


Abstract
Whole genome sequencing has become one of the routine methods in molecular epidemiological practice. In this study, we present BacWGSTdb (http://bacdb.org/BacWGSTdb), a bacterial whole genome sequence typing database which is designed for clinicians, clinical microbiologists and hospital epidemiologists. This database borrows the population structure from the current multi-locus sequence typing (MLST) scheme and adopts a hierarchical data structure: species, clonal complex and isolates. When users upload the pre-assembled genome sequences to BacWGSTdb, it offers the functionality of bacterial genotyping at both traditional MLST and whole-genome levels. More importantly, users are told which isolates in the public database are phylogenetically close to the query isolate, along with their clinical information such as host, isolation source, disease, collection time and geographical location. In this way, BacWGSTdb offers a rapid and convenient platform for worldwide users to address a variety of clinical microbiological issues such as source tracking bacterial pathogens.
INTRODUCTION
Based on the premise that similar isolates may share similar medical trait, a prima facie concern shared by the global medical community is: ‘Have we seen the particular pathogen before? Where, when and what kind of disease is it associated with?’ To address this concern, a variety of genotyping methods have been developed, of which multi-locus sequence typing (MLST) is considered the gold standard for many bacterial pathogens for over a decade (1,2). While the performance of MLST is good enough in inter-lineage genotyping, this technology lacks enough discriminatory capability to differentiate tightly linked bacterial isolates (3,4). The advent of next-generation sequencing technologies has made it possible to obtain the entire bacterial genome at relatively modest cost and effort. Because its extremely high resolution could afford source tracking of the same bacterial clone isolated from different patients, areas or periods, whole genome sequencing (WGS) has been increasingly used to solve a wide range of research problems concerning bacterial epidemiology, drug resistance, pathogenicity and evolution (5–9). To date, US Food and Drug Administration (FDA) has granted WGS the marketing authorization for investigating food-borne outbreaks in USA. It is therefore expected in the near future that WGS would become a routine tool not only for basic research but also for clinical diagnostics and surveillance.
Nevertheless, the development of sequencing technology itself is not sufficient to achieve this goal. An easy-to-use public database is also required in order for international exchange of whole genome sequence typing (WGST) information of bacteria. Although Sequence Read Archive (SRA) and European Nucleotide Archive (ENA) have already offered a platform for storing the raw reads of WGS (10,11), it is far from convenient for investigators to deploy the raw data, especially those clinicians with limited bioinformatics skills. More importantly, the provenance for many isolates, such as host, isolation source, disease, collection time and geographical location, has not always been submitted along with the genome sequences. Consequently, medical significance could not be predicted even if a highly similar genome can be found. From this perspective, a new tool that is designed for the clinicians, clinical microbiologists and hospital epidemiologists monitoring the emergence and outbreak of important bacterial pathogens is urgently needed.
Generally, two strategies scale well to handling the genomic comparison of thousands of bacterial isolates: gene-by-gene genomic analysis and a reference genome-based single nucleotide polymorphism (SNP) strategy (12). The former is actually a MLST-like approach called whole-genome MLST (wgMLST), which is based on indexing alleles for all coding sequences in the genome, and therefore providing a highly scalable means of studying the sequence variation encoded within it. In theory, the gene-by-gene strategy is suitable for typing bacteria at a wide range of resolutions and might present a much more accurate phylogenetic relationship than traditional MLST (13,14). Currently the Bacterial Isolate Genome Sequence Database (BIGSdb), which was developed following this strategy, has been integrated into the PubMLST database (http://pubmlst.org) for a few species. Users can choose either the traditional MLST scheme or the new whole genome-based one according to their specific typing purpose (14–16). However, the phylogenetic analysis performed by this strategy is quite time-consuming and usually demands considerable computational resources, especially when manipulating hundreds of genome sequences together.
