


Abstract
PhytoPath (www.phytopathdb.org) is a resource for genomic and phenotypic data from plant pathogen species, that integrates phenotypic data for genes from PHI-base, an expertly curated catalog of genes with experimentally verified pathogenicity, with the Ensembl tools for data visualization and analysis. The resource is focused on fungi, protists (oomycetes) and bacterial plant pathogens that have genomes that have been sequenced and annotated. Genes with associated PHI-base data can be easily identified across all plant pathogen species using a BioMart-based query tool and visualized in their genomic context on the Ensembl genome browser. The PhytoPath resource contains data for 135 genomic sequences from 87 plant pathogen species, and 1364 genes curated for their role in pathogenicity and as targets for chemical intervention. Support for community annotation of gene models is provided using the WebApollo online gene editor, and we are working with interested communities to improve reference annotation for selected species.
INTRODUCTION
PhytoPath provides access to all sequenced and annotated plant pathogen genomes that have been submitted to the International Nucleotide Sequence Database Nucleotide Consortium (1) (GenBank (2), the European Nucleotide Archive (3) (ENA) and the DNA Databank of Japan (4)), and is updated approximately four times per year; see http://www.phytopathdb.org/pathogens_eg for a complete list. With each release, all newly submitted data are incorporated in the Ensembl Fungi, Protist and Bacterial portals (5); mapped to the annotations in the Pathogen-Host Interactions database (http://www.phi-base.org) (6); and incorporated into the PhytoPath search tool.
A COMPLETE RESOURCE FOR ALL SEQUENCED PLANT PATHOGENS
The number of plant pathogen genomes in PhytoPath has increased from 14, when the PhytoPath resource was launched in January 2012, to a current total of 135, reflecting the increasingly widespread use of genome sequencing; the list includes the 10 most important fungal plant pathogens according to global scientific importance (7). Other pathogens included in the resource include several major current causes of concern to global food and feed security, for example, the Ug99 lineage of Puccinia graminis f. sp. tritici (a virulent agent of wheat stem rust in sub-Saharan Africa), the oomycete Phytophthora infestans (the causative agent of potato late blight disease) and various species and strains of Fusarium. The breakdown by kingdom is shown in Table 1.
Table 1. Total number of phytopathogenic genomes by kingdom.
| Fungi | Protists | Bacteria | Total | |
|---|---|---|---|---|
| Species | 62 | 15 | 10 | 87 |
| Genome sequences | 88 | 23 | 24 | 135 |
Genomic data for each species are available through the Ensembl Genomes portals. Available data include: genome sequence, structural and functional annotation of protein coding and non-protein coding genes, variants and regulatory features and repeat features, DNA and peptide-based alignments and evolutionary histories for gene families. Access is provided via programmatic tools, including Perl and RESTful Application Program Interfaces, public MySQL database instances, FTP for bulk data downloads and an interactive genome browser. The browser gives a number of different views provided at various levels of resolution: the ‘Location’ view offers a bird-eye view of a specific region of the assembly; while gene, transcript and variant views focus on specific annotated features. Where the community has performed more than one gene build on the same reference genome, the alternative models can be visualized in a supplemental track. Currently we provide alternative gene sets for the fungi Botrytis cinerea (the causative agent of gray mold disease on numerous host species) and Zymoseptoria tritici (the causative agents of Septoria tritici blotch disease on wheat).
Genes are routinely annotated with InterPro (8) and the Gene Ontology (9), and the functional consequences of known sequence variants are predicted. Users can incorporate their own data for interpretation in the context of the reference data, either by uploading their data, or by accessing it remotely over HTTP or FTP, using common file formats such as BAM, GFF and VCF. Tools for data analysis include BLAST (10) and a ‘Variant Effect Predictor’ (11) that will interpret new sequence variations in terms of their predicted functional consequences for the existing gene models.
To support more complex queries the data are packaged into gene and variant-centric data warehouses, constructed using the BioMart (12) system which allow users to filter large data sets and to select attributes for display and download. Access to BioMarts is provided via a web-based query tool and through SOAP and RESTful web services.
