

ABSTRACT
Viruses of the family Flaviviridae are important pathogens of humans and other animals and are currently classified into four genera. To better understand their diversity, evolutionary history, and genomic flexibility, we used transcriptome sequencing (RNA-seq) to search for the viruses related to the Flaviviridae in a range of potential invertebrate and vertebrate hosts. Accordingly, we recovered the full genomes of five segmented jingmenviruses and 12 distant relatives of the known Flaviviridae (“flavi-like” viruses) from a range of arthropod species. Although these viruses are highly divergent, they share a similar genomic plan and common ancestry with the Flaviviridae in the NS3 and NS5 regions. Remarkably, although these viruses fill in major gaps in the phylogenetic diversity of the Flaviviridae, genomic comparisons reveal important changes in genome structure, genome size, and replication/gene regulation strategy during evolutionary history. In addition, the wide diversity of flavi-like viruses found in invertebrates, as well as their deep phylogenetic positions, suggests that they may represent the ancestral forms from which the vertebrate-infecting viruses evolved. For the vertebrate viruses, we expanded the previously mammal-only pegivirus-hepacivirus group to include a virus from the graceful catshark (Proscyllium habereri), which in turn implies that these viruses possess a larger host range than is currently known. In sum, our data show that the Flaviviridae infect a far wider range of hosts and exhibit greater diversity in genome structure than previously anticipated.
IMPORTANCE The family Flaviviridae of RNA viruses contains several notorious human pathogens, including dengue virus, West Nile virus, and hepatitis C virus. To date, however, our understanding of the biodiversity and evolution of the Flaviviridae has largely been directed toward vertebrate hosts and their blood-feeding arthropod vectors. Therefore, we investigated an expanded group of potential arthropod and vertebrate host species that have generally been ignored by surveillance programs. Remarkably, these species contained diverse flaviviruses and related viruses that are characterized by major changes in genome size and genome structure, such that these traits are more flexible than previously thought. More generally, these data suggest that arthropods may be the ultimate reservoir of the Flaviviridae and related viruses, harboring considerable genetic and phenotypic diversity. In sum, this study revises the traditional view on the evolutionary history, host range, and genomic structures of a major group of RNA viruses.
INTRODUCTION
Since the discovery of the virological cause of yellow fever in the 1920s, viruses of the family Flaviviridae have been well documented as the cause of major vector-borne and hepatic diseases in humans (1). The family as currently classified comprises four genera: Flavivirus, Hepacivirus, Pestivirus, and the newly proposed genus Pegivirus (2). Despite the extensive divergence between these viruses, they share important similarities in genome structure, virion morphology, and life cycle. The “classical” Flaviviridae genome is an unsegmented, single-stranded, and positive-sense RNA molecule between 9.6 and 12.3 kb in length. This encodes a single polyprotein with multiple transmembrane domains that is cleaved, by both host and viral proteases, into structural and nonstructural (NS) proteins. Among the nonstructural protein products, the locations and sequences of NS3 and NS5, which contain motifs essential for polyprotein processing and RNA replication, are relatively well conserved across the family and hence are valuable for phylogenetic analysis.
Viruses of the Flaviviridae have a wide host range that includes both vertebrates and invertebrates. However, until recently, the only recognized invertebrate hosts for the flaviviruses were mosquitoes and ticks, which contained viruses exclusively found within the genus Flavivirus. In addition, our understanding of arthropods has largely focused on their role as vectors for vertebrate viruses, although this view has recently been challenged by the discovery of phylogenetically divergent arthropod-specific viruses (3, 4). The remaining genera in the family—Hepacivirus, Pegivirus, and Pestivirus—are exclusively found in mammals, and their diversity has greatly expanded with recent virus discoveries in various mammalian species, including bats, dogs, horses, pigs, ruminants, and rodents (5–13).
Recently, two “flavi-like” viruses (i.e., distant relatives of the known Flaviviridae)—the Gentian Kobu-sho-associated virus (GKaV) (14) and soybean cyst nematode virus 5 (SbCNV-5) (15)—were discovered from plants of the genus Gentiana and soybean cyst nematode, respectively. Despite possessing exceptionally long genomes (23 and 19 kb, respectively), these two viruses have the basic flavi-like genome/proteome structure, as well as sequence homology in the serine protease, helicase, and RNA-dependent RNA polymerase (RdRp) domains of the polyprotein. Although GKaV was reported to be a double-stranded RNA (dsRNA) virus (14), the RNA digestion experiment was performed on crude plant extracts where the dsRNA is more likely to be the replication complex rather than the genome itself (15). More remarkable was the recent demonstration that Jingmen tick virus (JMTV) and Toxocara canis larva agent (TCLA) are clearly related to flaviviruses but possess a very different genomic plan comprising four distinct segments (11), in turn raising questions about the evolutionary link between segmented and unsegmented genomes. Since these segmented flavi-like viruses have not been formally classified, we tentatively refer to them as the Jingmenvirus group.
To obtain a deeper understanding of the diversity and evolution of this important group of RNA viruses, we screened a number of arthropod and vertebrate species that have generally been ignored by surveillance programs, and characterized the genomic structure and evolutionary history of a wider range of Flaviviridae.
MATERIALS AND METHODS
Sample preparation and sequencing.
The present study was based on the analysis of twelve pools of arthropod samples and one pool of vertebrate samples. The sample preparation and sequencing of the several pools—“insects, mix 1”; “insects, mix 4”; “spiders”; “true flies”; “ticks (general)”; “ticks (H. asiaticum)”; and “water striders”—have been described previously (16). The remaining four pools comprised different arthropod species and one of fish liver tissue from China, as well as one pool of mosquitoes from the Republic of Panama in Central America (Table 1).
TABLE 1.
Host and geographic information and data output for each pool of animal samples
| Pool | No. of units | Species in the pool | Location(s) (country: city) | Data generated (no. of bases) |
|---|---|---|---|---|
| True flies | 24 | Atherigona orientalis, Chrysomya megacephala, Lucilia sericata, Musca domestica, Sarcophaga dux, S. peregrina, Sarcophaga sp. | China: Hubei | 6,574,954,320 |
| Water striders | 12 | Gerridae sp. | China: Hubei | 3,154,714,200 |
| Insects, mix 1 | 6 | Abraxas tenuisuffusa, Hermetia illucens, Chrysopidae sp., Coleoptera sp., Psychoda alternata, Diptera sp., Stratiomyidae sp. | China: Zhejiang | 7,745,172,660 |
| Insects, mix 4 | 12 | Psychoda alternata, Velarifictorus micado, Crocothemis servilia, Phoridae sp., Lampyridae sp., Aphelinus sp., Hyalopterus pruni, Aulacorthum magnolia | China: Hubei | 6,882,491,800 |
| Ticks (general) | 16 | Dermacentor marginatus, Dermacentor sp., Hemaphysalis doenitzi, H. longicornis, Hemaphysalis sp., H. formosensis, Hyalomma asiaticum, Rhipicephalus microplus, Argas miniatus | China: Hubei, Zhejiang, Beijing, and Xinjiang | 24,708,479,580 |
| Ticks (Hyalomma asiaticum) | 1 | Hyalomma asiaticum | China: Xinjiang | 2,006,000,100 |
| Spiders | 32 | Neoscona nautica, Parasteatoda tepidariorum, Plexippus setipes, Pirata sp., Araneae sp. | China: Hubei | 11,361,912,300 |
| Arthropods, mix Hubei | 5 | Ctenocephalides felis, Psychodidae sp., Cicadellidae sp., Heteroptera sp., Scutigeridae sp., Tetragnatha maxillosa | China: Hubei | 7,768,505,600 |
| Barnacle mix Beihai | 12 | Amphibalanus rhizophorae | China: Beihai | 6,150,114,400 |
| Fish liver tissue mix Beihai | 8 | Proscyllium habereri, Urolophus aurantiacus, Rajidae sp., Eptatretus burgeri, Heterodontus zebra | China: Wenling | 4,406,962,000 |
| 48 | Dasyatis bennetti, Acanthopagrus latus, Epinephelus awoara, Conger japonicus, Siganus canaliculatus, Glossogobius circumspectus, Halichoeres nigrescens, Boleophthalmus pectinirostris | China: Beihai | 4,406,962,000 | |
| Mosquitoes, mix Panama | NAa | Unidentified mosquitoes | Republic of Panama: Gamboa | 370,413,306 |
| Myriapoda, mix Hubei | 24 | Diplopoda sp., Otostigmus scaber, Scolopocryptops sp., Otostigmus scaber, Myriapoda sp. | China: Hubei | 7,337,861,800 |
| Orthoptera, mix Hubei | 12 | Orthoptera sp., Conocephalus sp., Gryllidae sp. | China: Hubei | 7,042,417,800 |
NA, not applicable.
