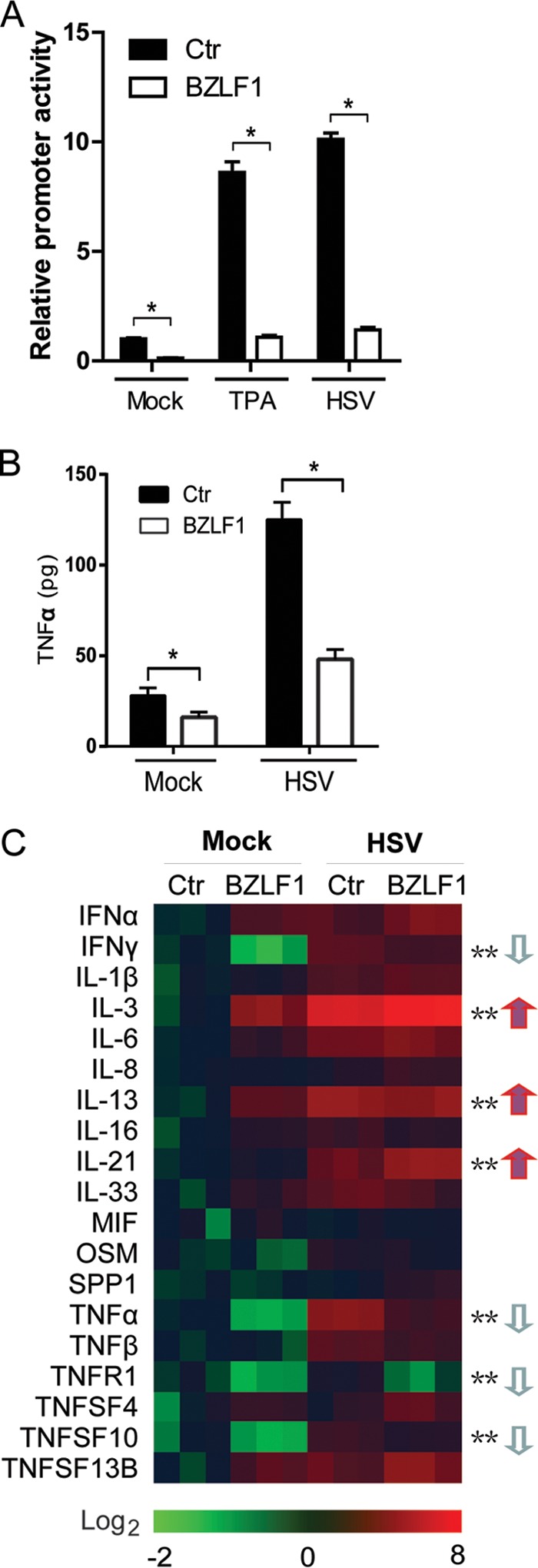
BZLF1 reduces TNF-α promoter activity, transcription, and expression. (A) pEGFP-BZLF1 or vector was cotransfected into 293T cells with TNF-α-luc and pRL-TK. After 24 h, 100 ng/ml TPA or HSV-1 (MOI = 10) was added, and the dual-luciferase assay was performed after 8 h of treatment. Ctr, control. (B and C) CNE-1 cells were transfected with pEGFP-BZLF1 or vector control. At 24 h after transfection, cells were left untreated or treated with HSV-1 (MOI = 10) for 8 h. The supernatants were used for the TNF-α ELISA (B), and RNA was extracted, reverse transcribed, and quantified by the inflammatory factor real-time PCR array (C). Values are shown as the means ± standard deviations of the results of three independent experiments. Statistical analysis was performed using one-way ANOVA followed by comparisons performed using the Bonferroni method. *, P < 0.05; **, P < 0.01. The arrows indicate down- or upregulation.
To further investigate whether BZLF1 regulates the inflammatory response, the expression of a series of inflammatory factors was determined using a real-time PCR-based inflammatory factor array. BZLF1 was introduced into the CNE-1 human NPC cell line following HSV-1 infection. Inflammatory factor arrays revealed that transcription of TNF-α was significantly reduced in BZLF1-transfected cells regardless of the presence or absence of HSV infection (Fig. 1C). In addition to inhibition of TNF-α and the TNFR1 receptor, similar levels of inhibition of expression of the inflammatory factors IFN-γ and TNFSF10 (also known as TNF-related apoptosis-inducing ligand [TRAIL]) by BZLF1 were observed, whereas IL-3, IL-13, and IL-21 levels were elevated. These results suggest that BZLF1 preferentially acts as a negative regulator of proinflammatory factors by inhibiting their transcription.
Mapping the key region of BZLF1 required for inhibition.
Several key functional domains of BZLF1, including the transcriptional activator (TA), the DNA binding domain (DB), the coiled-coil (CC) domain, and the C-terminal accessory domain, have been characterized previously (10). To further determine the mechanism of BZLF1 inhibition, we constructed a series of BZLF1 mutants (Fig. 2A). Deletion of the TA domain (BZLF1ΔTA) did not affect the inhibition of TNF-α expression compared with that seen with wild-type BZLF1, whereas deletion of the DB (BZLF1ΔDB) abolished its inhibition almost completely. Notably, a 4-amino-acid (aa) KSSE deletion in the CC domain (BZLF1Δ207-210) resulted in a loss of TNF-α inhibition (Fig. 2B and C). We also determined the effect of BZLF1 and the BZLF1 mutants on other inflammatory cytokines, including IFN-γ, IL-1β, and IL-8. During HSV-1 infection, wild-type BZLF1 and BZLF1ΔTA induced similar levels of inhibition of IFN-γ transcription, whereas inhibition was lost in both BZLF1Δ207-210 and BZLF1ΔDB constructs (Fig. 2D). Neither BZLF1 nor the mutants inhibited IL-1β or IL-8 expression (Fig. 2E and F). Notably, BZLF1Δ207-210 maintained the full capability of activating the EBV lytic promoters (BMLF1 and BMRF1) similarly to wild-type BZLF1, while BZLF1ΔTA and BZLF1ΔDB lost the transcriptional activity (Fig. 2G and H). None of the mutations affected the activity of latent promoter EBNA1 (Fig. 2I). These results suggest that the DNA binding and protein interaction properties of BZLF1 but not its transcriptional activation properties are specifically required for the BZLF1 inhibition of both TNF-α expression and IFN-γ expression.
FIG 2.

