


Abstract
KEGG (http://www.kegg.jp/ or http://www.genome.jp/kegg/) is an integrated database resource for biological interpretation of genome sequences and other high-throughput data. Molecular functions of genes and proteins are associated with ortholog groups and stored in the KEGG Orthology (KO) database. The KEGG pathway maps, BRITE hierarchies and KEGG modules are developed as networks of KO nodes, representing high-level functions of the cell and the organism. Currently, more than 4000 complete genomes are annotated with KOs in the KEGG GENES database, which can be used as a reference data set for KO assignment and subsequent reconstruction of KEGG pathways and other molecular networks. As an annotation resource, the following improvements have been made. First, each KO record is re-examined and associated with protein sequence data used in experiments of functional characterization. Second, the GENES database now includes viruses, plasmids, and the addendum category for functionally characterized proteins that are not represented in complete genomes. Third, new automatic annotation servers, BlastKOALA and GhostKOALA, are made available utilizing the non-redundant pangenome data set generated from the GENES database. As a resource for translational bioinformatics, various data sets are created for antimicrobial resistance and drug interaction networks.
INTRODUCTION
Thanks to the advancement of sequencing technologies, it is now a routine task to determine the genome sequence of an organism or an environmental sample that contains multiple organisms. However, it still remains a challenging task to fully understand biological meanings encoded in the genome. In 1995, we initiated the KEGG (Kyoto Encyclopedia of Genes and Genomes) database project as part of the Japanese Human Genome Program, in anticipation of the need for a reference knowledge base for biological interpretation of genome sequence data (1). The main objective of KEGG has been to establish links from collective sets of genes in the genome to high-level functions of the cell and the organism. We have developed, among others, the KEGG PATHWAY database as a representation of high-level functions, the KEGG GENES database as a collection of completely sequenced genomes, and the KO (KEGG Orthology) database for linking genes to high-level functions. With the arrival of high-throughput biology KEGG has become one of the most widely used biological databases in the world.
Genome annotation in KEGG is done differently from most other databases. First, molecular functions are stored in the KO database and associated with ortholog groups in order to enable extension of experimental evidence in a specific organism to other organisms. Annotation of individual genes in the GENES database is simply to create links to the KO database by assigning KO entry identifiers called K numbers. No updates are made to original data, such as gene names and descriptions in the GENES database, even if they are inconsistent with the KO assignment. Second, ortholog groups are defined in the context of KEGG pathway maps and other molecular networks, which are all created as networks of K number nodes. Thus, the genome annotation procedure to convert a gene set in the genome to a K number set leads to automatic reconstruction of KEGG pathways and other networks, enabling interpretation of high-level functions. Obviously, the quality of the KO database is critical in this procedure. Over the last two years, major efforts have been made to improve its quality.
In early 2015, we decided to remove the restriction of complete genomes in the KEGG GENES database. We first added the categories of viruses and plasmids, which are important in the analysis of metagenomes and antimicrobial resistance, respectively, as described below. We then introduced the addendum category where, for the first time, we started collecting protein sequence data from published literature rather than just importing complete genome sequences from RefSeq or GenBank. This is necessary because a pathway map created from literature information sometimes contains genes and proteins from organisms whose genome sequences are not known and would never be known. By expanding this addendum category it is now possible to capture all knowledge about gene/protein functions that can be associated with sequence data.
OVERVIEW AND NEW DEVELOPMENTS
Overview of KEGG
KEGG is an integrated database resource consisting of 16 main databases, which are categorized into systems, genomic, chemical and health information as shown in Table 1. The PATHWAY, BRITE and MODULE databases in the systems information category contain KEGG pathway maps, BRITE hierarchy and table files and KEGG modules, respectively, as representations of high-level functions. They are all manually created based on published literature. The BRITE table file is a newly introduced representation, which can be compared with the multi-column BRITE hierarchy file. When the data size is not large it is much easier to capture the overall relationship in a tabular form with a few columns optionally used for representation of hierarchy. BRITE table files are mainly used for drug classifications and for presenting various relationships involving diseases and drugs.