Arthropod samples in China were captured alive and stored at −80°C for RNA extraction. The fish samples were obtained on a fishing vessel where they were stored at −70°C before being transferred to our laboratory for dissection. The samples from China were initially identified by experienced field biologists and later confirmed by analyzing sequences of the mitochondrial cytochrome c oxidase subunit I gene. After being transferred to the laboratory, the arthropods (whole individuals) and fish liver tissue (200 mg) were washed with phosphate-buffered saline (PBS) and homogenized with the Mixer mill MM400 (Restsch). Subsequently, total RNA was extracted from homogenates using TRIzol LS reagent (Invitrogen), followed by purification by using an E.Z.N.A total RNA kit (Omega). RNA solutions for individual homogenates were then merged into pools as shown in Table 1. The transcriptome sequencing (RNA-seq) library preparation follows the standard protocol except for the rRNA removal step, for which we used a Ribo-Zero Magnetic Gold kit (Epidemiology) instead of the original mRNA purification procedures (16). Paired-end (100 bp) sequencing of the RNA library was performed on the HiSeq 2000 platform (Illumina). All library preparation and sequencing were performed by BGI Tech (Shenzhen, China).
In the case of the mosquito pool, samples were collected in 2012 by CO2-baited CDC light traps placed within a secondary dry tropical forest near Pipeline Road in the proximity of the town of Gamboa, Panama Province, Republic of Panama. An aliquot of the mosquito pool homogenate was passed twice in C6/36 cells (Aedes albopictus) until a cytopathic effect was observed. Virus harvest and isolation of vRNA for next-generation genome sequencing were undertaken as described previously (17).
Discovery and assembly of flavi-like genomes.
Sequencing reads were assembled de novo using the Trinity program (18). Assembled reads were compared using the BLASTX program against all Flaviviridae polyproteins downloaded from GenBank with a threshold E value of <1e–5, which provides high sensitivity while minimizing the false-positive rate. Putative flavi-like contigs were then blasted against the GenBank nonredundant (nr) database to filter those of non-Flaviviridae origin. The confirmed flavi-like contigs were merged by identifying unassembled overlaps between neighboring contigs using the SeqMan program implemented in the Lasergene software package v7.1 (DNAstar, Madison, WI). The remaining gaps were filled by aligning the reads to contigs with Bowtie2 (19). In cases where the assembly did not cover the complete open reading frame (ORF), we culled reads with a high percentage identity to both termini of the contigs and used them as seeds for extension until coverage at the termini was below 5-fold. Extensions and gaps were later reconfirmed with Sanger sequencing. To verify the assembly, reads were mapped back to the final full-length genome using Bowtie2, and the subsequent alignments were visualized with the Integrated Genomics Viewer (20). For Wuhan cricket virus, we further determined the 5′ and 3′ end of the genome as previously described (16). We also performed termini sequencing using RNA circularization (16) to verify that the virus has no poly(A) tail.
Identification of potential hosts.
For each verified flavi-like genome, we used nested reverse transcription-PCR and Sanger sequencing to examine the individual extractions from the unit sample before pooling, utilizing two sets of primers designed based on different regions of the deep-sequencing results. If a sample was found to contain viral RNA, the sample organism was regarded as putative virus host, whose information was also incorporated into the virus name. In a few cases where viruses were found in units with multiple host species, we used common names from a higher taxonomic grouping that includes all of the hosts in which that virus RNA was found.
Quantification of relative transcript abundances.
The relative abundance of each viral transcript was presented as the number of transcripts per million (TPM), a measure that corrects for the total number of reads as well as for transcript length (21). Estimates of TPM were performed as described previously (16). Briefly, we first removed rRNA-associated reads (22) from all data sets. The remaining reads from each library were then mapped to the assembled transcripts and analyzed with RSEM (21) as implemented in the Trinity program (18).
Prediction of protein domains and functions.
For each of the predicted polyprotein sequences, we used SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/sosui_submit.html) (23), Phobius (http://phobius.sbc.su.se/instructions.html) (24), and TMHMM v2.0c (http://www.cbs.dtu.dk/services/TMHMM/) (25) to predict the location of transmembrane domains. Similarly, we used SignalP v4.1 (http://www.cbs.dtu.dk/serv-ices/SignalP/) (26) to determine signal peptide or cellular cleavage sites, with a P value cutoff at 0.5. These predictions were first performed on reference sequences to examine their reliability. The test predictions suggested no false-positive estimations in any of the analyses. However, false-negative results are common in the case of transmembrane estimation, even if the results from all three programs are combined.
Phylogenetic analyses.
The predicted viral proteins discovered in the present study were aligned with representative reference proteins from the Flaviviridae using the E-INS-i algorithm implemented in MAFFT version 7 (27). Since the viruses were extremely divergent, only conserved domains within the NS3 (peptidase and helicase) and NS5 (RdRp) proteins produced alignments robust enough to be used in phylogenetic analysis. After removing all ambiguously aligned regions using TrimAl (28), the final lengths of the NS3 and NS5 alignments were 318 and 347 amino acids, respectively. The best-fit model of amino acid sequence evolution was identified using Prot-Test 3.4 (29) and found to be LG+Γ in both cases. Phylogenetic trees were then inferred using the maximum-likelihood method (ML) implemented in PhyML version 3.0 (30), employing a Subtree Pruning and Regrafting (SPR) topology searching algorithm. Statistical support for specific groupings in the tree was assessed using the approximate likelihood-ratio test (aLRT) with a Shimodaira-Hasegawa-like procedure. In addition, we inferred phylogenetic trees using MrBayes version 3.2.5 (31) and used the substitution model described above. In this case, we used two simultaneous runs of Markov chain Monte Carlo sampling, with the runs terminated when convergence was achieved (standard deviations of the split frequencies < 0.01). The phylogenies were then summarized from both runs with an initial burn-in of 10%.
Accession numbers.
All new sequence reads generated have been uploaded onto the NCBI Sequence Read Archive (SRA) database under the BioProject accession SRP058599. All virus genome sequences generated in this study have been deposited in GenBank under the accession numbers KR902709 to KR902741.
RESULTS
Virus discovery.