Mapping BZLF1 functional domains required for the inhibition of TNF-α. (A) Diagrams of wild-type and mutant BZLF1 are shown, and the expression levels of GFP-tagged BZLF1 constructs in 293T cells were detected by Western blotting. TA, transcriptional activator domain; DB, DNA binding domain; CC, coiled-coil domain. (B) Wild-type and mutant BZLF1 constructs were cotransfected with TNF-α-luc promoter reporters with GAPDH promoter-driven Renilla luciferase expression vector pRL-GAPDH as the internal control. (C) Different doses (0 ng, 10 ng, 50 ng, 100 ng, and 200 ng) of wild-type or mutated BZLF1-expressing plasmids were cotransfected with the TNF-α reporter and pRL-GAPDH in 293T cells. (D to I) Wild-type and mutant BZLF1 constructs were cotransfected with IFN-γ-luc, IL-1β-luc, pIL-8-luc, pBMLF1-luc, pBMRF1-luc, or pEBNA1-luc promoter reporters and pRL-GAPDH as an internal control. At 24 h after transfection, cells were left untreated or infected by HSV-1 (MOI = 10) for 8 h or treated with TPA (100 ng/ml) for 8 h or starved overnight and stimulated with 20% (vol/vol) FBS for 0.5 h. The luciferase assays were performed as described above. The values are shown as the means ± standard deviations of triplicate analyses of data from three independent experiments. *, P < 0.05.
Next, we investigated whether the BZLF1Δ207-210 mutant showed altered biochemical properties compared with BZLF1. We first performed CHX chase experiments to compare the half-life of BZLF1 to the half-life of BZLF1Δ207-210 and found that BZLF1Δ207-210 exhibited the same half-life as BZLF1 (Fig. 3A). Furthermore, the homodimerization of BZLF1Δ207-210 was detected, and similar patterns of oligomerization were observed for BZLF1Δ207-210 and wild-type BZLF1 (Fig. 3B). Finally, the results of an electrophoretic mobility shift assay performed using a BMLF1 probe showed that the BZLF1Δ207-210 mutation did not exhibit altered DNA binding to the ZRE compared with BZLF1 (Fig. 3C). Combining those data with its full activity for EBV lytic transcription (Fig. 2G to I), we concluded that BZLF1Δ207-210 retains its ability to activate EBV lytic transcription while losing the inhibitory effects on TNF-α and IFN-γ expression.
FIG 3.

Deletion of aa 207 to 210 does not affect BZLF1 biochemical properties. (A) GFP-tagged BZLF1 and BZLF1Δ207-210 were transfected into 293T cells. At 24 h after transfection, 20 μg/ml cycloheximide was added and the reaction mixtures were incubated for the different times as indicated; then, the cells were collected, and cell extracts were subjected to Western blotting with anti-BZLF1 antibody. The results of examination of the intensity of the BZLF1 bands were quantified, imaged, and analyzed using Li-COR Odyssey and are shown as the means of data from duplicate experiments. (B) GFP-fused BZLF1 and BZLF1Δ207-210 were transfected in 293T cells. After 36 h, the cell lysates were left untreated or incubated with 5 mM cross-linker disuccinimidyl suberate (DSS) at room temperature for 10 min, followed by Western blotting with an anti-BZLF1 antibody. (C) Different amounts of GFP-fused BZLF1 and BZLF1Δ207-210 were transfected in 293T cells for 48 h; then, the nuclear protein fractions were extracted, quantified, and validated with anti-GFP and anti-PCNA antibodies. The same amounts of nuclear proteins were incubated with Cys5.5-labeled BMLF1 promoter probes for 20 min; then, the mixtures were subjected to EMSA, and the images were visualized with Li-COR Odyssey.
BZLF1Δ207-210 exhibits no NF-κB inhibition.
Transcription factor binding site prediction showed that the TNF-α promoter contains multiple NF-κB elements, an AP-1 element, and a C/EBP binding site; however, BZLF1Δ207-210 did not suppress AP-1 element-driven transcription (data not shown). In addition, none of the single CC mutations of BZLF1, S208E, and N211G, which occur at residues required for C/EBP transactivation and EBV replication (39), significantly affected TNF-α inhibition (Fig. 2B). Because NF-κB is a well-known partner of BZLF1 (24, 25), NF-κB element-driven luciferase reporters were used to examine the role of the NF-κB elements in BZLF1-mediated TNF-α inhibition. Both BZLF1 wild-type constructs and BZLF1ΔTA constructs significantly reduced NF-κB element transcriptional activity, whereas BZLF1Δ207-210 did not (Fig. 4A). Similarly, phosphorylation of the NF-κB p65 subunits was also dramatically decreased by the BZLF1 wild-type and BZLF1ΔTA constructs but not by BZLF1Δ207-210 under both normal culture conditions and serum stimulation conditions (Fig. 4B). The levels of phosphorylated IκB-α, total p65, and IκB-α were not affected. Furthermore, BZLF1 wild-type and BZLF1ΔTA constructs interacted with the p65 subunit, whereas BZLF1Δ207-210 lost the interaction (Fig. 4C). As result, BZLF1 wild-type and BZLF1ΔTA constructs inhibited DNA binding of the p65 subunit, while BZLF1Δ207-210 reduced the inhibition (Fig. 4D). Finally, we deleted the NF-κB elements in the TNF-α promoter and found that deletion of any NF-κB element greatly decreased the transcriptional activity of the TNF-α promoter and that deletion of all NF-κB elements completely abolished TNF-α expression and BZLF1 inhibition (Fig. 4E and F), indicating the essential role of NF-κB in TNF-α transcription. Given the important role of NF-κB in inflammation, we conclude that BZLF1 inhibits TNF-α transcription through the disruption of NF-κB activation.
FIG 4.