Table 1. The KEGG resource including drug labels.
| Category | Database name | Content |
|---|---|---|
| Systems Information | KEGG PATHWAY | KEGG pathway maps |
| KEGG BRITE | BRITE functional hierarchies and BRITE tables | |
| KEGG MODULE | KEGG modules | |
| Genomic Information | KEGG ORTHOLOGY | KEGG Orthology (KO) groups |
| KEGG GENOME | KEGG organisms (complete genomes) | |
| KEGG GENES | Gene catalogs of KEGG organisms, viruses, plasmids and addendum category | |
| Chemical Information (KEGG LIGAND) | KEGG COMPOUND | Metabolites and other small molecules |
| KEGG GLYCAN | Glycans | |
| KEGG REACTION | Biochemical reactions | |
| KEGG RPAIR | Reactant pairs | |
| KEGG RCLASS | Reaction class | |
| KEGG ENZYME | Enzyme nomenclature | |
| Health Information (KEGG MEDICUS) | KEGG DISEASE | Human diseases |
| KEGG DRUG | Drugs | |
| KEGG DGROUP | Drug groups | |
| KEGG ENVIRON | Crude drugs and health-related substances | |
| JAPICa | Drug labels in Japan | |
| DailyMedb | Drug labels in the USA (links only) |
The genomic information category contains the GENOME and GENES databases for collections of organisms with complete genomes and their gene catalogs, which are mostly taken from RefSeq (2) and GenBank (3) databases. As mentioned, the GENES database now contains additional gene sets not related to complete genomes. There are also other databases not listed in Table 1: computationally generated sequence similarity database SSDB and auxiliary gene catalog databases DGENES and MGENES for draft genomes and metagenomes, respectively. The KO database containing ortholog groups associated with molecular functions is a hub for linking genomic information to systems information through the KEGG mapping procedure and also to chemical information through the dual aspect of the metabolic network (4).
The COMPOUND, GLYCAN, REACTION, RPAIR, RCLASS and ENZYME databases in the chemical information category contain chemical substances and reactions and are collectively called KEGG LIGAND for historical reasons. The ENZYME database originates from the database of Enzyme Nomenclature (5). There is also a small data set of reaction modules (1,4), which can be used for annotation of enzyme genes.
The health information category consists of the DISEASE, DRUG, DGROUP and ENVIRON databases for disease and drug information. DGROUP is a newly added database, which is being developed for grouping functionally identical or similar drugs in the drug interaction networks. KEGG MEDICUS is an interface for the general public integrating these internally developed databases with drug labels (package inserts) of all marketed drugs in Japan and the USA. The Japanese version of KEGG MEDICUS is especially advanced in this integration, and heavily accessed mostly through web search engines.
Experimental evidence for each KO
The development of the KO database is tightly coupled with the development of KEGG molecular networks including KEGG pathway maps, BRITE functional hierarchies and KEGG modules. Ideally a KO represents a single sequence similarity group with an appropriate level of similarity. In reality, there are a number of complications. A single KO may consist of multiple sequence similarity groups. A small group with a high similarity threshold is a subset of a larger group with a lower similarity threshold, in which case two KOs are defined as the small group and the large group excluding the small group part. As long as the constituent sequence similarity groups are well defined including these examples, the KOALA (KEGG Orthology and Links Annotation) program (1) to computationally assign K numbers works well. However, there are still a small number of legacy KOs converted from Enzyme Commission (EC) number groups, whose associated sequence data are not well defined.
Internally KO grouping is constantly updated in the manual verification part of the KOALA annotation procedure (1). For outside users the basis of KO grouping and its correspondence to molecular function should be made clear by experimental evidence. Thus, major efforts have been initiated to annotate individual KOs with reference information reporting experiments on functional characterization of genes and proteins and, whenever possible, protein sequence data used in the experiments, such as those submitted to the INSDC (DDBJ/ENA/GenBank) database or those stated in the reference. As of September 2015, references (PubMed links) and sequence data (GENES links) are included in 76% and 45%, respectively, of about 19 000 KO entries. The sequence data listed in the KO entry can now be considered as the core sequence(s) from which an ortholog group has been defined.
New additions to GENES database
For many years the KEGG GENES database was created from NCBI's RefSeq database. Since mid-2014, newly sequenced prokaryotic genomes are taken from GenBank, and since mid-2015, existing prokaryotic genomes, excluding the NCBI reference genomes, are updated using GenBank, for the current RefSeq entries produced by the NCBI Prokaryotic Genome Annotation Pipeline (6) are very different from previous versions. No changes have been made to eukaryotic genomes. The data source of KEGG GENES is summarized in Table 2.