CURRENT DATA IN PHYTOPATH
Comparative analysis
For each species, a representative genome is chosen, and all genes annotated on this reference are included in a protein-based comparative analysis, together with the genes of non-phytopathogenic relatives, which is especially useful when a related species has been studied in greater depth than the phytopathogen itself. Genes are first clustered according to sequence similarity, after which alignments are constructed and the ancestral history of the gene family (a ‘gene tree’) is deduced (13). Each tree is given a unique identifier and can be downloaded and published. These analyses are provided per kingdom (i.e. fungi, protists, bacteria); an additional analysis is available that shows the relationships between widely conserved genes from a more limited selection of species from across the taxonomic space. Six phytopathogenic species (Zymoseptoria tritici, Puccinia graminis f. sp. tritici, Ustilago maydis, Phytophthora infestans, Ralstonia solanacearum and Xanthomonas campestris p.v. campestris) have been included in this broad range analysis, alongside three other fungi, seven other protists and a total of 167 other genomes.
For closely related species, pairwise genomic alignments are computed using the tools lastZ (14) combined with the use of the UCSC chain/net algorithm (15). The available alignments are listed in Table 2. For pairs of species with assembled chromosomes, broader range data about large syntenic regions are also available.
Table 2. Whole genome alignments in PhytoPath.
| Clade | Aligned species (all against all) |
|---|---|
| Hypocreales | Fusarium graminearuma |
| Fusarium pseudograminearum | |
| Fusarium oxysporuma | |
| Fusarium fujikuroia | |
| Fusarium solania | |
| Fusarium verticillioidesa | |
| Trichoderma virens | |
| Trichoderma reeseib | |
| Pucciniales | Puccinia graminis f. sp. tritici CRL 75-36-700-3 |
| Puccinia graminis f.sp. tritici Ug99 | |
| Puccinia triticina | |
| Melampsora larici-populina | |
| Saccharomycetales | Ashbya gossipia |
| Saccharomyces cerevisiaea,b | |
| Glomerellales | Colletotrichum graminicola |
| Verticillium dahliae JR2a | |
| Protists | Albugo laibachi |
| Hyaloperonospora arabidopsidis | |
| Phytophthora infestans | |
| Phytophthora kernoviae | |
| Phytophthora lateralis | |
| Phytophthora parasitica | |
| Phytophthora ramorum | |
| Phytophthora sojae | |
| Pythium arrhenomanes | |
| Pythium aphanidermatum | |
| Pythium irregulare | |
| Pythium iwayamai | |
| Pythium ultimum | |
| Pythium vexans | |
| (Plus 19 non-phytopathogenic species) |
aSpecies with fully assembled chromosomes.
bNon-phytopathogenic species.
Alignment to other sequence data
The Ensembl framework can display alignments to various sequencing-based experiments as tracks. These data can be used to validate current gene models or identify alternative isoforms and splice sites and also to indicate gene expression under different experimental conditions. Whenever there are data in dbEST (16) for a PhytoPath species, alignments are performed against the reference assembly using Exonerate (17) and incorporated into the database. EST alignments are available for the following species: Fusarium graminearum (18) Fusarium verticillioides, Fusarium oxysporum, Fusarium solani, Phaeosphaeria nodorum, Puccinia triticina, Ustilago maydis, Magnaporthe oryzae, Leptosphaeria maculans, Melampsora larici-populina, Zymoseptoria tritici, Colletotrichum graminicola, Phytophthora infestans, Phytophthora sojae and Pythium ultimum. High throughput sequencing data are also available: with each release, the ENA is scanned for available resequencing data, which is mapped to the genome using the Burrows-Wheeler Alignment Tool (19). Supplemental Table S1 gives a brief description of the available data set samples (20–24).