Effect of BZLF1 constructs on NF-κB activity. (A) Wild-type and mutant BZLF1 constructs were cotransfected into 293T cells with the NF-κB-luc reporter. At 24 h after transfection, the medium was replaced with DMEM supplemented with 1% FBS for starvation conditions. After incubation overnight, 20% FBS was added for 15 min as a serum stimulation. Cell lysates were subjected to the dual-luciferase assay. Relative luciferase activities from three independent experiments performed in triplicate are shown. (B) 293T cells were transfected with wild-type and mutant BZLF1 constructs. After 24 h, the cells were left untreated or treated as described above, and the cell extracts were subjected to Western blotting with antibodies as indicated; one representative Western blot image is shown. (C) GST-tagged wild-type and mutant BZLF1 constructs (in pEBG vector) were transfected into 293T cells. At 48 h after transfection, cell lysates were subjected to immunoprecipitation (IP) with anti-GST beads. Immunoprecipitated samples and input samples were analyzed by Western blotting (WB) as indicated. (D) GST-tagged wild-type and mutant BZLF1 constructs were cotransfected with myc-tagged NF-κB p65 plasmid into 293T cells. At 48 h after transfection, nuclear extracts were subjected to ChIP assay with anti-myc beads and analyzed by real-time PCR. Representative results of three independent experiments are shown. (E) Diagram of NF-κB binding sites in TNF-α promoter. (F) The wild-type and different NF-κB binding site-mutated TNF-α-luc promoter reporters were cotransfected into 293T cells with BZLF1 construct and pRL-GAPDH. After 24 h, the cells with treated with 100 ng/ml TPA for 8 h, and the dual-luciferase assay was performed. *, P < 0.05.
BZLF1 binds the TNF-α promoter.
Because a typical ZRE is not present in the TNF-α promoter, we performed a ChIP assay to determine whether BZLF1 binds the TNF-α promoter. EBV-harboring P3HR-1 cells were treated with TPA and butyrate to induce lytic replication and BZLF1 expression; at the 24-h peak, the cells were collected and subjected to a ChIP assay with an anti-BZLF1 antibody, with the GAPDH promoter as a negative control. We observed a strong interaction of BZLF1 with the TNF-α promoter during reactivation and a weaker interaction when the cells were not induced (Fig. 5A). A similar result was obtained for the lytic promoters of BMLF1 and BSRF1, while no interaction was observed for the latent EBNA1 promoter or the GAPDH promoter. Furthermore, BZLF1Δ207-210 was completely unable to bind the TNF-α promoter, whereas binding of BZLF1ΔTA was not reduced compared with that of wild-type BZLF1 (Fig. 5B). In combination with the inhibition of NF-κB activation by BZLF1, BZLF1 impaired the recruitment and activity of NF-κB p65 to the TNF-α promoter. These results suggest that BZLF1 blocks TNF-α expression by direct disruption of NF-κB-dependent transcription at the TNF-α promoter.
FIG 5.

Determination of activity of BZLF1 binding to the TNF-α promoter. (A) P3HR-1 cells were left untreated or induced by TPA and butyrate as described in Materials and Methods. The cells were harvested, and a ChIP assay was performed with an anti-BZLF1 antibody. Real-time PCR assays were used to detect the promoter binding of TNF-α, EBNA1, BMLF1, BSRF1, and the GAPDH negative control in the ChIP samples. The relative levels of DNA binding were normalized to the input, and the means of the results of three independent experiments are shown. (B) GST-tagged wild-type and mutant BZLF1 constructs were transfected into CNE-1 cells for 48 h, and the ChIP assay was performed using anti-GST beads. Promoter binding of TNF-α and GAPDH was detected using real-time PCR. *, P < 0.05.
BZLF1 inhibits TNF-α and IFN-γ expression during early lytic replication.
As expected, LMP1-mediated TNF-α expression was inhibited by wild-type BZLF1 but not BZLF1Δ207-210 (Fig. 6A). To exclude the inhibition of endogenous BZLF1, BZLF1-KO EBV-harboring BJAB cells were established and transduced with wild-type BZLF1- and BZLF1Δ207-210-expressing lentiviruses, and then equal levels of BZLF1 and BZLF1Δ207-210 expression were observed (Fig. 6B). Latent BZLF1-KO EBV-harboring cells secreted TNF-α, and ectopic BZLF1 expression significantly reduced TNF-α production within 2 days, while this production was slightly inhibited by BZLF1Δ207-210 (Fig. 6C). Although equal ectopic BZLF1Δ207-210 expression induced the same level of expression of viral genes as wild-type BZLF1 (see below) (Fig. 6B), BZLF1Δ207-210 exhibited a substantially lower ability to inhibit TNF-α during the IE and early stages (12 h and 24 h) in EBV-infected cells (Fig. 6D). TNF-α expression was dramatically decreased by BZLF1, whereas BZLF1Δ207-210 exhibited a reduced inhibitory ability during the IE and early stages and showed a lower ability to inhibit TNF-α expression than wild-type BZLF1 (Fig. 6D). Although ectopic BZLF1Δ207-210 itself did not inhibit TNF-α expression, it induced the expression of other inhibitory viral proteins that gradually led to the secondary inhibition during the late stage (48 h). These results suggest that BZLF1 itself mainly plays an inhibitory role in TNF-α expression during the IE and early stages of the EBV lytic life cycle.
FIG 6.

BZLF1Δ207-210 reduces the inhibition of TNF-α and IFN-γ expression during the IE and early EBV lytic replication. (A) The control vector, BZLF1, or BZLF1Δ207-210 was cotransfected into 293T cells with LMP1-expressing plasmids and the TNF-α promoter-luciferase reporter, and the luciferase activities were determined at 24 h after transfection. (B to F) BZLF1-KO EBV BAC-harboring BJAB cells were transduced with the control, BZLF1, or BZLF1Δ207-210 using the lentiviral system (MOI = 10), the expression of viral genes was detected by Western blotting (B), TNF-α (C) and IFN-γ (E) secretions were detected by ELISA, and the relative TNF-α (D) and IFN-γ (F) mRNA levels at different time points were determined by real-time PCR. *, P < 0.05.
Furthermore, the expression of IFN-γ in BZLF1-KO EBV-harboring BJAB cells was measured in the presence of wild-type BZLF1 or BZLF1Δ207-210 expression. BZLF1-induced lytic replication in these cells triggered significant IFN-γ secretion within 2 days, while much higher IFN-γ production was observed in cells with ectopic BZLF1Δ207-210 expression (Fig. 6E). Similarly, ectopic BZLF1Δ207-210 expression induced much more IFN-γ mRNA in the cells than wild-type BZLF1 expression, where approximately 3-fold-increased IFN-γ mRNA levels were detected in the cells with BZLF1Δ207-210 expression at early and late stages (24 and 48 h postransduction) compared to the cells with wild-type BZLF1 expression (Fig. 6F). These results indicate that BZLF1 inhibits IFN-γ expression and production during EBV lytic replication.
Loss of TNF-α inhibition by BZLF1 does not inhibit viral gene expression but decreases EBV replication.