We performed a total 12 RNA-seq runs on pools of arthropod and vertebrate samples, generating 96 Gb of total sequence data. Through a BLASTX search with the assembled genome sequences we discovered one novel hepacivirus, 12 distant relatives of the Flaviviridae (flavi-like viruses), and 5 novel segmented jingmenviruses in our animal pools (Table 2). With the exception of Wenling shark virus (WLSV), whose polypeptide aligned well with viruses in the genus Hepacivirus (27.9 to 29.3% overall identity), all of the other viruses only matched the NS3 and NS5 genomic regions in the BLASTX analysis and with extremely low amino acid identity (21 to 31%) even in the most conserved domains (Table 3). The host distributions of these newly discovered viruses were diverse. Although the jingmenviruses and flavi-like viruses were identified in an extensive range of arthropods, the new hepacivirus was only found in vertebrate liver tissue, being present in 2 of the 56 samples, with both positives identified in the graceful catshark (Proscyllium habereri) (Table 2). For all of the viruses described here, we obtained the complete or near complete genome encompassing the entire coding region, and by mapping the original reads to these genomes we obtained good coverage across their full lengths (Table 2).
TABLE 2.
Summary of the viruses discovered in this study
| Classification and name (abbreviation) | Library/pool | Host | Genome length (bp) | Abundance (TPM) | Coverage (fold) |
|---|---|---|---|---|---|
| Jingmenvirus | |||||
| Shuangao insect virus 7 (SAIV7) | Insects, mix 1 | Chrysopidae sp., Psychoda alternata, Diptera sp. | 3,040 (seg1) | 243 | 2,438 |
| 1,940 (seg2) | 3,031 | ||||
| 2,756 (seg3) | 953 | ||||
| 2,670 (seg4) | 592 | ||||
| Wuhan flea virus (WHFV) | Arthropods, mix Hubei | Ctenocephalides felis | 3,170 (seg1) | 32 | 200 |
| 2,236 (seg2) | 224 | ||||
| 2,863 (seg3) | 65 | ||||
| 2,698 (seg4) | 58 | ||||
| Wuhan aphid virus 1 (WHAV1) | Insects, mix 4 | Hyalopterus prun | 3,156 (seg1) | 47 | 139 |
| 2,166 (seg2) | 250 | ||||
| 2,841 (seg3) | 52 | ||||
| 2,829 (seg4) | 64 | ||||
| Wuhan aphid virus 2 (WHAV2) | Insects, mix 4 | Hyalopterus pruni, Aulacorthum magnoliae | 3,053 (seg1) | 910 | 1,833 |
| 2,169 (seg2) | 2,911 | ||||
| 2,818 (seg3) | 878 | ||||
| 2,852 (seg4) | 1,050 | ||||
| Wuhan cricket virus (WHCV) | Orthoptera, mix Hubei | Conocephalus sp. | 3,135 (seg1) | 354 | 3,365 |
| 1,846 (seg2) | 6,892 | ||||
| 2,771 (seg3) | 1,533 | ||||
| 2,736 (seg4) | 3,459 | ||||
| Hepacivirus | |||||
| Wenling shark virus (WLSV) | Fish liver tissue mix Beihai | Proscyllium habereri | 9,653 | 8 | 34 |
| Distant members of the Flaviviridaea | |||||
| Shayang fly virus 4 (SYFV4) | True flies | Musca domestica, Sarcophaga sp. | 16,053 | 10 | 49 |
| Shuangao lacewing virus 2 (SALV2) | Insects, mix 1 | Chrysopidae sp. | 18,554 | 27 | 127 |
| Xingshan cricket virus (XSCV) | Insects, mix 4 | Velarifictorus micado | 21,779 | 16 | 40 |
| Gamboa mosquito virus (GMV) | Mosquitoes, mix South America | Culicidae sp. | 26,315 | NAb | NA |
| Sanxia water strider virus 6 (SXWSV6) | Water striders | Gerridae sp. | 22,879 | 291 | 105 |
| Wuhan centipede virus (WHCeV) | Myriapoda, mix Hubei | Otostigmus scaber, Scolopocryptops sp. | 23,677 | 102 | 352 |
| Tacheng tick virus 8 (TCTV8) | Ticks | Dermacentor marginatus | 19,537 | 54 | 164 |
| Bole tick virus 4 (BLTV4) | Ticks (Hyalomma asiaticum), ticks (general) | Hyalomma asiaticum | 16,249 | 125 | 166 |
| Xinzhou spider virus 2 (XZSV2) | Spiders | Unknown Araneae, Neoscona nautica | 24,521 | 10 | 55 |
| Shayang spider virus 4 (SYSV4) | Spiders | Neoscona nautica | 21,414 | 34 | 203 |
| Xinzhou spider virus 3 (XZSV3) | Spiders | Neoscona nautica, Parasteatoda tepidariorum | 20,433 | 24 | 141 |
| Beihai barnacle virus 1 (BHBV1) | Barnacle mix Beihai | Amphibalanus rhizophorae | 18,697 | 511 | 732 |
That is, flavi-like viruses.
NA, not applicable.
TABLE 3.
Pairwise amino acid identities of the NS3 (upper right) and NS5 (lower left) regions of all viruses related to the Flaviviridae studied herea
| Virus |
Flavivirus |
Jingmenvirus |
Flavi-like virus |
Pestivirus |
Hepacivirus |
Pegivirus |
|||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| General | Specific | WNV | CFAV | TABV | SAIV7 | WHFV | WHAV1 | WHAV2 | WHCV | TCLA | JMTV | SYFV4 | SALV2 | XSCV | GMV | SXWSV6 | WHCeV | TCTV8 | BLTV4 | XZSV2 | XZSV3 | SYSV4 | BHBV | SbCNV-5 | GKaV | BVDV1 | NrPV | WLSV | HCV | GBV-A | GBV-B |