Table 2. Data Source of KEGG GENES.
| Category | Primary data source | Genome identifier | Gene identifier | |
|---|---|---|---|---|
| Eukaryotes | RefSeq | RefSeq releasea (complete) | T0 numbers (three or four letter organism codes) | GeneID |
| Prokaryotes | RefSeq | NCBI reference genomesb | Locus_tag | |
| GenBank | Other complete genomes listed in prokaryotes.txtc | Locus_tag | ||
| Viruses | RefSeq | RefSeq releasea (viral) | T40000 (vg) | GeneID |
| Plasmids | RefSeq | RefSeq releasea (plasmid) | T20000 (pg) | GeneID |
| Addendum | PubMed | Functionally characterized genes | T10000 (ag) | ProteinID |
Eukaryotes and prokaryotes with complete genomes constitute KEGG organisms identified by three- or four-letter organism codes. As shown in this table, there are three additional categories, viruses, plasmids and addendum, with two-letter codes of vg, pg and ag, respectively. The viruses and plasmids categories are taken from RefSeq collections. The annotation (K-number assignment) rate is very low for viruses, about 7% compared to 46% for KEGG organisms, but this category is useful in metagenome annotation. Many plasmids are included in the complete genomes of KEGG organisms, and the remaining ones are selected and stored in the plasmids category.
The addendum category is a collection of manually created protein sequence entries. In the KEGG pathway maps, there used to be cases where no corresponding genes could be found in KEGG organisms, thus, only links to UniProt (7) were given. In order to associate them with sequence data and K numbers, addendum entries are created using the original sequence data with International Nucleotide Sequence Database Collaboration (INSDC) protein accession numbers. In addition, there are two focused areas where sequence records are being created. One is Enzyme Nomenclature. As we do for each KO entry, we believe that each EC number entry should be linked to the sequence data used in the original experiment, so that the sequence similarity based extension of EC number assignment can safely be done. Thus, we are trying to create a list of protein sequences from the reference list of the Enzyme Nomenclature database (5). Another focused area is antimicrobial resistance (AMR), which will be discussed later.
Taxonomy and pangenomes
The increasing number of sequenced genomes, especially those of closely related bacterial strains, poses problems of how to process and represent them in the database. Attempts are made to define selected sets of genomes, such as reference and representative genomes in RefSeq (6) and reference proteomes in UniProt (7), which include both well-studied organisms and taxonomic diversity. In KEGG, such reference genomes are not explicitly defined, but the ordering of KEGG organisms contains preference of genomes. The KEGG organisms ordering is consistent with that of the NCBI taxonomy (8), but members in each taxonomic rank are manually ordered, not alphabetically ordered. The first genome in each taxonomic rank is considered as a reference genome, which is used for generating the following pangenome data sets.
As an organism's functional capacity is represented by the set of assigned K numbers (KO content), the functional capacity of an organism group is represented by the combined set of assigned K numbers. A pangenome data set, as we define it here, is created by removing similar organisms, but retaining the KO content, at the species, genus or family level. When multiple members are present in each species/genus/family group, the first genome in the KEGG organisms order is taken as a representative genome. When the other members in the group contain different K numbers that are not present in the representative genome, those genes are added as if they are present in additional chromosomes or plasmids.
BlastKOALA and GhostKOALA
By the genome annotation procedure in KEGG, the GENES database becomes structured in terms of the KO (K number) groups. This facilitates the processing of sequence similarity search results against the GENES database, which is simply to assign the most appropriate K numbers, as implemented in the automatic annotation services of KAAS (9) and newly released BlastKOALA and GhostKOALA. As shown in Table 3, BlastKOALA and GhostKOALA utilize the pangenome data set, which can be viewed as a non-redundant GENES database after removing similar sequences in similar organisms, but retaining the KO content and the taxonomic diversity. The reduced database size was 55% and 24% for prokaryotes at the species and genus levels, respectively, and 81% and 59% for eukaryotes at the genus and family levels, respectively, as of this writing.
Table 3. BlastKOALA and GhostKOALA for genome and metagenome annotation.
| Program | KOALA | BlastKOALA | GhostKOALA |
|---|---|---|---|
| URL | www.kegg.jp/blastkoala/ | www.kegg.jp/ghostkoala/ | |
| Purpose | Internal GENES annotation | Genome annotation | Metagenome annotation |
| Search program | SSEARCH | BLASTP | GHOSTX |
| Scoring | Weighted sum of SW scores (KOALAa) | Weighted sum of BLAST bit scores (Modified KOALA) | Unweighted sum of GHOSTX scores |
| Database | All KEGG organisms | Pangenomes | Pangenomes + Viruses |
aKOALA scoring includes: SW (Smith-Waterman) score, best-best flag, overlap of alignment, ratio of query and DB sequences, taxonomic category and Pfam domains.