In contrast, the reference genome-based SNP strategy borrows the population structure from the current MLST schemes and requires a reference genome for each of the clonal complexes. The genomes of bacterial isolates are compared against the reference genome and the derived SNP data are used for further phylogenetic analysis. This algorithm assigns a higher identity between sequences differing in only one single nucleotide and a lower identity between sequences with multiple differences. Although it is unsuited to some highly diverse bacterial species such as Pseudomonas aeruginosa, or for comparing relationship among remotely related lineages, the reference genome-based SNP strategy offers a satisfactory resolution to differentiate isolates belonging to the same clone and is therefore suitable for analyzing the clonal structure of isolates emerging in an outbreak.
In this study, we introduced a new database which offers the ability to extract MLST information from a bacterial genome sequence. More importantly, it would also help find isolates in the public database that are phylogenetically close to the query isolate, along with their clinical information. We believe this function is vital for source tracking bacterial pathogens during outbreak investigation in the era of genomic epidemiology.
DATABASE DESCRIPTION
Bacterial Whole Genome Sequence Typing Database (BacWGSTdb, http://bacdb.org/BacWGSTdb) aims to provide genotyping at both traditional MLST and WGST level. For this purpose, we borrowed the population structure from the current MLST schemes and specified a reference genome for each of the clonal complexes. The clonal complex is defined herein to be a set of Sequence Types (STs) that differ by one or two alleles. The isolates stored in our database are firstly genotyped according to the MLST scheme, and an appropriate reference genome is chosen by the typing result. The complete or draft genome of the isolates continues to be compared against the specified reference genome. The obtained SNP information is stored in the database as well as the clinical information of isolates, including host, isolation source, disease, collection time and geographical location. Figures 1 and 2 list the infrastructure and the general workflow of data processing of BacWGSTdb, respectively.
Figure 1.
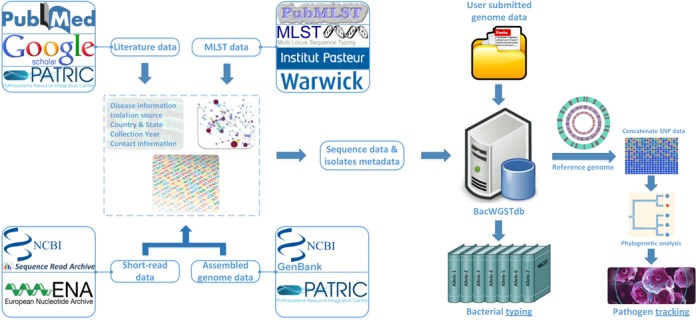
Database structure.
Figure 2.
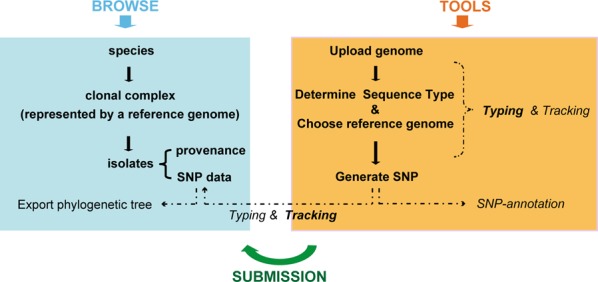
Workflow of data processing. BacWGSTdb contains three main sections: Browse, Tools and Submission. They all adopt a hierarchical infrastructure: species, clonal complex (represented by a reference genome) and isolates. SNP data is the key component of the database and connects the three sections together.
Construction of the phylogenetic tree in BacWGSTdb relies on Neighbor-Joining (NJ) algorithm. Although NJ is less accurate than other algorithms such as Maximum-Parsimony or Maximum-Likelihood, this disadvantage is minimized to a great extent when the compared strains are very close to each other. Meanwhile, the NJ algorithm runs significantly faster than the others, especially when manipulating the genome of hundreds of isolates together (17,18). Thus, the speed of retrieval in BacWGSTdb is fast, which fulfills the need of real-time monitoring and identification of bacterial outbreaks.
BacWGSTdb has been implemented using MySQL 5.6 (http://www.mysql.org), PHP 5.5 (http://www.php.net) and Apache 2.4 (http://www.apache.org) on a Red Hat Enterprise Linux Server 6.0. The interface component consists of webpages designed and implemented in HTML/CSS in a Linux environment. It has been tested in the Google Chrome, Mozilla Firefox, Apple Safari, Internet Explorer and Microsoft Edge web browsers.