Because BZLF1 is a key immediate early transcriptional activator that switches viral gene expression from latent program to lytic program, we examined whether the loss of BZLF1 inhibitory activity in the TNF-α proinflammatory response affects viral lytic replication. GFP-tagged wild-type BZLF1, BZLF1ΔTA, and BZLF1Δ207-210 were introduced into P3HR-1 cells as inducers of lytic replication. Whereas the proportion of GFP-positive P3HR-1 cells that were observed after infection with lentiviruses that express the BZLF1 wild type, BZLF1ΔTA, or BZLF1Δ207-210 was more than 80%, similar levels of GFP-tagged BZLF1 wild-type and mutated protein were detected (Fig. 7A). Upon transduction of equal levels of BZLF1 and BZLF1Δ207-210, similar levels of endogenous BZLF1 and BMRF1 expression were induced, whereas BZLF1ΔTA did not show this induction (Fig. 7A). We next detected viral gene expression, which revealed that BZLF1ΔTA did not induce the expression of latent and lytic genes, whereas wild-type BZLF1 and BZLF1Δ207-210 comparably activated expression of the lytic BMRF1, BALF4, BcLF1, and BLLF1 genes (Fig. 7B) by several hundredfold, and both constructs increased the expression of the latent EBNA1 and LMP1 genes by 10-fold (Fig. 7C). No differences in the cell cycle or cell death were observed upon BZLF1Δ207-210 or BZLF1 transduction (data not shown). Interestingly, decreased viral DNA replication and decreased virion production were observed in BZLF1Δ207-210-transduced P3HR-1 cells compared with BZLF1-transduced cells (Fig. 7D and E). These results indicate that BZLF1Δ207-210 sustains its complete activity in lytic viral transcription but reduces its activity in lytic replication.
FIG 7.

Analysis of EBV lytic replication and gene expression induced by mutant BZLF1 in P3HR1 cells. Lentivirus-based BZLF1 wild-type and mutated constructs were introduced into P3HR-1 cells. (A) After lentiviral infection, the cells were collected at the different time points, and cell lysates were analyzed by Western blotting with antibodies as indicated. (B and C) Two days after lentiviral infection, total RNAs were extracted and reverse transcribed, and the expression levels of lytic genes (B) and latent genes (C) were detected by real-time PCR. (D and E) Cells and supernatants were harvested at the time points indicated, and genomic and virion DNA were extracted and quantified by real-time PCR for viral DNA replication (D) and virion particles (E), respectively. *, P < 0.05.
To further exclude the effect of endogenous BZLF1 expression induced by BZLF1Δ207-210, BZLF1-KO EBV BAC-harboring BJAB cells were established. Following treatment with equal titers of wild-type BZLF1- and BZLF1Δ207-210-expressing lentivirus infections, similar (>90%) percentages of BZLF1-positive cells were observed in BZLF1-KO EBV BAC-harboring BJAB cells (Fig. 8A), and equal ectopic expression levels of wild-type BZLF1 and BZLF1Δ207-210 expression were detected (Fig. 8B). Wild-type BZLF1 and BZLF1Δ207-210 induced equal levels of BMRF1 expression in wild-type and BZLF1-KO EBV BAC-harboring cells that were similar to that activated by chemical inducer TPA and sodium butyrate, while no differences in the levels of EBNA1 and LMP2A expression were observed in cells with ectopic expression of wild-type BZLF1 or BZLF1Δ207-210 (Fig. 8B). The same levels of expression of viral EBNA1, LMP1, BMRF1, BALF4, BcLF1, and BLLF1 genes were observed in BZLF1-KO EBV BAC-harboring BJAB cells with wild-type BZLF1 or BZLF1Δ207-210 expression (Fig. 8C); however, BZLF1Δ207-210-induced levels of viral replication and virion production in both wild-type and BZLF1-KO EBV BAC-harboring BJAB cells were substantially lower than those induced by wild-type BZLF1 (Fig. 8D and E). Likely due to the lower original viral genomic DNA load in the cells, both wild-type BZLF1- and BZLF1Δ207-210-induced levels of viral replication and virion production in BZLF1-KO EBV BAC-harboring BJAB cells were lower than those seen with wild-type EBV BAC-harboring cells. Similarly, in wild-type and BZLF1-KO EBV BAC-harboring epithelial HNE-1 cells, equal levels of wild-type BZLF1 and BZLF1Δ207-210 ectopic expression induced similar levels of expression of the lytic BMRF1 gene that were equal to that induced by TPA plus sodium butyrate, and no difference in EBNA1 expression was observed (Fig. 8F); however, BZLF1Δ207-210-induced viral lytic replication was dramatically weaker than that induced by wild-type BZLF1 in wild-type and BZLF1-KO EBV BAC-harboring HNE-1 cells (Fig. 8G). These results indicate that the loss of BZLF1-mediated inhibition of the proinflammatory response does not influence viral transcription but reduces viral replication.
FIG 8.

Analysis of EBV lytic replication and gene expression induced by mutant BZLF1 in BZLF1-KO EBV BAC-harboring B cells and epithelial cells. (A to E) Wild-type and BZLF1-KO EBV BAC-harboring BJAB cells were infected with GFP-tagged wild-type BZLF1 or BZLF1Δ207-210-expressing lentiviruses (MOI = 10). (A) At 48 h postinfection, BZLF1-KO EBV BAC-harboring BJAB cells were collected and subjected to immunofluorescence (IF) staining with anti-BZLF1 antibody. (B and C) At 48 h postinfection, wild-type and BZLF1-KO EBV BAC-harboring BJAB cells were collected, whole-cell extracts were analyzed by Western blotting as indicated (B), and viral mRNAs were extracted, reverse transcribed, and analyzed for expression by reverse transcription-quantitative PCR (RT-qPCR) (C). (D and E) The cell pellets and supernatants were collected at different time points after lentiviral infection, and then intracellular viral genomic DNA (D) and extracellular virion DNA (E) were extracted and analyzed by real-time PCR. *, P < 0.05. (F and G) Wild-type and BZLF1-KO EBV-BAC-harboring HNE-1 cells were seeded in a 6-well plate and transfected with 2 μg control vector or GFP-BZLF1- or GFP-Δ207-210-expressing plasmids. The expression levels of viral proteins were determined by Western blotting 48 h posttransfection (F), and intracellular viral genomic DNA was extracted and analyzed 72 h after transfection (G). *, P < 0.05.