Variation data sets
PhytoPath provides access to publicly available variant feature calls, including the location of variant loci, alleles and their distribution amongst individuals and populations, linkage disequilibrium, phenotypic associations resulting from whole genome analysis and their functional consequence on coding genes. For some data sets, however, sequence read data from resequencing experiments are present in the ENA, but the variant calls have not been published. Therefore, with each release of PhytoPath, such data are identified, mapped to the genome using the Burrows-Wheeler Alignment Tool (19), and variants are identified using SAMtools (25). In the future, we will additionally incorporate all variant calls submitted to the recently established European Variation Archive (26), which should provide a scalable model for accommodating an increased flow of new data. The complete set of variation data available is listed in Supplemental Table S2.
LINKING GENOMES TO PHENOTYPIC DATA
PHI-base is a database of manual curated phenotypic data for genes that have been functionally investigated in pathogen-host studies (6,27). Version 3.8 contains more than 5000 interactions with over 60% involving plant-infecting pathogens. The integration of these data in Ensembl provides a way to view these data in their genomic context and perform complex analysis for the study of phytopathogens and the pathogenic process.
Where annotated genes have been identified in the context of a reference genome, mapping involves first picking up the taxon identifier from the PHI-base database, then activating the mapping process at that taxon level and finally searching for the matching UniProt accession in all the genomes belonging to that clade. However, many of the older annotations in PHI-base were derived from studies performed before a wide genome level annotation was performed in that species and are identified using a different database and/or some sequence. Other studies use a gene set that comes from a different gene build version, which can completely rename all genes with no mapping between both versions available in the public domain. In other cases, annotations in PHI-base have been associated with a species level pathogen while in Ensembl Genomes sequences may exist for several strains. Ambiguous mappings are completed using taxonomic identifiers, transitive cross-references to the UniProt Knowledgebase and direct sequence alignment. A summary of mapped alignments is provided in Table 3.
Table 3. Number of annotated interactions and individually annotated genes per species on PHI-base and Ensembl.
| PHI-base genes | Interaction outcomes curated | Mapped genes on Ensembl | |
|---|---|---|---|
| Fungal species: | |||
| Botrytis cinerea | 86 | 210 | 0 |
| Fusarium graminearum | 966 | 1078 | 864 |
| Fusarium oxysporum f.sp. lycopersici | 58 | 85 | 35 |
| Fusarium solani | 9 | 9 | 4 |
| Leptosphaeria maculans | 17 | 21 | 4 |
| Magnaporthe oryzae | 423 | 662 | 237 |
| Phaeosphaeria nodoruma | 40 | 46 | 24 |
| Sclerotina sclerotiorum | 9 | 22 | 2 |
| Ustilago maydis | 197 | 252 | 141 |
| Zymoseptoria tritici | 41 | 42 | 12 |
| Protist species: | |||
| Hyaloperonospora arabidopsidis | 55 | 67 | 2 |
| Phytophthora infestans | 22 | 26 | 9 |
| Phytophthora sojae | 16 | 30 | 9 |
| Bacterial species: | |||
| Burkholderia glumae | 14 | 23 | 5 |
| Erwinia amylovora | 20 | 33 | 0 |
| Pantoea stewartii | 6 | 6 | 0 |
| Pseudomonas syringae pv tomato | 8 | 12 | 4 |
| Pseudomonas syringae pv tabaci | 4 | 6 | 0 |
| Xanthomonas campestris pv. campestris | 5 | 5 | 1 |
| Xanthomonas oryzae pv. oryzae | 12 | 132 | 3 |
aSynonym Parastagonospora nodorum.
DISPLAY OF AND ACCESS TO PHENOTYPIC DATA
PHI-base annotations contain information about the host, the experimental condition and the resulting phenotype. The link to the peer reviewed published article has also been imported. Phytopathogenic genes are highlighted in the browser, highlighting pathogenicity islands. If a gene has a single annotation or all the annotations confirm the same phenotype, it is highlighted with a color specific to the phenotype. A pop-up menu provides an immediate link to each of the PHI-base annotations associated with that gene, and a link to the gene page in PHI-base where additional information can be accessed through new design of the PHI-base website. These include details that may influence the observed interaction phenotype, such as the host strain, or chemistry that may act to induce or prevent a pathogenic outcome. Figure 1 shows a typical region view with one PHI-base annotated gene.