| Flavivirus | WNV | 0.419 | 0.335 | 0.325 | 0.329 | 0.293 | 0.325 | 0.315 | 0.277 | 0.306 | 0.211 | 0.215 | 0.231 | 0.228 | 0.211 | 0.221 | 0.217 | 0.227 | 0.211 | 0.204 | 0.223 | 0.236 | 0.235 | 0.206 | 0.255 | 0.239 | 0.224 | 0.249 | 0.253 | 0.275 | |
| CFAV | 0.561 | 0.334 | 0.332 | 0.359 | 0.343 | 0.326 | 0.306 | 0.300 | 0.291 | 0.214 | 0.224 | 0.248 | 0.228 | 0.220 | 0.228 | 0.240 | 0.228 | 0.221 | 0.217 | 0.217 | 0.201 | 0.245 | 0.235 | 0.252 | 0.240 | 0.218 | 0.263 | 0.238 | 0.225 | ||
| TABV | 0.406 | 0.380 | 0.306 | 0.333 | 0.335 | 0.304 | 0.294 | 0.282 | 0.289 | 0.212 | 0.251 | 0.213 | 0.236 | 0.234 | 0.210 | 0.219 | 0.235 | 0.187 | 0.225 | 0.256 | 0.193 | 0.216 | 0.224 | 0.266 | 0.278 | 0.212 | 0.241 | 0.205 | 0.225 | ||
| Jingmenvirus | SAIV7 | 0.416 | 0.392 | 0.359 | 0.605 | 0.585 | 0.547 | 0.482 | 0.372 | 0.360 | 0.209 | 0.232 | 0.223 | 0.220 | 0.222 | 0.210 | 0.219 | 0.223 | 0.216 | 0.206 | 0.206 | 0.212 | 0.263 | 0.205 | 0.244 | 0.248 | 0.212 | 0.267 | 0.245 | 0.232 | |
| WHFV | 0.408 | 0.393 | 0.357 | 0.651 | 0.556 | 0.572 | 0.450 | 0.315 | 0.345 | 0.208 | 0.231 | 0.238 | 0.215 | 0.233 | 0.228 | 0.237 | 0.218 | 0.221 | 0.211 | 0.208 | 0.218 | 0.252 | 0.216 | 0.268 | 0.259 | 0.208 | 0.256 | 0.221 | 0.240 | ||
| WHAV1 | 0.381 | 0.355 | 0.371 | 0.616 | 0.674 | 0.607 | 0.438 | 0.339 | 0.335 | 0.191 | 0.198 | 0.196 | 0.196 | 0.194 | 0.233 | 0.204 | 0.208 | 0.201 | 0.207 | 0.207 | 0.194 | 0.228 | 0.196 | 0.232 | 0.229 | 0.217 | 0.210 | 0.204 | 0.214 | ||
| WHAV2 | 0.413 | 0.378 | 0.388 | 0.637 | 0.689 | 0.738 | 0.449 | 0.339 | 0.366 | 0.194 | 0.217 | 0.205 | 0.189 | 0.204 | 0.214 | 0.194 | 0.208 | 0.188 | 0.207 | 0.201 | 0.188 | 0.257 | 0.190 | 0.236 | 0.207 | 0.191 | 0.223 | 0.204 | 0.224 | ||
| WHCV | 0.367 | 0.329 | 0.319 | 0.567 | 0.564 | 0.564 | 0.599 | 0.318 | 0.325 | 0.191 | 0.208 | 0.224 | 0.208 | 0.213 | 0.188 | 0.194 | 0.208 | 0.211 | 0.213 | 0.213 | 0.204 | 0.231 | 0.215 | 0.252 | 0.242 | 0.207 | 0.236 | 0.217 | 0.220 | ||
| TCLA | 0.340 | 0.382 | 0.361 | 0.381 | 0.366 | 0.352 | 0.360 | 0.332 | 0.349 | 0.184 | 0.185 | 0.186 | 0.168 | 0.188 | 0.203 | 0.191 | 0.224 | 0.206 | 0.223 | 0.195 | 0.199 | 0.196 | 0.186 | 0.238 | 0.206 | 0.185 | 0.224 | 0.192 | 0.179 | ||
| JMTV | 0.349 | 0.346 | 0.376 | 0.458 | 0.399 | 0.417 | 0.426 | 0.414 | 0.436 | 0.205 | 0.209 | 0.232 | 0.210 | 0.199 | 0.222 | 0.228 | 0.203 | 0.193 | 0.218 | 0.180 | 0.202 | 0.248 | 0.207 | 0.252 | 0.252 | 0.199 | 0.222 | 0.206 | 0.206 | ||
| Flavi-like virus | SYFV4 | 0.299 | 0.279 | 0.269 | 0.298 | 0.293 | 0.292 | 0.301 | 0.270 | 0.321 | 0.297 | 0.353 | 0.329 | 0.335 | 0.270 | 0.262 | 0.265 | 0.274 | 0.274 | 0.226 | 0.280 | 0.275 | 0.235 | 0.311 | 0.255 | 0.277 | 0.259 | 0.252 | 0.253 | 0.250 | |
| SALV2 | 0.283 | 0.304 | 0.254 | 0.294 | 0.283 | 0.267 | 0.282 | 0.286 | 0.321 | 0.293 | 0.478 | 0.351 | 0.343 | 0.278 | 0.256 | 0.252 | 0.237 | 0.282 | 0.237 | 0.233 | 0.247 | 0.245 | 0.308 | 0.249 | 0.259 | 0.234 | 0.231 | 0.254 | 0.234 | ||
| XSCV | 0.287 | 0.276 | 0.251 | 0.280 | 0.282 | 0.274 | 0.298 | 0.255 | 0.291 | 0.273 | 0.459 | 0.451 | 0.357 | 0.278 | 0.276 | 0.253 | 0.267 | 0.289 | 0.237 | 0.218 | 0.232 | 0.252 | 0.283 | 0.256 | 0.253 | 0.244 | 0.244 | 0.251 | 0.251 | ||
| GMV | 0.296 | 0.284 | 0.267 | 0.298 | 0.293 | 0.280 | 0.310 | 0.314 | 0.288 | 0.261 | 0.478 | 0.475 | 0.472 | 0.320 | 0.273 | 0.277 | 0.283 | 0.295 | 0.244 | 0.256 | 0.273 | 0.289 | 0.316 | 0.266 | 0.259 | 0.254 | 0.257 | 0.248 | 0.299 | ||
| SXWSV6 | 0.234 | 0.222 | 0.198 | 0.247 | 0.246 | 0.221 | 0.226 | 0.234 | 0.248 | 0.237 | 0.363 | 0.350 | 0.351 | 0.342 | 0.237 | 0.227 | 0.284 | 0.300 | 0.248 | 0.255 | 0.249 | 0.267 | 0.317 | 0.264 | 0.280 | 0.256 | 0.274 | 0.288 | 0.263 | ||
| WHCeV | 0.302 | 0.290 | 0.267 | 0.298 | 0.311 | 0.301 | 0.310 | 0.290 | 0.292 | 0.264 | 0.399 | 0.369 | 0.393 | 0.378 | 0.319 | 0.253 | 0.247 | 0.316 | 0.221 | 0.237 | 0.272 | 0.251 | 0.287 | 0.278 | 0.297 | 0.228 | 0.237 | 0.209 | 0.244 | ||
| TCTV8 | 0.260 | 0.248 | 0.278 | 0.294 | 0.274 | 0.270 | 0.261 | 0.271 | 0.286 | 0.289 | 0.357 | 0.351 | 0.339 | 0.372 | 0.276 | 0.419 | 0.247 | 0.272 | 0.220 | 0.259 | 0.266 | 0.292 | 0.264 | 0.275 | 0.249 | 0.231 | 0.224 | 0.225 | 0.234 | ||
| BLTV4 | 0.262 | 0.244 | 0.236 | 0.248 | 0.241 | 0.258 | 0.264 | 0.271 | 0.267 | 0.258 | 0.313 | 0.298 | 0.295 | 0.308 | 0.238 | 0.337 | 0.347 | 0.269 | 0.262 | 0.287 | 0.275 | 0.275 | 0.290 | 0.309 | 0.322 | 0.272 | 0.266 | 0.273 | 0.283 | ||
| XZSV2 | 0.298 | 0.260 | 0.287 | 0.279 | 0.275 | 0.279 | 0.300 | 0.237 | 0.299 | 0.266 | 0.371 | 0.368 | 0.374 | 0.333 | 0.297 | 0.462 | 0.400 | 0.349 | 0.237 | 0.278 | 0.272 | 0.271 | 0.325 | 0.271 | 0.293 | 0.250 | 0.244 | 0.235 | 0.244 | ||