TNF-α depletion restores viral DNA replication induced by BZLF1Δ207-210.
To confirm that inhibition of inflammatory factors by BZLF1 is required for EBV replication, P3HR-1 cells that secrete a high level of TNF-α were transfected with stable TNF-α shRNA and then transiently infected with equal titers of lentiviruses expressing BZLF1 and BZLF1Δ207-210. The secretion of TNF-α in control, BZLF1-, and BZLF1Δ207-210-transduced P3HR-1 cells was dramatically depleted by stable transfection of TNF-α shRNA (Fig. 8A). Equal levels of ectopic BZLF1 and BZLF1Δ207-210 expression were observed in the cells transduced with control or TNF-α shRNA, as well as the expression of viral BMRF1 and EBNA1 genes (Fig. 8B). Viral DNA replication was detected under conditions of TNF-α depletion, and TNF-α shRNA influenced neither latent DNA replication nor the lytic viral replication induced by wild-type BZLF1. However, TNF-α depletion accelerated the DNA replication induced by BZLF1Δ207-210 (Fig. 8C) and augmented the production of EBV virion induced by BZLF1Δ207-210 without elevation by wild-type BZLF1 (Fig. 8D). Furthermore, BZLF1-KO EBV BAC-harboring BJAB cells were also transduced with stable TNF-α shRNA followed by infection with control and BZLF1- and BZLF1Δ207-210-expressing lentiviruses. The secretion of TNF-α in control and BZLF1Δ207-210-expressing cells was greatly depleted by TNF-α shRNA, and that in BZLF1-expressing cells was dramatically decreased by BZLF1 regardless of whether or not transduction with TNF-α shRNA occurred (Fig. 9E). Similarly, TNF-α shRNA affected neither ectopic BZLF1 expression and BZLF1Δ207-210 expression nor the expression of the viral BMRF1 and EBNA1 genes (Fig. 9F). Because TNF-α was already inhibited, TNF-α shRNA did not promote viral lytic replication in BZLF1-expressing cells; however, intracellular viral DNA replication and virion production were elevated appropriately 2-fold by TNF-α depletion in BZLF1Δ207-210-expressing cells (Fig. 9G and H). Because IFN-γ and other inflammatory factors that contribute to the inflammatory response were also suppressed (Fig. 1C), TNF-α depletion restored half of the viral replication and virion production. These results suggest that BZLF1 decreases TNF-α secretion to facilitate viral DNA replication.
FIG 9.

Depletion of TNF-α restores EBV lytic replication induced by BZLF1Δ207-210. (A to D) Three stable TNF-α shRNAs were introduced into P3HR-1 cells by the pLKO.1 lentiviral system. Each cell line was then infected with BZLF1- or BZLF1Δ207-210-expressing lentiviruses (MOI = 10). (A) After 24 h, supernatants were collected, and TNF-α was quantified by ELISA. (B) The expression of BZLF1 and viral genes was detected by Western blot analyses as indicated at 48 h postlentivirus transduction. (C and D) Levels of DNA replication inside the cells (C) and virion particles in the supernatants (D) were determined as described above. (E to H) BZLF1-KO EBV-harboring BJAB cells were transfected with stable shTNF-α1 and infected with BZLF1- or BZLF1Δ207-210-expressing lentiviruses as described above, and the levels of TNF-α secretion (E), expression of viral genes (F), intracellular viral DNA replication (G), and extracellular virion particles (H) were detected. Statistical analysis was performed by ANOVA for replicate measures. *, P < 0.05.
Neutralizing TNF-α and IFN-γ completely recovers viral DNA replication.
To determine the cooperative role of TNF-α and IFN-γ in viral replication, anti-TNF-α- and -IFN-γ-neutralizing antibodies were used to deplete cytokine activity. Incubation with IgG or antibodies themselves induced only very weak lytic replication, and neither treatment affected the equal levels of wild-type BZLF1 and BZLF1Δ207-210 ectopic expression as well as the expression of BMRF1 and EBNA1 (Fig. 10A). Viral replication and virion production were elevated by anti-TNF-α-neutralizing antibodies alone in BZLF1Δ207-210-expressing P3HR-1 cells, whereas anti-IFN-γ-neutralizing antibodies did not increase virion production and increased viral DNA replication only slightly (Fig. 10B and C). The combination of the two antibodies augmented viral replication and virion production and restored viral replication to the same level as that seen with wild-type BZLF1 after incubation for 3 days and virion production after 5 days.
FIG 10.

Neutralization of TNF-α and IFN-γ promotes EBV lytic replication induced by BZLF1Δ207-210. (A to C) P3HR-1 cells were transduced with lentivirus-based BZLF1 or BZLF1Δ207-210 or control vector. After 6 h, BZLF1- or BZLF1Δ207-210-transduced P3HR-1 cells were incubated with anti-TNF-α- or anti-IFN-γ-neutralizing antibodies or isotype IgG in the different combinations and at the different time points as indicated. (A) After incubation for 48 h, expression of viral genes was detected by Western blot analyses as indicated. (B and C) Then, the intracellular DNA genome (B) and the extracellular virion particles (C) were collected and levels were determined as described in Materials and Methods. (D to F) BZLF1-KO EBV BAC-harboring BJAB cells were infected with GFP-tagged wild-type BZLF1- or BZLF1Δ207-210-expressing lentiviruses (MOI = 10). At 6 h after lentiviral infection, anti-TNF-α- or anti-IFN-γ-neutralizing antibodies or isotype IgG was added as indicated. (D) Levels of expression of viral genes were detected by Western blot analyses after incubation with antibodies for 48 h. (E) After incubation for 3 days, the cells were collected and intracellular viral genomic DNAs were extracted and analyzed by real-time PCR for intracellular viral DNA replication. (F) After incubation for 5 days, the supernatants were collected, and virion DNAs were extracted and analyzed. *, P < 0.05 (compared with isotype IgG). The x-axis data for panels E and F are as indicated for panels B and C, respectively.