Figure 1.
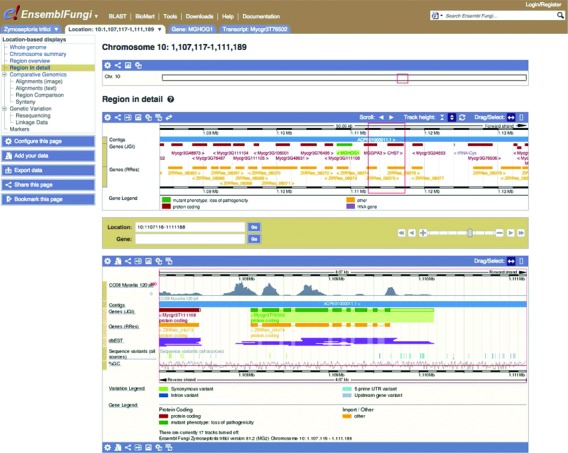
Zymoseptoria tritici region view in the Ensembl Genomes browser. Two gene build sets are represented in red and orange. The gene highlighted in green has a PHI-base annotation with a ‘loss of pathogenicity’ mutant phenotype. Expression data for mycelia are shown in the lower panel in gray alongside an EST track in purple and sequence variants represented by colored bands. The visibility of each track is chosen using the ‘Configure this page’ panel on the left.
All the PHI-base attributes stored in Ensembl are included in the BioMart (12) query tool. Ensembl BioMarts are typically constructed to provide access on a per-species basis, but through PhytoPath, a new interface is available supporting multi-species queries (http://www.phytopathdb.org/query/builder), and allowing users to navigate a taxonomic tree to select groups of related phytopathogens to query. Successive queries can be combined using set algebra and both gene annotation and sequences can be downloaded for further use. Figure 2 shows the results for a query consisting on the intersection of two filters, one regarding one PHI-base field and the other a GO annotation. There is the possibility to show GO data and protein domains.
Figure 2.
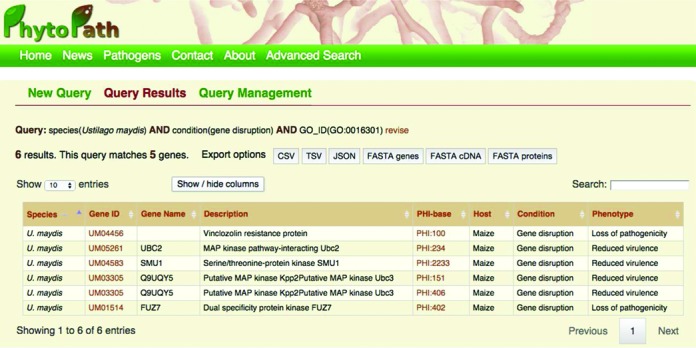
Typical results table displayed when using the PhytoPath advanced search.
FUTURE DIRECTIONS
Tools for community-led curation
We are working to support the community annotation of gene models. For interested communities, we can provide access to a private site, allowing groups to share data prior to publication and utilize the PhytoPath toolset while completing their annotation. In addition, we can provide access to the online editing tool WebApollo (28) for community members to directly submit new or modified gene models, which are immediately visible in public as an additional track and which are incorporated (following appropriate quality control) into the official gene set at periodic intervals. Groups seeking this support should contact us on http://www.phytopathdb.org/content/contact-us.