| XZSV3 | 0.311 | 0.276 | 0.217 | 0.319 | 0.287 | 0.260 | 0.268 | 0.267 | 0.284 | 0.275 | 0.340 | 0.355 | 0.308 | 0.311 | 0.271 | 0.317 | 0.330 | 0.301 | 0.283 | 0.434 | 0.227 | 0.251 | 0.276 | 0.283 | 0.305 | 0.240 | 0.256 | 0.256 | 0.294 | ||
| SYSV4 | 0.292 | 0.268 | 0.268 | 0.264 | 0.280 | 0.252 | 0.279 | 0.268 | 0.314 | 0.280 | 0.326 | 0.322 | 0.347 | 0.326 | 0.284 | 0.335 | 0.313 | 0.317 | 0.308 | 0.575 | 0.272 | 0.235 | 0.292 | 0.296 | 0.321 | 0.262 | 0.268 | 0.256 | 0.304 | ||
| BHBV | 0.247 | 0.220 | 0.214 | 0.231 | 0.247 | 0.240 | 0.251 | 0.238 | 0.250 | 0.240 | 0.299 | 0.314 | 0.263 | 0.299 | 0.218 | 0.341 | 0.311 | 0.402 | 0.316 | 0.319 | 0.301 | 0.255 | 0.294 | 0.300 | 0.281 | 0.228 | 0.279 | 0.248 | 0.286 | ||
| SbCNV-5 | 0.284 | 0.287 | 0.269 | 0.277 | 0.296 | 0.274 | 0.268 | 0.278 | 0.308 | 0.294 | 0.373 | 0.373 | 0.373 | 0.393 | 0.280 | 0.405 | 0.393 | 0.314 | 0.431 | 0.320 | 0.329 | 0.288 | 0.319 | 0.290 | 0.300 | 0.294 | 0.245 | 0.256 | 0.275 | ||
| GKaV | 0.284 | 0.273 | 0.249 | 0.292 | 0.273 | 0.277 | 0.277 | 0.276 | 0.258 | 0.288 | 0.367 | 0.351 | 0.349 | 0.352 | 0.304 | 0.405 | 0.345 | 0.317 | 0.418 | 0.308 | 0.297 | 0.305 | 0.417 | 0.270 | 0.283 | 0.264 | 0.293 | 0.285 | 0.271 | ||
| Pestivirus | BVDV1 | 0.239 | 0.254 | 0.269 | 0.277 | 0.263 | 0.246 | 0.277 | 0.257 | 0.314 | 0.276 | 0.264 | 0.257 | 0.289 | 0.274 | 0.212 | 0.289 | 0.272 | 0.261 | 0.282 | 0.323 | 0.330 | 0.256 | 0.280 | 0.282 | 0.667 | 0.271 | 0.315 | 0.256 | 0.297 | |
| NrPV | 0.221 | 0.242 | 0.266 | 0.255 | 0.245 | 0.249 | 0.243 | 0.245 | 0.295 | 0.260 | 0.276 | 0.266 | 0.264 | 0.267 | 0.192 | 0.273 | 0.269 | 0.252 | 0.278 | 0.322 | 0.305 | 0.226 | 0.295 | 0.254 | 0.605 | 0.278 | 0.287 | 0.260 | 0.266 | ||
| Hepacivirus | WLSV | 0.234 | 0.225 | 0.185 | 0.220 | 0.213 | 0.187 | 0.199 | 0.188 | 0.238 | 0.237 | 0.199 | 0.188 | 0.175 | 0.209 | 0.165 | 0.196 | 0.210 | 0.182 | 0.196 | 0.202 | 0.183 | 0.181 | 0.195 | 0.184 | 0.196 | 0.187 | 0.511 | 0.465 | 0.487 | |
| HCV | 0.241 | 0.220 | 0.198 | 0.212 | 0.202 | 0.182 | 0.209 | 0.196 | 0.215 | 0.216 | 0.207 | 0.205 | 0.210 | 0.201 | 0.167 | 0.170 | 0.183 | 0.187 | 0.194 | 0.198 | 0.190 | 0.164 | 0.224 | 0.219 | 0.185 | 0.204 | 0.378 | 0.471 | 0.509 | ||
| Pegivirus | GBV-A | 0.225 | 0.247 | 0.206 | 0.192 | 0.213 | 0.189 | 0.208 | 0.203 | 0.223 | 0.230 | 0.245 | 0.218 | 0.210 | 0.207 | 0.200 | 0.207 | 0.211 | 0.194 | 0.219 | 0.230 | 0.219 | 0.176 | 0.204 | 0.219 | 0.206 | 0.197 | 0.367 | 0.351 | 0.453 | |
| GBV-B | 0.215 | 0.221 | 0.199 | 0.220 | 0.212 | 0.207 | 0.207 | 0.206 | 0.224 | 0.211 | 0.196 | 0.203 | 0.211 | 0.193 | 0.186 | 0.211 | 0.200 | 0.186 | 0.210 | 0.202 | 0.201 | 0.193 | 0.204 | 0.205 | 0.189 | 0.196 | 0.403 | 0.442 | 0.379 | ||
Phylogenetic history of the family Flaviviridae and jingmenviruses.
With the inclusion of the viruses newly discovered here, we were able to greatly expand the biodiversity of the Flaviviridae, with ML and Bayesian methods producing highly congruent topologies (Fig. 1). Importantly, rather than adding new members to existing genera, these viruses often filled in major phylogenetic “gaps” that exist between these genera. Interestingly, based on the NS3 and NS5 proteins, the segmented jingmenviruses formed a well-supported monophyletic group that are closely related to the “classic” flaviviruses, suggesting that they may share a single common ancestor in these genes. The remaining arthropod viruses (i.e., flavi-like viruses) fell between the flavivirus-jingmenvirus clade, pestivirus clade, and the hepacivirus-pegivirus clade (Fig. 1). Notably, these viruses are highly divergent to the existing clades as well as to each other. Although some viruses (e.g., XZSV3 andSYSV4) clustered with pestiviruses or hepaci-pegiviruses (Fig. 1), this inferred phylogenetic relationship may not be reliable because (i) the arthropod and vertebrate viruses were separated by very long branches, and (ii) phylogenetic relationships differed between the NS3 and NS5 phylogenies. Interestingly, among the flavi-like arthropod viruses was a potential plant virus, GKaV (14), and a nematode virus, SbCNV-5 (15). This indicates that the host range of the Flaviviridae is likely to be far wider than previously anticipated. In addition, the single vertebrate virus discovered in the present study—WLSV—was found in the hepacivirus-pegivirus clade (Fig. 1). However, this virus represents a distinct lineage—as revealed in the hepacivirus-pegivirus-only phylogeny—that was more closely related to the hepaciviruses than the pegiviruses (Fig. 2).
FIG 1.