To further confirm that BZLF1-mediated inhibition of TNF-α and IFN-γ promotes viral lytic replication, anti-TNF-α- and anti-IFN-γ-neutralizing antibodies were used to deplete TNF-α and IFN-γ secretion in BZLF1-KO EBV BAC-harboring BJAB cells after infection with equal titers of wild-type BZLF1- and BZLF1Δ207-210-expressing lentiviruses. Equal levels of wild-type BZLF1 and BZLF1Δ207-210 ectopic expression were observed, and incubation of either of the antibodies did not affect their expression levels (Fig. 10D). Equal levels of BMRF1 expression were induced by wild-type BZLF1 and BZLF1Δ207-210 expression, and no difference in EBNA1 expression was observed, regardless of the conditions of incubation with or without either antibody (Fig. 10D). The levels of viral replication and virion production were augmented by both anti-TNF-α- and anti-IFN-γ-neutralizing antibodies in both cells, and optimal levels of replication and yield of progeny virus were induced under conditions that included the combination of the two antibodies (Fig. 10E and F). Although the replication of viral genomic DNA was substantially lower in BZLF1-KO EBV BAC-harboring BJAB cells, the combination of the two antibodies increased viral DNA replication and virion yield approximately 4-fold under conditions of BZLF1Δ207-210 transduction (Fig. 10E and F). Notably, wild-type BZLF1 and BZLF1Δ207-210 induced nearly identical levels of viral DNA replication and virion production under the conditions that included the combination of the two antibodies (Fig. 10E and F). These results suggest that blockade of TNF-α and IFN-γ cooperatively abolished the antiviral inflammatory response and facilitated optimal replication during EBV lytic replication.
DISCUSSION
In the present study, we demonstrated that BZLF1 suppresses TNF-α and IFN-γ proinflammatory responses through disrupting NF-κB activation, thereby facilitating optimal viral replication and virion production. BZLF1 mainly suppresses proinflammatory factors and alters the inflammatory microenvironment during the IE and early stages of EBV lytic replication, representing a novel strategy for the evasion of the inflammatory response during the EBV lytic life cycle.
Experimental and clinical studies have revealed that levels of inflammatory factors are elevated in EBV-infected cells and lesions (40–45). The majority contribute to expression of latent viral proteins, such as LMP1, which activate inflammatory factors through the participation of several cellular partners (46, 47). However, the pattern of inflammatory factors is altered following the switch from the latent EBV life cycle to the lytic life cycle. Previous studies have shown that BZLF1 regulates inflammation (10, 48), and our present study provided new evidence indicating that BZLF1 attenuates TNF-α and IFN-γ expression as well as expression of a series of other inflammatory factors. In addition, BZLF1 abolishes the response to TNF-α and IFN-γ stimuli by inhibiting the expression of the major TNF receptor TNFR1 and the IFN-γ receptor, respectively (21–23). These results indicate that BZLF1 mediates the evasion of the antiviral inflammatory response during the EBV lytic life cycle by inhibiting proinflammatory factors and receptors.
The C/EBP homologous motif in BZLF1 is required for EBV replication; however, the BZLF1 A204D and S208E mutations in this motif, which abolish TNFR1 inhibition and EBV replication (23, 39), respectively, do not affect the inhibition of TNF-α (Fig. 2B), indicating that this distinct mechanism of TNF-α inhibition is C/EBP independent. We confirmed that the inhibition is due to the disruption of the NF-κB pathway (Fig. 4), which is frequently involved in inflammation and cancer (49, 50). Previous and current studies have revealed that BZLF1 directly targets the NF-κB p65 subunit and prevents the recruitment of NF-κB to cellular promoters (see references 24 and 25 and Fig. 4). This inhibition does not require the domains of BZLF1 DNA binding and transcriptional activation but requires the coiled-coil domain of BZLF1 protein-protein interactions and dimerization. This domain is required for interaction with the NF-κB p65 subunit (24) and is sensitive to mutations because many conserved residues are critical for BZLF1 DNA binding and viral DNA replication (39). This region is an α-helix that forms a coiled-coil structure (51), and a single deletion disrupts the α-helix structure. BZLF1Δ207-210 with 4-aa KSSE residues deleted (one single spiral coil in the α-helix) may not have an altered global conformation. It exhibits nearly identical biochemical properties and transactivation (Fig. 2 and 3), while losing the interaction with the NF-κB p65 subunit (Fig. 4). It is presumable that the truncated coiled-coil domain of BZLF1Δ207-210 is sufficient for homodimerization and DNA binding to ZREs, while deletion of the KSSE coil may disrupt the binding motif of the NF-κB p65 subunit or the truncation of the α-helix may cause the shift of the C-terminal tail to block the approach of the NF-κB p65 subunit.
Our results demonstrate that the inhibition of the inflammatory factors during the lytic life cycle promotes viral replication and virion production in lytic EBV-infected cells. The BZLF1Δ207-210-mediated loss of inhibition of proinflammatory responses does not reduce either lytic or latent EBV gene expression, whereas DNA replication and virion production are dramatically decreased (Fig. 7 and 8). However, depletion of TNF-α restores viral replication by half, indicating that TNF-α is one of the key suppressors of the antiviral inflammatory response, which is primarily blocked by BZLF1 during the IE and early lytic cycles (Fig. 6). Furthermore, the use of TNF-α- and IFN-γ-neutralizing antibodies confirms the prominent function of TNF-α and the cooperative role of IFN-γ in the antiviral inflammatory response during EBV lytic replication (Fig. 10).
The expression of the TNF-α and IFN-γ proinflammatory factors is essential for the activation of antiviral inflammatory responses, which restrict viral infection and replication as well as directly triggering cell death (17, 20). The results of previous studies and the present study show that BZLF1 is a key inhibitor in suppression of TNF-α and IFN-γ signaling, by which BZLF1 induces intracellular and microenvironmental alterations to promote viral lytic replication. Expression of BZLF1Δ207-210 triggers viral lytic gene expression, while TNF-α and IFN-γ expression levels are not suppressed during the IE and early stages of lytic replication; then, the proinflammatory responses counteract viral replication. Depletion of TNF-α by shRNA or neutralization of TNF-α and IFN-γ by antibodies eliminates the antiviral effect and recovery of viral replication (Fig. 9 and 10). Thus, early inhibition of the TNF-α and IFN-γ responses by BZLF1 is an efficient and important strategy utilized by EBV to evade antiviral inflammatory responses and facilitate optimal viral replication.
In the EBV life cycle, BZLF1-mediated proinflammatory factors might function in lytic cells in an autocrine manner and in latent or uninfected cells in a paracrine manner. BZLF1 inhibition of the autocrine proinflammatory response is required for optimal viral DNA replication and progeny virion production. In contrast, the inhibition of proinflammatory factors alleviates the antiviral states in latent and uninfected cells to enhance secondary infection and viral maintenance. In addition to EBV replication and infection, BZLF1 inhibition of proinflammatory factors inhibits paracrine senescence in neighboring epithelial cells (unpublished data). This senescence is also due to proinflammatory factors TNF-α and IFN-γ, whose presence is sufficient to induce senescence and attenuate viral infection (16, 52–54).