Linking pathogen and host genomes
Pathogens and their hosts are directly engaged in communication and molecular warfare using secreted effector molecules and proteinaceous toxins. Both animal and plant pathogens aim to suppress early host detection by camouflage and to weaken the host's immune system once detected (29). Many pathogen delivered molecules have been shown to interact directly with one to a few extracellular or intracellular host target molecules. A current priority is to associate the host target genes in genomes available at Ensembl with the pathogen genes in PHI-base. The 231 pathogenic organisms in PHI-base have a total of 84 associated host species, of which eight plants are already available in Ensembl. Table 4 lists the host organisms associated with bacterial, fungal and protist pathogens in PHI-base Version 3.8.
Table 4. Host plants in PHI-base infected by two or more phytopathogenic species also in PHI-base.
| Host species | Pathogen type | ||
|---|---|---|---|
| Bacteria | Fungi | Protists | |
| Dicotyledonous plants | |||
| Arabidopsis thaliana* | 2 | 8 | 2 |
| Brassica napus | 0 | 3 | 0 |
| Capsicum annuum | 4 | 1 | 1 |
| Cucumis sativus | 1 | 4 | 0 |
| Citrus sinensis | 1 | 2 | 0 |
| Glycine max | 2 | 2 | 1 |
| Lactuca sativa | 1 | 0 | 1 |
| Malus domestica | 1 | 2 | 0 |
| Nicotiana benthamiana | 2 | 2 | 3 |
| Nicotiana tabacum | 1 | 1 | 2 |
| Olea europaea | 1 | 0 | 0 |
| Phaseolus vulgaris | 2 | 4 | 0 |
| Pisum sativum | 1 | 2 | 0 |
| Pyrus communis | 0 | 3 | 0 |
| Solanum lycopersicum* | 5 | 11 | 3 |
| Solanum tuberosum* | 7 | 2 | 2 |
| Vitis vinifera* | 1 | 1 | 0 |
| Monocotyledonous plants | |||
| Hordeum vulgare* | 0 | 9 | 0 |
| Oryza sativa* | 2 | 3 | 0 |
| Secale cereale | 0 | 3 | 0 |
| Triticum aestivum* | 0 | 10 | 0 |
| Zea mays* | 1 | 10 | 0 |
*Species present on Ensembl Plants.
Acknowledgments
The authors would like to thank all the species experts who contributed additional database entries from their field of expertise into PHI-base; and the members of our Scientific Advisory Board (Michael Csukai, Jonathan Jones, Leighton Pritchard and Pietro Spanu). Drs Arathi Ragunath and Rashmi Pant from Molecular Connections Private Limited, Basavanagudi, Bangalore 560 004, India, are thanked for expert curation of PHI-base accessions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
United Kingdom Biotechnology and Biological Sciences Research Council (BBSRC) [BB/I000488/1, BB/I/001077/1, BB/K020056/1]. PHI-base receives additional support from the BBSRC as a National Capability [BB/J/004383/1 to K.H.K.]. Specific funding for the inclusion of Puccinia graminis f. sp. tritici within Ensembl Fungi has been approved by a grant from the Bill and Melinda Gates Foundation [OPPGD1491 to P.K.]. Funding for open access charge: Research Councils UK—Block Funding.
Conflict of interest statement. None declared.