BlastKOALA is suitable for annotating fully sequenced genomes, while GhostKOALA, which uses GHOSTX (10) and runs 100 times faster, is suitable for annotating large data sets such as metagenomes. Both assign K numbers to query amino acid sequences and allow KEGG mapping for interpretation of high-level functions. In BlastKOALA most appropriate K numbers are determined by a method similar to the KOALA program internally used for annotation of KEGG organisms (1). In GhostKOALA only the top scores are examined for K number assignment. One additional feature of GhostKOALA is the assignment of taxonomic compositions. For this purpose the pangenome data set for GhostKOALA is supplemented by sequences selected from CD-HIT clusters (11), adding sequences without K numbers in each taxonomic rank and viral sequences, thus representing the sequence diversity of the GENES database.
TRANSLATIONAL BIOINFORMATICS
Antimicrobial resistance (AMR)
AMR is a universal problem in the management of infectious diseases and complications. Traditionally, the KEGG database contains various contents for infectious diseases and antimicrobial drugs, including KEGG disease pathway maps for infectious diseases, KEGG metabolic pathway maps for biosynthesis of antibiotics, KEGG drug structure maps for the history of antimicrobial drug development and KEGG DRUG entries for all drugs currently in use. Knowledge on AMR mechanisms is now organized in KEGG pathway maps and KEGG modules (Table 4). Furthermore, to meet the practical needs for combating AMR, we have started developing signature modules and signature KOs that can be used to characterize AMR from pathogen genome sequences. Signature modules are a class of KEGG modules, which can be used for linking units of genes in the genome, represented by sets of K numbers, to phenotypic features. Signature modules of drug resistance in pathogens are treated separately with annotation of threat levels defined by CDC (Table 4).
Table 4. KEGG contents for antimicrobial resistance.
| Category | Content | Example |
|---|---|---|
| Pathway map | Drug resistance pathway | map01501 beta-Lactam resistance |
| map01502 Vancomycin resistance | ||
| Signature modulea | Resistance caused by: (i) altered target site | M00625 Methicillin resistance |
| (ii) enzymatic inactivation | M00627 beta-Lactam resistance, Bla system | |
| (iii) decreased penetration | M00745 Imipenem resistance, repression of porin OprD | |
| (iv) increased efflux | M00704 Tetracycline resistance, efflux pump Tet38 | |
| Signature KOa | Resistance gene mutation groups | Tight KOs for beta-lactamases |
| Sequence data | Amino acid sequences of known resistance genes | GENES addendum data listed in BRITE table files: br08453 beta-lactamases br08456 tetracyclin resistance genes br08454 aminoglycoside resistance genes br08455 macrolide resistance genes |
aSignature modules and signature KOs are annotated with the threat level defined by CDC
(http://www.cdc.gov/drugresistance/threat-report-2013/) and shown in the BRITE table file
There are also cases where mutations of a single gene play direct roles for AMR. Beta-lactams, the major class of antibiotics, have a long history of newly appearing resistant strains, and in Gram-negative bacteria this is mainly due to mutations of beta-lactamase genes. There have been efforts to collect and classify beta-lactamase mutations (12). We examined about 1200 sequences and concluded that they can be represented by finely classified KOs, named tight KOs, because clear phylogenetic relationships exist for groups of mutated genes. Signature KOs are tight KOs that can be linked to phenotypic features, in this case resistant drug groups. The addendum category of the GENES database now contains beta-lactamase sequences, as well as protein sequences of tetracycline, aminoglycoside and macrolide resistance genes. Figure 1 shows taxonomic distributions of signature KOs for beta-lactamases that are linked to carbapenem resistance, according to the current GENES database. A tool called Pathogen Checker is being developed as a specialized version of the BlastKOALA server for comparing a query pathogen genome against a subset of the GENES database containing sequences of signature KOs and signature modules.
Figure 1.
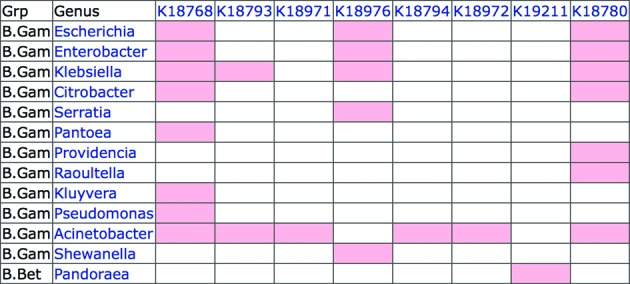
Eight signature KOs for beta-lactamases that represent carbapenem resistance are shown indicating which organisms at the genus level contain which genes. This table is generated by the Module Table interface in the KEGG Annotation page (http://www.kegg.jp/kegg/annotation/). The K numbers correspond to the following gene groups: K18768 (KPC), K18793 (OXA-23), K18971 (OXA-24), K18976 (OXA-48), K18794 (OXA-51), K18972 (OXA-58), K19211 (OXA-62) and K18780 (NDM).