In summary, we have demonstrated that BZLF1 acts as a novel transcriptional silencer of proinflammatory factors by directly binding to the TNF-α promoter and disrupting NF-κB-dependent transcription. This inhibition facilitates EBV lytic infection and replication, which may represent a strategy to evade the antiviral inflammatory response during the EBV lytic life cycle.
ACKNOWLEDGMENTS
We thank Hui Zhang, Musheng Zeng, and Xi Huang for critical discussions and all members of our laboratory for helpful assistance.
This work was supported by National Natural Science Foundation of China grants (81371792 and U1301121), the Guangdong Innovative Research Team Program (no. 2009010058), and the Sun Yat-sen University Setup fund (to E.K.).
REFERENCES
- 1.Kutok JL, Wang F. 2006. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol 1:375–404. doi: 10.1146/annurev.pathol.1.110304.100209. [DOI] [PubMed] [Google Scholar]
- 2.Busson P, Keryer C, Ooka T, Corbex M. 2004. EBV-associated nasopharyngeal carcinomas: from epidemiology to virus-targeting strategies. Trends Microbiol 12:356–360. doi: 10.1016/j.tim.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Allday MJ. 2008. The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol 6:913–924. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- 4.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 6.Liu P, Fang X, Feng Z, Guo YM, Peng RJ, Liu T, Huang Z, Feng Y, Sun X, Xiong Z, Guo X, Pang SS, Wang B, Lv X, Feng FT, Li DJ, Chen LZ, Feng QS, Huang WL, Zeng MS, Bei JX, Zhang Y, Zeng YX. 2011. Direct sequencing and characterization of a clinical isolate of Epstein-Barr virus from nasopharyngeal carcinoma tissue by using next-generation sequencing technology. J Virol 85:11291–11299. doi: 10.1128/JVI.00823-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, Delecluse HJ. 2013. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5:458–470. doi: 10.1016/j.celrep.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Zeng MS, Li DJ, Liu QL, Song LB, Li MZ, Zhang RH, Yu XJ, Wang HM, Ernberg I, Zeng YX. 2005. Genomic sequence analysis of Epstein-Barr virus strain GD1 from a nasopharyngeal carcinoma patient. J Virol 79:15323–15330. doi: 10.1128/JVI.79.24.15323-15330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odumade OA, Hogquist KA, Balfour HH Jr. 2011. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev 24:193–209. doi: 10.1128/CMR.00044-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Li D, Guo N. 2009. Regulation of cellular and viral protein expression by the Epstein-Barr virus transcriptional regulator Zta: implications for therapy of EBV associated tumors. Cancer Biol Ther 8:987–995. doi: 10.4161/cbt.8.11.8369. [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 12.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. 2006. Inflammation and cancer: how hot is the link? Biochem Pharmacol 72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Torchinsky MB, Garaude J, Blander JM. 2010. Infection and apoptosis as a combined inflammatory trigger. Curr Opin Immunol 22:55–62. doi: 10.1016/j.coi.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poeck H, Ruland J. 2012. From virus to inflammation: mechanisms of RIG-I-induced IL-1beta production. Eur J Cell Biol 91:59–64. doi: 10.1016/j.ejcb.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Zheng D, Chen H, Bartee MY, Williams J, Davids JA, Huang E, Moreb J, Lucas A. 2012. Virus-derived anti-inflammatory proteins: potential therapeutics for cancer. Trends Mol Med 18:304–310. doi: 10.1016/j.molmed.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Wong GH, Goeddel DV. 1986. Tumour necrosis factors alpha and beta inhibit virus replication and synergize with interferons. Nature 323:819–822. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 17.Rahman MM, McFadden G. 2006. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog 2:e4. doi: 10.1371/journal.ppat.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faber M, Bette M, Preuss MA, Pulmanausahakul R, Rehnelt J, Schnell MJ, Dietzschold B, Weihe E. 2005. Overexpression of tumor necrosis factor alpha by a recombinant rabies virus attenuates replication in neurons and prevents lethal infection in mice. J Virol 79:15405–15416. doi: 10.1128/JVI.79.24.15405-15416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lundberg P, Welander PV, Edwards CK III, van Rooijen N, Cantin E. 2007. Tumor necrosis factor (TNF) protects resistant C57BL/6 mice against herpes simplex virus-induced encephalitis independently of signaling via TNF receptor 1 or 2. J Virol 81:1451–1460. doi: 10.1128/JVI.02243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sedý JR, Spear PG, Ware CF. 2008. Cross-regulation between herpesviruses and the TNF superfamily members. Nat Rev Immunol 8:861–873. doi: 10.1038/nri2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison TE, Mauser A, Klingelhutz A, Kenney SC. 2004. Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J Virol 78:544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison TE, Mauser A, Wong A, Ting JP, Kenney SC. 2001. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity 15:787–799. doi: 10.1016/S1074-7613(01)00226-6. [DOI] [PubMed] [Google Scholar]
- 23.Bristol JA, Robinson AR, Barlow EA, Kenney SC. 2010. The Epstein-Barr virus BZLF1 protein inhibits tumor necrosis factor receptor 1 expression through effects on cellular C/EBP proteins. J Virol 84:12362–12374. doi: 10.1128/JVI.00712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutsch DE, Holley-Guthrie EA, Zhang Q, Stein B, Blanar MA, Baldwin AS, Kenney SC. 1994. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol Cell Biol 14:1939–1948. doi: 10.1128/MCB.14.3.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrison TE, Kenney SC. 2004. BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 328:219–232. doi: 10.1016/j.virol.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 26.de Oliveira DE, Ballon G, Cesarman E. 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol 18:248–257. doi: 10.1016/j.tim.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Hui KF, Ho DN, Tsang CM, Middeldorp JM, Tsao GS, Chiang AK. 2012. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int J Cancer 131:1930–1940. doi: 10.1002/ijc.27439. [DOI] [PubMed] [Google Scholar]