At the background of BacWGSTdb, BLAST 2.2.26 is used for comparing the query genome with the MLST allele sequences (19). MUMmer 3.22 is used for alignment with the reference genome and the subsequent SNP identification (20). Indels and adjacent mismatches are not considered as true SNPs and are pruned by self-developed Perl scripts. The phylogenetic tree was generated by Clearcut 1.0, a fast and open source implementation for the relaxed NJ algorithm and displayed as Scalable Vector Graphics (SVG) in the web browser using TreeVector (21,22).
BacWGSTdb currently encompasses nine bacterial organisms of medical importance, i.e. Acinetobacter baumannii, Bacillus anthracis, Escherichia coli, Klebsiella pneumoniae, Mycobacterium tuberculosis, Salmonella enterica, Staphylococcus aureus, Streptococcus pneumoniae and Yersinia pestis, all of which can be described by a clonal population structure. At the present stage, genome sequences from GenBank and PATRIC databases have been deployed for preparing BacWGSTdb (23,24). The genome sequences from SRA and ENA databases with detailed strain information have also been incorporated (10,11). When the genome assembly is not available, the raw sequence reads were de novo assembled into the draft genome, which was further mapped to the reference genome. The obtained SNP data were stored in BacWGSTdb. The database will be updated periodically and new species can be easily added.
USAGE OF BacWGSTdb
Use of BacWGSTdb includes two major parts: TOOLS and BROWSE. One of the most important tools, Typing & Tracking, is designed for users who have sequenced the genome of their query isolates. After uploading a pre-assembled complete or draft genome sequence, users are told the MLST information of the isolate as well as the recommended reference genome. The query genome is then aligned against the reference genome and the SNP data are provided for download, which is in standard Variant Call Format (VCF). Next, the SNP data would be automatically compared with those deposited in the database, and the most similar isolates to the query one will be displayed (Figure 3). At the bottom of the resulting page, a phylogenetic tree is displayed in order to better reveal the phylogenetic relationship between the query isolate and the listed close isolates.
Figure 3.
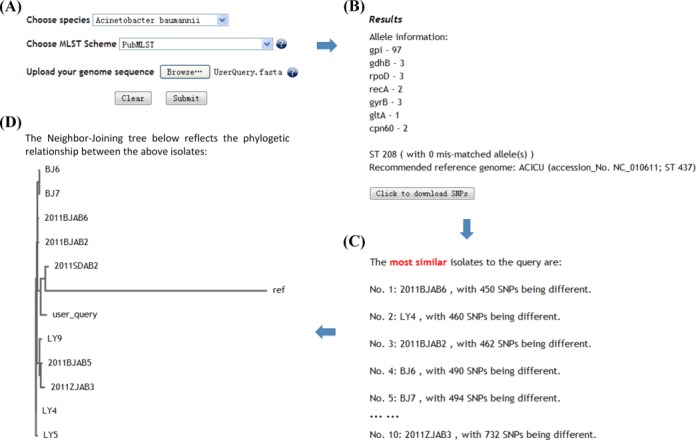
Usage example of the tool Typing & Tracking. Panel (A) shows the entry page of Typing & Tracking, in which users choose species, MLST scheme and upload a query genome sequence. Panel (B–D) are the results of Typing & Tracking: Panel (B) lists the MLST information, the suggested reference genome and the SNP file for download; Panel (C) lists the ten most similar isolates to the query one based on the number of different SNPs; Panel (D) shows the phylogenetic relationship between the query and the ten most similar isolates.
We have also developed a series of tools to serve as effective supplementations to the key tool Typing & Tracking, including:
SNP-annotation: this tool facilitates users to predict the outcome of SNPs. Based on the genomic annotation stored within the database, each of the SNPs uploaded by users will be judged whether it is synonymous, non-synonymous or intergenic.
Choose-refgenome-by-ST: by using this tool, users input the ST number and are told which reference genome they should use.