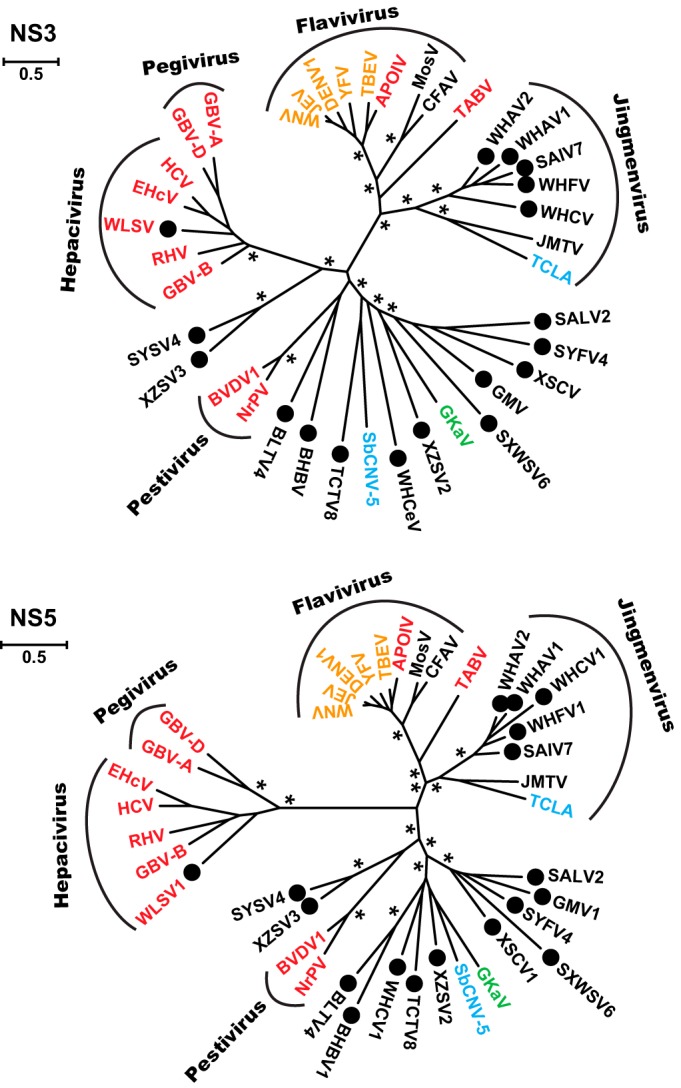
Phylogenetic tree (unrooted) of the family Flaviviridae and jingmenviruses based on conserved domains in the NS3 (upper panel) and NS5 (lower panel). Viruses discovered in the present study are marked with solid black circles. Based on their host types, the virus names are shaded red (vertebrate only), yellow (vertebrate and arthropod), green (plant), blue (nonarthropod invertebrates), or black (arthropod only). The names of previously or newly defined genera/phylogenetic groups are labeled. The trees shown here were inferred using an ML method, although highly congruent topologies were obtained using a Bayesian approach. Asterisks indicate well-supported nodes by both the approximate likelihood-ratio test (aLRT) in the ML method (>0.9) and by posterior probabilities in the Bayesian approach.
FIG 2.

Phylogenetic tree of the hepacivirus and pegivirus group based on the NS3 (left panel) and NS5B (right panel) alignments and midpoint rooted for clarity only. Viruses discovered in the present study are marked with solid black circles. The names of previously defined genera are labeled to the right of the phylogenies. The trees shown here were inferred using an ML method, although highly congruent topologies were obtained using a Bayesian approach. Asterisks indicate well supported nodes by both the approximate likelihood-ratio test (aLRT) in the ML method (>0.9) and by posterior probabilities in the Bayesian approach.
Genomes and proteomes of the newly discovered viruses.
One of the most striking features of these newly discovered viruses is the extent of variation in genome length within the Flaviviridae. The genera Hepacivirus and Pegivirus had the shortest genomes (9 kb), followed by viruses of the genus Flavivirus and the jingmenviruses (10 kb) and then those of the genus Pestivirus (12 to 13 kb). Remarkably, the remaining flavi-like viruses all exhibited far longer genomes than “classic” members of the Flaviviridae. Indeed, their genome lengths varied from ca. 15 to 26 kb (Table 2) and, accordingly, their polyprotein lengths ranged from 5,175 to 8,572 amino acids (Fig. 3), making them some of the longest of all RNA viruses (32). Furthermore, given their position in the phylogeny, it seems that long genomes might have evolved early in the history of the family Flaviviridae.
FIG 3.

Comparison of polyprotein structures among the family Flaviviridae and jingmenviruses, which includes 14 newly discovered viruses and 5 prototypical members of the Flaviviridae described previously. A unified length scale is used for all of the polyproteins. Within each protein, regions containing the key domains for serine protease, RNA helicase, and RdRp are colored blue, orange, and green, respectively. Predicted or known transmembrane domains (gray vertical bars) and cleavage sites for the host signalase (red arrow), viral protease (brown arrow), and other proteases (black arrow) are indicated.
Despite this substantial variation in length, the genomic and proteomic structures of the flavi-like viruses resemble the prototypical genome of the family Flaviviridae. Each of the flavi-like genomes contains a single long ORF that can be translated as a polyprotein, and the N-terminal of the polyprotein contained multiple target sites for the host signalase (Fig. 3), which roughly defines the structural part of the polyprotein although no sequence homology can be detected. The remainder of the polyprotein contained a serine protease and a RNA helicase in the central part of the protein, as well as an RdRp toward the C-terminal end of the protein (Fig. 3) as seen in all previously described members of the family. The locations of predicted multiple transmembrane domains are also well conserved: one was located between the N-terminal structural proteins and the serine protease, while the other was located between the RNA helicase and RdRp. In the case of Bole tick virus 4 (BLTV4) and Beihai barnacle virus 1 (BHBV), multiple transmembrane domains also appeared at the very end (C terminal) of the polyprotein (Fig. 3).
The genome/proteome structure of WLSV is generally very similar to those of members of the genus Hepacivirus (Fig. 3). The polypeptide starts with a nucleocapsid core protein (C) that bears a resemblance to that in hepaciviruses. It contains an N-terminal hydrophilic domain and a C-terminal hydrophobic domain. The latter is well conserved at the sequence level compared to other hepaciviruses. The remaining structural proteins include a relatively conserved E1 and a highly variable E2, and the boundaries for these structural proteins and for p7 are predicted based on target cleavage sites for host signalase. The nonstructural part of the polyprotein is also well conserved except for the N-terminal of NS4 protein, and the majority of NS5A protein for which only a zinc finger domain (NS5A-1a domain) can be identified. Finally, the boundaries for WLSV nonstructural proteins are not clear at this stage, largely due to little sequence similarity at the regions that define known viral cleavage sites.
In the phylogeny, the newly discovered segmented jingmenviruses, represented by Wuhan cricket virus (WHCV), formed a separate group that is distantly related to JMTV and TCLA. Notably, this new group lacked the poly(A) tail at the end of each segment. Despite these differences, the four segments of WHCV match those of JMTV and TCLA. The most obvious match, that of segments 1 and 3 which encode NS2b-NS3 and NS5, respectively, shared substantial structural and sequence similarity between WHCV and JMTV (Fig. 3). Conversely, segments 2 and 4 of WHCV have very limited sequence similarity to JMTV or TCLA, although there was a structural resemblance for segment 4 and its predicted proteins (Fig. 3). Interestingly, segment 4 of both WHCV and JMTV contained slippery heptanucleotide sequences that represent potential ribosomal frameshift signals at the end of the predicted first ORFs (Fig. 3), although this needs to be verified with future experiments. The most striking difference between WHCV and JMTV lies in segment 2. Compared to JMTV, WHCV had a shorter segment 2 that contained two overlapping ORFs. Both are predicted to have N-terminal signal peptides, and the second contained a transmembrane domain at the C-terminal (Fig. 3). No slippery heptanucleotide sequences were found in segment 2 for WHCV.
Differential expression of segment copy numbers.
For the segmented jingmenviruses we compared the abundance of four segments within each library after the removal of rRNA reads (Fig. 4). Abundance was presented as the number of transcripts per million (TPM). The exception was TCLA, for which we used the “frequency of transcripts” as in the original publication (33). Strikingly, abundance levels vary greatly for different segments in the newly discovered jingmenviruses, which include SAIV7, WHFV, WHAV1, WHAV2, and WHCV. Most notably, segment 2 is always significantly more abundant than the remaining segments (Fig. 4). No common patterns were observed among the other segments, except that segment 1 (encoding RdRp) tends to be the least abundant among the four. In addition, the contrast between segment 2 and segment 1 is highest in WHFV, with a ratio of 19.5. Such a dramatic contrast was not observed in JMTV and TCLA, whose four segments showed no consistent pattern of variation in abundance.