- 28.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol 79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prang NS, Hornef MW, Jager M, Wagner HJ, Wolf H, Schwarzmann FM. 1997. Lytic replication of Epstein-Barr virus in the peripheral blood: analysis of viral gene expression in B lymphocytes during infectious mononucleosis and in the normal carrier state. Blood 89:1665–1677. [PubMed] [Google Scholar]
- 30.Tomkinson B, Robertson E, Yalamanchili R, Longnecker R, Kieff E. 1993. Epstein-Barr virus recombinants from overlapping cosmid fragments. J Virol 67:7298–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cotter M, Callahan J, Aster J, Robertson E. 2000. Intracellular forms of human NOTCH1 functionally activate essential Epstein-Barr virus major latent promoters in the Burkitt's lymphoma BJAB cell line but repress these promoters in Jurkat cells. J Virol 74:1486–1494. doi: 10.1128/JVI.74.3.1486-1494.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scientia Sinica. 1978. Establishment of an epitheloid cell line and a fusiform cell line from a patient with nasopharyngeal carcinoma. Sci Sin 21:127–134. [PubMed] [Google Scholar]
- 33.Yao KT, Zhang HY, Zhu HC, Wang FX, Li GY, Wen DS, Li YP, Tsai CH, Glaser R. 1990. Establishment and characterization of two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus and derived from nasopharyngeal carcinomas. Int J Cancer 45:83–89. doi: 10.1002/ijc.2910450116. [DOI] [PubMed] [Google Scholar]
- 34.Kong QL, Hu LJ, Cao JY, Huang YJ, Xu LH, Liang Y, Xiong D, Guan S, Guo BH, Mai HQ, Chen QY, Zhang X, Li MZ, Shao JY, Qian CN, Xia YF, Song LB, Zeng YX, Zeng MS. 2010. Epstein-Barr virus-encoded LMP2A induces an epithelial-mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog 6:e1000940. doi: 10.1371/journal.ppat.1000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Spandidos A, Wang H, Seed B. 2012. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holden NS, Tacon CE. 2011. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods 63:7–14. doi: 10.1016/j.vascn.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J 19:3080–3089. doi: 10.1093/emboj/19.12.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald CM, Petosa C, Farrell PJ. 2009. Interaction of Epstein-Barr virus BZLF1 C-terminal tail structure and core zipper is required for DNA replication but not for promoter transactivation. J Virol 83:3397–3401. doi: 10.1128/JVI.02500-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foss HD, Herbst H, Hummel M, Araujo I, Latza U, Rancso C, Dallenbach F, Stein H. 1994. Patterns of cytokine gene expression in infectious mononucleosis. Blood 83:707–712. [PubMed] [Google Scholar]
- 41.Klein SC, Kube D, Abts H, Diehl V, Tesch H. 1996. Promotion of IL8, IL10, TNF alpha and TNF beta production by EBV infection. Leuk Res 20:633–636. doi: 10.1016/0145-2126(96)00029-X. [DOI] [PubMed] [Google Scholar]
- 42.Ho JW, Liang RH, Srivastava G. 1999. Differential cytokine expression in EBV positive peripheral T cell lymphomas. Mol Pathol 52:269–274. doi: 10.1136/mp.52.5.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D'Addario M, Ahmad A, Morgan A, Menezes J. 2000. Binding of the Epstein-Barr virus major envelope glycoprotein gp350 results in the upregulation of the TNF-alpha gene expression in monocytic cells via NF-kappaB involving PKC, PI3-K and tyrosine kinases. J Mol Biol 298:765–778. doi: 10.1006/jmbi.2000.3717. [DOI] [PubMed] [Google Scholar]
- 44.Hernádi K, Gyöngyösi E, Mészáros B, Szakács L, Szalmás A, Csoma E, Mogyorósi R, Czompa L, Veress G, Varga I, Márton IJ, Kónya J. 2013. Elevated tumor necrosis factor-alpha expression in periapical lesions infected by Epstein-Barr virus. J Endod 39:456–460. doi: 10.1016/j.joen.2012.12.028. [DOI] [PubMed] [Google Scholar]
- 45.Wada T, Muraoka M, Yokoyama T, Toma T, Kanegane H, Yachie A. 2013. Cytokine profiles in children with primary Epstein-Barr virus infection. Pediatr Blood Cancer 60:E46–E48. doi: 10.1002/pbc.24480. [DOI] [PubMed] [Google Scholar]
- 46.Li HP, Chang YS. 2003. Epstein-Barr virus latent membrane protein 1: structure and functions. J Biomed Sci 10:490–504. doi: 10.1007/BF02256110. [DOI] [PubMed] [Google Scholar]
- 47.Morris MA, Dawson CW, Young LS. 2009. Role of the Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the pathogenesis of nasopharyngeal carcinoma. Future Oncol 5:811–825. doi: 10.2217/fon.09.53. [DOI] [PubMed] [Google Scholar]
- 48.Guenther JF, Cameron JE, Nguyen HT, Wang Y, Sullivan DE, Shan B, Lasky JA, Flemington EK, Morris GF. 2010. Modulation of lung inflammation by the Epstein-Barr virus protein Zta. Am J Physiol Lung Cell Mol Physiol 299:L771–L784. doi: 10.1152/ajplung.00408.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiDonato JA, Mercurio F, Karin M. 2012. NF-kappaB and the link between inflammation and cancer. Immunol Rev 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 50.Ben-Neriah Y, Karin M. 2011. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 51.Petosa C, Morand P, Baudin F, Moulin M, Artero JB, Muller CW. 2006. Structural basis of lytic cycle activation by the Epstein-Barr virus ZEBRA protein. Mol Cell 21:565–572. doi: 10.1016/j.molcel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Beyne-Rauzy O, Recher C, Dastugue N, Demur C, Pottier G, Laurent G, Sabatier L, Mansat-De Mas V. 2004. Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene 23:7507–7516. doi: 10.1038/sj.onc.1208024. [DOI] [PubMed] [Google Scholar]
- 53.Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, Ranta F, Ullrich S, Mocikat R, Braungart K, Mehra T, Fehrenbacher B, Berdel J, Niessner H, Meier F, van den Broek M, Häring HU, Handgretinger R, Quintanilla-Martinez L, Fend F, Pesic M, Bauer J, Zender L, Schaller M, Schulze-Osthoff K, Röcken M. 2013. T-helper-1-cell cytokines drive cancer into senescence. Nature 494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki M, Ikeda H, Sato Y, Nakanuma Y. 2008. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of ATM pathway: a culture study. Free Radic Res 42:625–632. doi: 10.1080/10715760802244768. [DOI] [PubMed] [Google Scholar]