Generate-SNP-by-genome: when users are willing to choose other reference genomes instead of the recommended one, they can upload both the reference and the query genome and download the resulting SNP data.
Coordinate-conversion: if users don't generate the SNP data by the recommended reference genome at the very beginning, but they are willing to use or submit the SNP data into our database, they can make the coordinate conversion with this tool. Users are required to upload their SNP data and specify the two reference genomes. Pairwise alignment between the two genomes runs at the backend server and then the converted coordinates will be provided.
The BROWSE function is designed for visualizing and comparing isolates deposited in the database. When users browse BacWGSTdb, they need to choose a reference genome first (each reference genome represents a clonal complex) and further choose isolates of interest based on ST, host, clinical outcome, geographical location or any other attributes (Figure 4). According to the SNP data against the same reference genome, a NJ unrooted tree is provided for guidance, which reflects the phylogenetic relationship between the selected isolates. Users can also upload their own SNP data (e.g., produced by Typing & Tracking) and compare it with those in the database to figure out the phylogenetic position of their query isolate among the selected isolates. In addition to view the SVG formatted phylogenetic tree in the browser directly, users can also choose to download the Newick-formatted tree files for examination and/or annotation in external tree-drawing applications.
Figure 4.
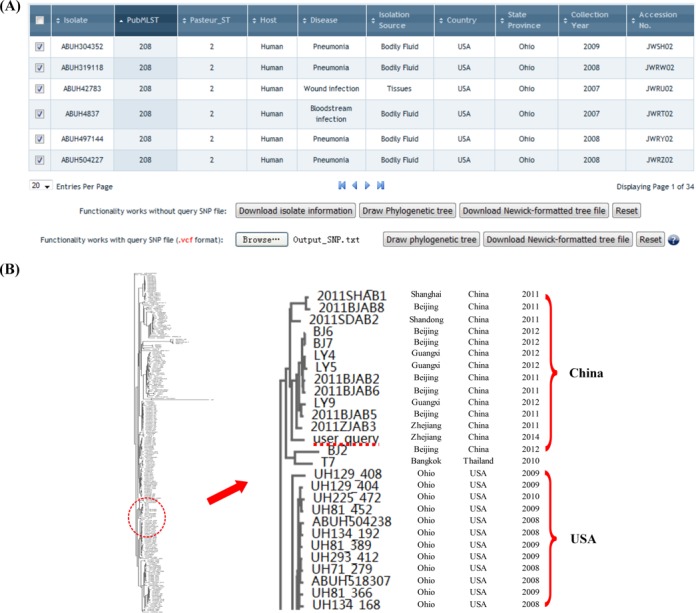
Usage example of Browse. Panel (A), a snapshot of isolate information in Browse Page. When users want to incorporate their query SNP data into the phylogenetic analysis, the uploaded SNP file should follow the same reference genome to the selected isolates. Panel (B), a phylogenetic tree based on the SNP data, which contains all ST208OD isolates and user query in this case.
In the SUBMISSION page, users are encouraged to submit their own data to BacWGSTdb. They need to fill in some basic information of their isolate and upload the SNP data (e.g. produced by Typing & Tracking). The uploaded SNP data will appear in the BROWSE page 24 h after submission. Users can also contact the administrator to make the curation. The SNP data could be prepared in two ways: users can directly map the raw WGS reads to a reference genome, or align the de novo-assembled contigs to the reference genome. We recommend the latter way because the SNP data stored in BacWGSTdb are prepared in this way.