FIG 4.

Variation in levels of abundance for the four segments of the jingmenviruses. Abundance is measured as the frequency of transcripts for TCLA and as transcripts per million (TPM) for the remaining viruses. The abundance of JMTV segments are quantified within two libraries: ticks R. microplus (11) and ticks (16), respectively.
DISCUSSION
We describe here the discovery and characterization of 18 novel flavi-like and jingmenviruses, most of which were sampled from arthropods. The abundance levels for most of the viruses are relatively high (>10; Table 2), suggesting the presence of large quantities of viral genetic material within the host environment, although we were unable to demonstrate active replication with these data. It is also apparent that abundance levels vary considerably. One source of such variation is sample pooling. Specifically, higher abundance levels are usually associated with pools that contain fewer samples with relatively pure host population/species (e.g., pools Ticks Hyalomma asiaticum, Water striders, and Barnacle mix Beihai), whereas lower abundance occurs in pools that contain large number of samples from a complex host population/species background, such as the “fish liver tissue mix Beihai” pool that contains 56 RNA samples from 13 species (Table 2). Since WLSV is only detected in two samples, the true abundance of this virus should be at least 20 times higher. Viral load and/or replication strategy may also contribute to differences in abundance. Indeed, it is clear that the jingmenviruses tend to have higher abundance levels than the flavi-like viruses (Table 2), although this needs to be reexamined using purer host backgrounds.
The discovery of flavi-like viruses in invertebrates, including those described here, revises our understanding of the host range and genomic organization of the Flaviviridae. In particular, it is now clear that viral genetic diversity in invertebrates (largely arthropods) exceeds that of vertebrates, such that they are likely the major reservoir for genetic diversity (Fig. 1). Indeed, invertebrates are associated with the genus Flavivirus, as well as multiple divergent viral lineages, each of which may be distinct enough to be defined as a novel genus. It is striking that even our limited sampling in arthropods could yield such distinctive and phylogenetically diverse viruses. In contrast, lower levels of virus diversity are found in vertebrates, despite the previous studies of pathogen discovery in these species (34). In addition, since the vertebrate viruses tend to form paraphyletic groups in the phylogenetic trees (Fig. 1), it is likely that they represent independent transfers from invertebrate ancestors.
Before this study, all members of the genera Hepacivirus and Pegivirus were described in mammals, incorporating viruses from primates, pigs, ruminants, horses, dogs, rodents and bats, and many use liver as their common target tissue (5, 7, 9, 10, 12, 13). The discovery of WLSV therefore marks the expansion of the hepaciviris-pegivirus clade from warm-blooded mammalian species to cold-blooded cartilage fish in an aquatic environment. Although currently only represented by a single sequence, the presence of hepacivirus in such a basal vertebrate species suggests that this group may also be present in a far larger range of hosts such as reptiles, amphibians, and fish, to which relatively little attention has been paid to date.
The discovery of more jingmenviruses and their close relationship to the classic flaviviruses in the phylogeny indicates that segmentation has played an important role in the evolutionary history of these viruses. Furthermore, all segmented viruses formed a monophyletic group, indicating that, on the current sample at least, genome segmentation evolved only once. Interestingly, once formed, the original segmented genomic plan is relatively well conserved, with the exception of a few minor differences, such as the presence of poly(A) tail in each of the segments and the frame structures of segment 2. The segmented genome organization also appears to be well adapted to a wide variety of hosts, including various phytophagous insects, as well as external and internal parasites of vertebrates. This is surprising given that the only other known segmented positive-sense RNA viruses are found in plants (35) or generated under laboratory conditions (36, 37), although this likely reflects a lack of comprehensive sampling. Finally, it remains to be determined whether the remaining genes in the jingmenviruses have a separate evolutionary history or are simply are too divergent for sequence-based homology determination. More efforts are still required to study the origin and evolutionary history of flaviviruses and jingmenviruses.
A notable feature of the newly discovered jingmenviruses is that the abundance of genome segments is seemingly differentially regulated, such that a much higher copy number is always observed in the segment that encodes a predicted structural (envelope) protein. This reflects the common requirement of essentially all viruses to produce more structural proteins than nonstructural ones, which is achieved through a variety of ways, including subgenomic RNA, ribosomal frame shifting, and transcriptional gradients, as well as genome segmentation (38). However, for many segmented viruses of animals, gene regulation is often expected to be mediated through translation, while the copy number of each segment generally remains similar during the replication process. Although it is uncommon to see such dramatic variation in segment copy numbers as that described here, the regulation of segment copy numbers is reported in various multipartite plant viruses and is potentially a general feature for those viruses (39). Although the mechanisms for the control of segment numbers in the viruses described here remain unclear, it only seems to be associated with the cluster of viruses comprising SAIV7, WHFV, WHAV1, WHAV2, and WHCV and not the distantly related JMTV and TCLA, suggesting that these latter two viruses may use a different replication strategy. However, on the data provided here it is impossible exclude that a bias has been introduced during amplification or sequencing. Further confirmation requires qPCR quantification of viral segments in less complex host backgrounds, such as cell culture.
The genomes of the all flavi-like viruses identified here are exceptionally long and comparable to those in the order Nidovirales that possess the largest genomes among RNA viruses. A unique feature of viruses of the order Nidovirales is that the longer genomes (>20 kb) are always accompanied by an RNA 3′-5′ exoribonuclease (ExoN) (40), which has been proposed to offer a repair function that will reduce mutation rates and in turn allow these viruses to attain longer genomes (i.e., by reducing the genomic load of deleterious mutations) (32, 41). However, we did not identify any known error proof-reading domains in the flavi-like viruses, such that it is unclear whether they possess another type of error-proofing mechanism or that there is an additional reason why their genome sizes are so large. Moreover, although it seems that viruses of the order Nidovirales have expanded their genomes through evolutionary history (41), our results suggest this may not be the case in the Flaviviridae. In particular, the longer genomes are found in arthropod viruses that are the presumed ancestral hosts, while the shorter genomes are found in the “derived” vertebrate viruses. The causes for this change in genome size clearly merit additional investigation.
Finally, there were several differences between the topologies of NS3 and NS5 trees. For example, the branching order and branch lengths of the hepacivirus-pegivirus group differed between the phylogenies based on these two genes. It is currently unclear whether these differences are due to long branch attraction (42), rate variation reflecting very different selection pressures, or recombination. Unfortunately, the extensive sequence divergence among these viruses precludes additional analyses of the causes of these topological differences.
In summary, by describing novel arthropod and vertebrate members of the family Flaviviridae and the jingmenviruses we are able to provide a fuller depiction of the diversity and evolutionary history of this important group of viruses than that previously based on viruses sampled predominantly from mammals. In addition, we highlight the central role played by invertebrates in the evolution of the Flaviviridae and the jingmenviruses and describe a remarkable diversity of both genome structures and genome lengths. Evidently, such broad taxonomic sampling similarly has the potential to transform our understanding of the diversity and evolutionary history of additional viral groups.