REFERENCES
- 1.Nakamura Y., Cochrane G., Karsch-Mizrachi I., International Nucleotide Sequence Database Collaboration The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2015;43:D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silvester N., Alako B., Amid C., Cerdeño-Tárraga A., Cleland I., Gibson R., Goodgame N., Hoopen P., Ten Kay S., Leinonen R., et al. Content discovery and retrieval services at the European Nucleotide Archive. Nucleic Acids Res. 2015;43:D23–D29. doi: 10.1093/nar/gku1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tateno Y., Imanishi T., Miyazaki S., Fukami-Kobayashi K., Saitou N., Sugawara H., Gojobori T. DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002;30:27–30. doi: 10.1093/nar/30.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kersey P.J., Allen J.E., Christensen M., Davis P., Falin L.J., Grabmueller C., Hughes D.S.T., Humphrey J., Kerhornou A., Khobova J., et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urban M., Pant R., Raghunath A., Irvine A.G., Pedro H., Hammond-Kosack K.E. The Pathogen-Host Interactions database (PHI-base): additions and future developments. Nucleic Acids Res. 2015;43:D645–D655. doi: 10.1093/nar/gku1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean R., Van Kan J.A.L., Pretorius Z.A., Hammond-Kosack K.E., Di Pietro A., Spanu P.D., Rudd J.J., Dickman M., Kahmann R., Ellis J., et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012;13:414–430. doi: 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell A., Chang H.-Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–D221. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 11.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinforma. Oxf. Engl. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasprzyk A. BioMart: driving a paradigm change in biological data management. Database J. Biol. Databases Curation. 2011 doi: 10.1093/database/bar049. doi:10.1093/database/bar049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris R.S. The Pennsylvania State University; 2007. Improved Pairwise Alignment of Genomic DNA. PhD thesis. [Google Scholar]
- 15.Kent W.J., Baertsch R., Hinrichs A., Miller W., Haussler D. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boguski M.S., Lowe T.M., Tolstoshev C.M. dbEST–database for ‘expressed sequence tags’. Nat. Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 17.Slater G.S., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King R., Urban M., Hammond-Kosack M.C.U., Hassani-Pak K., Hammond-Kosack K.E. The completed genome sequence of the pathogenic ascomycete fungus Fusarium graminearum. BMC Genomics. 2015;16:544. doi: 10.1186/s12864-015-1756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Connell R.J., Thon M.R., Hacquard S., Amyotte S.G., Kleemann J., Torres M.F., Damm U., Buiate E.A., Epstein L., Alkan N., et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012;44:1060–1065. doi: 10.1038/ng.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faino L., de Jonge R., Thomma B.P.H.J. The transcriptome of Verticillium dahliae-infected Nicotiana benthamiana determined by deep RNA sequencing. Plant Signal. Behav. 2012;7:1065–1069. doi: 10.4161/psb.21014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye W., Wang X., Tao K., Lu Y., Dai T., Dong S., Dou D., Gijzen M., Wang Y. Digital gene expression profiling of the Phytophthora sojae transcriptome. Mol. Plant-Microbe Interact. 2011;24:1530–1539. doi: 10.1094/MPMI-05-11-0106. [DOI] [PubMed] [Google Scholar]
- 23.Kemen E., Gardiner A., Schultz-Larsen T., Kemen A.C., Balmuth A.L., Robert-Seilaniantz A., Bailey K., Holub E., Studholme D.J., Maclean D., et al. Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol. 2011;9:e1001094. doi: 10.1371/journal.pbio.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lévesque C.A., Brouwer H., Cano L., Hamilton J.P., Holt C., Huitema E., Raffaele S., Robideau G.P., Thines M., Win J., et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 2010;11:R73. doi: 10.1186/gb-2010-11-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lappalainen I., Lopez J., Skipper L., Hefferon T., Spalding J.D., Garner J., Chen C., Maguire M., Corbett M., Zhou G., et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41:D936–D941. doi: 10.1093/nar/gks1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urban M., Irvine A.G., Cuzick A., Hammond-Kosack K.E. Using the pathogen-host interactions database (PHI-base) to investigate plant pathogen genomes and genes implicated in virulence. Front. Plant Sci. 2015;6 doi: 10.3389/fpls.2015.00605. doi:10.3389/fpls.2015.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee E., Helt G.A., Reese J.T., Munoz-Torres M.C., Childers C.P., Buels R.M., Stein L., Holmes I.H., Elsik C.G., Lewis S.E. Web Apollo: a web-based genomic annotation editing platform. Genome Biol. 2013;14:R93. doi: 10.1186/gb-2013-14-8-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammond-Kosack, Jones J.D. Chapter 21 - Responses to Plant Pathogens. In: Buchanan BB, Gruissem W, Jones RL, Jones BG, editors. Biochemistry & Molecular Biology of plants. Rockville, MD: American Soc. of Plant Physiologists; 2015. pp. 984–1050. [Google Scholar]