EXAMPLE
The following is an example of how to use BacWGSTdb. A.baumannii has emerged worldwide as an important nosocomial pathogen due to its global occurrence and the ability to develop antimicrobial resistance. Clonal dissemination is characteristic of this important bacterial pathogen as revealed by previous studies (25–30). Currently, there are two MLST schemes available for A. baumannii, namely MLST-OD (associated with Oxford Database, http://pubmlst.org) and MLST-IP scheme (developed by Institute Pasteur, http://bigsdb.web.pasteur.fr). The former has a higher resolution and the latter is relatively more conservative. In the year 2014, an outbreak of bacteremia caused by A. baumannii was detected in a tertiary hospital in Hangzhou, China. We therefore selected one isolate (ABMDR55) for WGS using Illumina Miseq sequencer and the raw reads were assembled into contigs by using CLC Genomics Workbench 8.0 software. Then we analyzed the draft genome by the tool Typing & Tracking in BacWGSTdb. The resulting page confirmed that ABMDR55 belonged to ST208OD/ST2IP according to the two MLST schemes and its appropriate reference genome was ACICU (ST437OD/ST2IP). The strains closely related to ABMDR55 in the database were all isolated from different Chinese cities and different time periods. The close relationship among these isolates indicated they probably belonged to the same clone which had been widely disseminated from a wide spatial and temporal range in China. The SNP file between ABMDR55 and ACICU was downloaded from the resulting page for further analysis. The entire analysis process took <30 s (Figure 3).
Then we went to the BROWSE page for obtaining the clinical information of isolates that were close to ABMDR55 (Figure 4). In this case, ST208OD is very likely to be a pandemic lineage since a total of 240 ST208OD A. baumannii genomic sequence data were compiled in BacWGSTdb. The strains belonging to ST208OD were isolated from different countries, such as Spain, Denmark, Czech, Iraq, Thailand, China, Japan and USA. The earliest strain was isolated in the year 2002, and the most recent one was in 2014. We selected all of the ST208OD isolates and meanwhile uploaded the SNP file of ABMDR55 to perform the phylogenetic analysis. Generating the phylogenetic tree took <15 s. According to the derived NJ tree, ABMDR55 and the Chinese ST208OD isolates were grouped into an independent branch (Figure 4), which was consistent with a wide clonal dissemination of A. baumannii in China (27).
CONCLUDING REMARKS AND PERSPECTIVES
In light of the rising threats of antimicrobial resistance and emerging virulence among bacterial pathogens, BacWGSTdb represents a rapid and convenient tool for monitoring the emergence or dissemination of new clones and also for global collaboration on the molecular epidemiological investigation of medically important bacterial pathogens. BacWGSTdb will continue to improve, and additional features for analyzing WGS data are also under development.
Acknowledgments
We thank Dr. Cheng-Hsun Chiu and Dr. Huan Chen for their valuable suggestions on construction of BacWGSTdb.
FUNDING
National Natural Science Foundation of China [81201248, 81401698]. Funding for open access charge: National Natural Science Foundation of China [81201248, 81401698].
Conflict of interest statement. None declared.
REFERENCES
- 1.Maiden M.C., Bygraves J.A., Feil E., Morelli G., Russell J.E., Urwin R., Zhang Q., Zhou J., Zurth K., Caugant D.A., et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Losada M., Cabezas P., Castro-Nallar E., Crandall K.A. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect. Genet. Evol. 2013;16:38–53. doi: 10.1016/j.meegid.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Aanensen D.M., Spratt B.G. The multilocus sequence typing network: mlst.net. Nucleic Acids Res. 2005;33:W728–W733. doi: 10.1093/nar/gki415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maiden M.C. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 5.Forde B.M., O'Toole P.W. Next-generation sequencing technologies and their impact on microbial genomics. Brief. Funct. Genomics. 2013;12:440–453. doi: 10.1093/bfgp/els062. [DOI] [PubMed] [Google Scholar]
- 6.Sabat A.J., Budimir A., Nashev D., Sa-Leao R., van Dijl J., Laurent F., Grundmann H., Friedrich A.W. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro. Surveill. 2013;18:20380. doi: 10.2807/ese.18.04.20380-en. [DOI] [PubMed] [Google Scholar]