REFERENCES
- 1.Lindenbach BD, Murray CL, Thiel H-J, Rice CM. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Wolters Kluwer, Philadelphia, PA. [Google Scholar]
- 2.Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. 2011. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol 92:233–246. doi: 10.1099/vir.0.027490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook S, Moureau G, Kitchen A, Gould EA, de Lamballerie X, Holmes EC, Harbach RE. 2012. Molecular evolution of the insect-specific flaviviruses. J Gen Virol 93:223–234. doi: 10.1099/vir.0.036525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grard G, Lemasson JJ, Sylla M, Dubot A, Cook S, Molez JF, Pourrut X, Charrel R, Gonzalez JP, Munderloh U, Holmes EC, de Lamballerie X. 2006. Ngoye virus: a novel evolutionary lineage within the genus Flavivirus. J Gen Virol 87:3273–3277. doi: 10.1099/vir.0.82071-0. [DOI] [PubMed] [Google Scholar]
- 5.Chandriani S, Skewes-Cox P, Zhong W, Ganem DE, Divers TJ, Van Blaricum AJ, Tennant BC, Kistler AL. 2013. Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc Natl Acad Sci U S A 110:E1407–E1415. doi: 10.1073/pnas.1219217110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corman VM, Grundhoff A, Baechlein C, Fischer N, Gmyl A, Wollny R, Dei D, Ritz D, Binger T, Adankwah E, Marfo KS, Annison L, Annan A, Adu-Sarkodie Y, Oppong S, Becher P, Drosten C, Drexler JF. 2015. Highly divergent hepaciviruses from African cattle. J Virol 89:5876–5882. doi: 10.1128/JVI.00393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, Coutard B, Adam A, Ritz D, Leijten LM, van Riel D, Kallies R, Klose SM, Gloza-Rausch F, Binger T, Annan A, Adu-Sarkodie Y, Oppong S, Bourgarel M, Rupp D, Hoffmann B, Schlegel M, Kummerer BM, Kruger DH, Schmidt-Chanasit J, Setien AA, Cottontail VM, Hemachudha T, Wacharapluesadee S, Osterrieder K, Bartenschlager R, Matthee S, Beer M, Kuiken T, Reusken C, Leroy EM, Ulrich RG, Drosten C. 2013. Evidence for novel hepaciviruses in rodents. PLoS Pathog 9:e1003438. doi: 10.1371/journal.ppat.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein JH, Quan PL, Briese T, Street C, Jabado O, Conlan S, Ali Khan S, Verdugo D, Hossain MJ, Hutchison SK, Egholm M, Luby SP, Daszak P, Lipkin WI. 2010. Identification of GBV-D, a novel GB-like flavivirus from Old World frugivorous bats (Pteropus giganteus) in Bangladesh. PLoS Pathog 6:e1000972. doi: 10.1371/journal.ppat.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, Conte JM, Ng J, Garcia J, Bhuva NP, Lee B, Che X, Quan PL, Lipkin WI. 2014. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 5:e01933-14. doi: 10.1128/mBio.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI. 2011. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 108:11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin XC, Shi M, Tian JH, Lin XD, Gao DY, He JR, Wang JB, Li CX, Kang YJ, Yu B, Zhou DJ, Xu J, Plyusnin A, Holmes EC, Zhang YZ. 2014. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc Natl Acad Sci U S A 111:6744–6749. doi: 10.1073/pnas.1324194111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, Osinubi MO, Recuenco S, Markotter W, Breiman RF, Kalemba L, Malekani J, Lindblade KA, Rostal MK, Ojeda-Flores R, Suzan G, Davis LB, Blau DM, Ogunkoya AB, Alvarez Castillo DA, Moran D, Ngam S, Akaibe D, Agwanda B, Briese T, Epstein JH, Daszak P, Rupprecht CE, Holmes EC, Lipkin WI. 2013. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A 110:8194–8199. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka T, Kasai H, Yamashita A, Okuyama-Dobashi K, Yasumoto J, Maekawa S, Enomoto N, Okamoto T, Matsuura Y, Morimatsu M, Manabe N, Ochiai K, Yamashita K, Moriishi K. 2014. Hallmarks of hepatitis C virus in equine hepacivirus. J Virol 88:13352–13366. doi: 10.1128/JVI.02280-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi K, Atsumi G, Iwadate Y, Tomita R, Chiba K, Akasaka S, Nishihara M, Takahashi H, Yamaoka N, Nishiguchi M, Sekine KT. 2013. Gentian Kobu-sho-associated virus: a tentative, novel double-stranded RNA virus that is relevant to gentian Kobu-sho syndrome. J Gen Plant Pathol 79:56–63. doi: 10.1007/s10327-012-0423-5. [DOI] [Google Scholar]
- 15.Bekal S, Domier LL, Gonfa B, McCoppin NK, Lambert KN, Bhalerao K. 2014. A novel flavivirus in the soybean cyst nematode. J Gen Virol 95:1272–1280. doi: 10.1099/vir.0.060889-0. [DOI] [PubMed] [Google Scholar]
- 16.Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. 2015. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife doi: 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasilakis N, Forrester NL, Palacios G, Nasar F, Savji N, Rossi SL, Guzman H, Wood TG, Popov V, Gorchakov R, Gonzalez AV, Haddow AD, Watts DM, da Rosa APAT, Weaver SC, Lipkin WI, Tesh RB. 2013. Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol 87:2475–2488. doi: 10.1128/JVI.00776-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen ZH, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29:644-U130. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357-U354. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. 2010. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA rRNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 24.Kall L, Krogh A, Sonnhammer ELL. 2007. Advantages of combined transmembrane topology and signal peptide prediction - the Phobius web server. Nucleic Acids Res 35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 26.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 27.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 31.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 32.Holmes EC. 2009. The evolution and emergence of RNA viruses. Oxford University Press, New York, NY. [Google Scholar]
- 33.Tetteh KKA, Loukas A, Tripp C, Maizels RM. 1999. Identification of abundantly expressed novel and conserved genes from the infective larval stage of Toxocara canis by an expressed sequence tag strategy. Infect Immun 67:4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Junglen S, Drosten C. 2013. Virus discovery and recent insights into virus diversity in arthropods. Curr Opin Microbiol 16:507–513. doi: 10.1016/j.mib.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. 2012. Virus taxonomy: 9th Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, Inc, San Diego, CA. [Google Scholar]
- 36.Garcia-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmis C. 2004. Evolutionary transition toward defective RNAs that are infectious by complementation. J Virol 78:11678–11685. doi: 10.1128/JVI.78.21.11678-11685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ojosnegros S, Garcia-Arriaza J, Escarmis C, Manrubia SC, Perales C, Arias A, Mateu MG, Domingo E. 2011. Viral genome segmentation can result from a trade-off between genetic content and particle stability. PLoS Genet 7:e1001344. doi: 10.1371/journal.pgen.1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes EC. 2013. Virus evolution, p 286–313. In Knipe DM, H PM (ed), Fields virology, 6th ed Wolters Kluwer, Philadelphia, PA. [Google Scholar]
- 39.Sicard A, Yvon M, Timchenko T, Gronenborn B, Michalakis Y, Gutierrez S, Blanc S. 2013. Gene copy number is differentially regulated in a multipartite virus. Nat Commun 4:2248. doi: 10.1038/ncomms3248. [DOI] [PubMed] [Google Scholar]
- 40.Nga PT, Parquet Mdel C, Lauber C, Parida M, Nabeshima T, Yu F, Thuy NT, Inoue S, Ito T, Okamoto K, Ichinose A, Snijder EJ, Morita K, Gorbalenya AE. 2011. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog 7:e1002215. doi: 10.1371/journal.ppat.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauber C, Goeman JJ, Parquet Mdel C, Nga PT, Snijder EJ, Morita K, Gorbalenya AE. 2013. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog 9:e1003500. doi: 10.1371/journal.ppat.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felsenstein J. 1978. Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool 27:401–410. doi: 10.2307/2412923. [DOI] [Google Scholar]