Drug interaction network
The KO database is our attempt to make limited experimental evidence applicable to many other data. Genes and proteins in the GENES database are considered as instances of functional orthologs represented by KOs. By organizing knowledge in terms of generalized (KO-based) networks, high-level functions of individual organisms can be inferred from gene sets in the genome. As shown in Table 5, there are two other network types that are organized in a similar way. One is the chemical reaction network. Enzymatic reactions in the REACTION database are grouped into reaction class (RC) in the RCLASS database, representing the same local structure transformation patterns for substrate-product pairs irrespective of overall structures (13). As previously reported (1,13) one-to-many relationships between reaction modules (ordered sets of RCs) and KEGG modules (sets of KOs) may help to annotate enzyme genes.
Table 5. Three types of molecular networks in KEGG.
| Type | Instance (Database) | Class (Database) | Abbreviation | Functional unit |
|---|---|---|---|---|
| Gene/protein network | Gene/protein (KEGG GENES) | Ortholog (KEGG ORTHOLOGY) | KO | KEGG module |
| Chemical reaction network | Reaction (KEGG REACTION) | Reaction class (KEGG RCLASS) | RC | Reaction module |
| Drug interaction network | Drug (KEGG DRUG) | Drug group (KEGG DGROUP) | DG | Interaction unit |
The other is the drug interaction network, which is generalized using the newly introduced drug groups (DGs) in the DGROUP database. There are multiple levels of drug groups, the lowest level being the chemical group for the same active ingredient with different salts or hydrates. Many drug interactions are caused by overlapping targets and metabolizing enzymes (14), and appropriate drug groups have been defined. The drug interaction data set in the KEGG DRUG database, which is based on known interactions listed in the drug labels of all marketed drugs in Japan, is being expanded with the DG representation. This will allow better detection of drug interactions associated with contraindications and precautions, as well as duplicate administration of drugs with the same or similar efficacy. Currently, the interaction is defined simply by the pair of D numbers (DRUG identifiers) or DG numbers (DGROUP identifiers). Attempts will be made to incorporate additional factors in the human genome, such as polymorphism of cytochrome P450 (CYP) enzymes and mutation of specific genes, for defining interaction units (Table 5), which may be used for interpretation of drug responses and drug interactions from personal genomes.
Accessing KEGG
KEGG is made available at both the KEGG main website (http://www.kegg.jp/) and the GenomeNet mirror website (http://www.genome.jp/kegg/). BlastKOALA and GhostKOALA are maintained in the main website, while KAAS, SIMCOMP and many other tools are maintained in the GenomeNet website, which also develops the LinkDB and MGENES databases.
Acknowledgments
Computational resources were provided by the Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan.
FUNDING
National Bioscience Database Center of the Japan Science and Technology Agency (partial). Funding for open access charge: National Bioscience Database Center of the Japan Science and Technology Agency.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson D.A., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2015;43:D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanehisa M. Chemical and genomic evolution of enzyme-catalyzed reaction networks. FEBS Lett. 2013;587:2731–2737. doi: 10.1016/j.febslet.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 5.McDonald A.G., Tipton K.F. Fifty-five years of enzyme classification: advances and difficulties. FEBS J. 2014;281:583–592. doi: 10.1111/febs.12530. [DOI] [PubMed] [Google Scholar]
- 6.Tatusova T., Ciufo S., Federhen S., Fedorov B., McVeigh R., O'Neill K., Tolstoy I., Zaslavsky L. Update on RefSeq microbial genomes resources. Nucleic Acids Res. 2015;43:D599–D605. doi: 10.1093/nar/gku1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Federhen S. The NCBI Taxonomy database. Nucleic Acids Res. 2012;40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moriya Y., Itoh M., Okuda S., Yoshizawa A., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki S., Kakuta M., Ishida T., Akiyama Y. GHOSTX: an improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS One. 2014;9:e103833. doi: 10.1371/journal.pone.0103833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 12.Bush K., Jacoby G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muto A., Kotera M., Tokimatsu T., Nakagawa Z., Goto S., Kanehisa M. Modular architecture of metabolic pathways revealed by conserved sequences of reactions. J. Chem. Inf. Model. 2013;53:613–622. doi: 10.1021/ci3005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takarabe M., Shigemizu D., Kotera M., Goto S., Kanehisa M. Network-based analysis and characterization of adverse drug-drug interactions. J. Chem. Inf. Model. 2011;51:2977–2985. doi: 10.1021/ci200367w. [DOI] [PubMed] [Google Scholar]