- 7.Bertelli C., Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin. Microbiol. Infect. 2013;19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 8.Torok M.E., Peacock S.J. Rapid whole-genome sequencing of bacterial pathogens in the clinical microbiology laboratory–pipe dream or reality. J. Antimicrob. Chemother. 2012;67:2307–2308. doi: 10.1093/jac/dks247. [DOI] [PubMed] [Google Scholar]
- 9.Didelot X., Bowden R., Wilson D.J., Peto T.E., Crook D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kodama Y., Shumway M., Leinonen R., International Nucleotide Sequence Database Collaboration The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40:D54–D56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silvester N., Alako B., Amid C., Cerdeno-Tarraga A., Cleland I., Gibson R., Goodgame N., Ten Hoopen P., Kay S., Leinonen R., et al. Content discovery and retrieval services at the European Nucleotide Archive. Nucleic Acids Res. 2015;43:D23–D29. doi: 10.1093/nar/gku1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiden M.C., Jansen van Rensburg M.J., Bray J.E., Earle S.G., Ford S.A., Jolley K.A., McCarthy N.D. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 2013;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jolley K.A., Maiden M.C. Using multilocus sequence typing to study bacterial variation: prospects in the genomic era. Future Microbiol. 2014;9:623–630. doi: 10.2217/fmb.14.24. [DOI] [PubMed] [Google Scholar]
- 14.Jolley K.A., Maiden M.C. Automated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplar. Euro. Surveill. 2013;18:20379. doi: 10.2807/ese.18.04.20379-en. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jolley K.A., Maiden M.C. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheppard S.K., Jolley K.A., Maiden M.C. A gene-by-gene approach to bacterial population genomics: whole genome MLST of campylobacter. Genes (Basel) 2012;3:261–277. doi: 10.3390/genes3020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamura K., Nei M., Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 19.Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheneman L., Evans J., Foster J.A. Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics. 2006;22:2823–2824. doi: 10.1093/bioinformatics/btl478. [DOI] [PubMed] [Google Scholar]
- 22.Pethica R., Barker G., Kovacs T., Gough J. TreeVector: scalable, interactive, phylogenetic trees for the web. PLoS One. 2010;5:e8934. doi: 10.1371/journal.pone.0008934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2015;43:D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wattam A.R., Abraham D., Dalay O., Disz T.L., Driscoll T., Gabbard J.L., Gillespie J.J., Gough R., Hix D., Kenyon R., et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams-Haduch J.M., Onuoha E.O., Bogdanovich T., Tian G.B., Marschall J., Urban C.M., Spellberg B.J., Rhee D., Halstead D.C., Pasculle A.W., et al. Molecular epidemiology of carbapenem-nonsusceptible Acinetobacter baumannii in the United States. J. Clin. Microbiol. 2011;49:3849–3854. doi: 10.1128/JCM.00619-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright M.S., Haft D.H., Harkins D.M., Perez F., Hujer K.M., Bajaksouzian S., Benard M.F., Jacobs M.R., Bonomo R.A., Adams M.D. New insights into dissemination and variation of the health care-associated pathogen Acinetobacter baumannii from genomic analysis. Mbio. 2014;5 doi: 10.1128/mBio.00963-13. doi:10.1128/mBio.00963-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruan Z., Chen Y., Jiang Y., Zhou H., Zhou Z., Fu Y., Wang H., Wang Y., Yu Y. Wide distribution of CC92 carbapenem-resistant and OXA-23-producing Acinetobacter baumannii in multiple provinces of China. Int. J. Antimicrob. Agents. 2013;42:322–328. doi: 10.1016/j.ijantimicag.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 28.Lee H.Y., Chen C.L., Wu S.R., Huang C.W., Chiu C.H. Risk factors and outcome analysis of acinetobacter baumannii complex bacteremia in critical patients. Crit. Care Med. 2014;42:1081–1088. doi: 10.1097/CCM.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 29.Wang N., Ozer E.A., Mandel M.J., Hauser A.R. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. Mbio. 2014;5 doi: 10.1128/mBio.01163-14. doi:10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen H., Wang K., Liu Y., Tay M., Lauro F.M., Huang H., Wu H., Liang H., Ding Y., Givskov M., et al. Population dynamics of an Acinetobacter baumannii clonal complex during colonization of patients. J. Clin. Microbiol. 2014;52:3200–3208. doi: 10.1128/JCM.00921-14. [DOI] [PMC free article] [PubMed] [Google Scholar]