

Abstract
We present here the rVarBase database (http://rv.psych.ac.cn), an updated version of the rSNPBase database, to provide reliable and detailed regulatory annotations for known and novel human variants. This update expands the database to include additional types of human variants, such as copy number variations (CNVs) and novel variants, and include additional types of regulatory features. Now rVarBase annotates variants in three dimensions: chromatin states of the surrounding regions, overlapped regulatory elements and variants’ potential target genes. Two new types of regulatory elements (lncRNAs and miRNA target sites) have been introduced to provide additional annotation. Detailed information about variants’ overlapping transcription factor binding sites (TFBSs) (often less than 15 bp) within experimentally supported TF-binding regions (∼150 bp) is provided, along with the binding motifs of matched TF families. Additional types of extended variants and variant-associated phenotypes were also added. In addition to the enrichment in data content, an element-centric search module was added, and the web interface was refined. In summary, rVarBase hosts more types of human variants and includes more types of up-to-date regulatory information to facilitate in-depth functional research and to provide practical clues for experimental design.
INTRODUCTION
The association between non-coding variants and human diseases has been of an increasing concern (1–3), and variants that are associated with gene expression abundance have been rapidly identified and accumulated in recent years. Annotating the regulatory features of human variants has been a practical requirement in clinical and basic research (1,4); multiple approaches have been developed to allow the functional annotation of non-coding variants (5–8). To provide reliable, comprehensive and user-friendly regulatory annotation of human single nucleotide polymorphisms (SNPs), we developed the rSNPBase database (9). In the past 2 years, burgeoning sequencing techniques have driven the identification of new disease-associated SNPs and additional types of variants, such as copy number variations (CNVs) and novel variants (10). Meanwhile, advancements in regulatory research have been made in the past few years. For example, the Roadmap project systematically characterized the epigenomic landscapes of representative primary human tissues and cells and then released the relevant data (11,12); new modes of regulation, such as long non-coding RNA (lncRNA) mediated regulation, have been studied in depth (13–16); and more expression quantitative trait loci (eQTLs) have been identified and analyzed (17). Therefore, there is a growing need to update the database to host more types of human variants and include more types of up-to-date regulatory information.
The updated rVarBase hosts human regulatory variants (known SNPs and CNVs); furthermore, it annotates novel variants. rVarBase describes a variant's regulatory features in three fields: chromatin states (in different tissues/cells), overlapped regulatory elements and potential target genes. rVarBase also provides an optional extended annotation for variants, including linkage disequilibrium (LD) proxies of known regulatory SNPs (rSNPs), SNPs that are located in regulatory CNVs (rCNVs) and traits (diseases and expression quantitative traits) that are associated with variants. A three-module (variant-centric, gene-centric and element-centric) search engine is provided to facilitate data navigation.
New features
rVarBase is consistent with the previous version in its utilization of experimentally supported regulatory information to make relevant annotations. As shown in Figure 1, genome-wide human variants were gotten and standardized with information from the NCBI dbSNP (build 142) (18), the dbVar (GRCh37) (19) and the UCSC (20). The regulatory features (chromatin states of the surrounding regions, overlapped and experimentally supported regulatory elements and potential target genes) of each variant were analyzed with reference to experimentally supported information. Known human SNPs and CNVs with regulatory features were stored as rSNPs and rCNVs, on which further extended analyses were performed. The reference data utilized for the regulatory feature analysis and extended analysis are shown in http://rv.psych.ac.cn/datacontent.do and Supplementary Tables S1 and S2. A summarized comparison of the current and previous versions is shown in Table 1.
Figure 1.
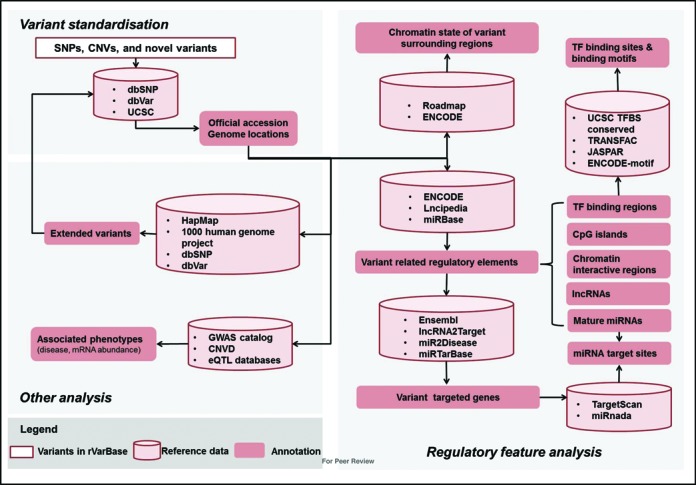
Data processing and data content of rVarBase.
Table 1. Data content of rVarBase (as of September 11, 2015) and rSNPBase.
| Data type | rSNPBase | rVarBase |
|---|---|---|
| Variants | ||
| rSNPsa | 22 846 898 | 87 345 304 |
| rCNVsb | – | 1 368 424 |
| Annotation for novel variants | No | Yes |
| Regulatory features | ||
| Chromatin states | No | Yes |
| Regulatory elements | ||
| CpG islands | Yes | Yes |
| TF binding regions | Yes | Yes |
| Matched TFBS and TF-binding matrixes | No | Yes |
| Interactive chromatin regions | Yes | Yes |
| lncRNAs | No | Yes |
| miRNAs | Yes | Yes |
| miRNA binding sites | No | Yes |
| Target genes | 56 869 | 82 640 |
| Extended variants | ||
| LD-proxies of rSNPs (non-rSNPs) | 2 281 874 | 1 626 737 |
| Non-rSNPs inside rCNVs | – | 21 797 660 |
| Associated traits | ||
| Diseases (variant-disease pairs) | – | 198 928 |
| eQTLs (SNP-mRNA pairs) | 2 428 727 | 4 201 218 |
aKnown human SNPs that have regulatory features were stored as rSNPs.
bKnown human CNVs that have regulatory features were stored as rCNVs.
CNVs and novel variants
In addition to accounting for the increased number of SNPs in dbSNP since the publication of rSNPBase 2 years ago, rVarBase provides annotations on more types of human variants, such as known CNVs, novel single-nucleotide variants (SNVs) and regions. Human CNVs were obtained from the dbVar database (19). To focus on regulatory features and to avoid including long CNVs that cover one or more protein-coding gene regions, only CNVs with a length of less than 1 Mb were analyzed. The analytical flow for CNVs and user-requested novel SNVs (with their chromosomal location information) is similar to that of known SNPs; it includes an analysis of the chromatin states of the surrounding regions, a comparison with experimentally supported elements according to their genomic locations and then a map of potential target genes with reference to the genomic proximity of the regulatory elements and transcript start sites (TSSs). For novel regions that are uploaded by users, we provide known regulatory variants that overlap with such regions.
Chromatin states
The Roadmap project provides 111 reference epigenomes and a 15-state model that is trained to generate genome-wide maps of chromatin state using the 111 epigenomes along with 16 epigenomes from the ENCODE project (11). The detailed chromatin state map was downloaded from the project's supplementary data repository web portal (http://egg2.wustl.edu/roadmap/web_portal/index.html). Eight active states (‘Active TSS’, ‘Flanking Active TSS’, ‘Transcr. at gene 5′ and 3′’, ‘Strong transcription’, ‘Weak transcription’, ‘Genic enhancers’, ‘Enhancers’ and ‘ZNF genes & repeats’) and three bivalent states (‘Bivalent/Poised TSS’, ‘Flanking Bivalent TSS/Enhancer’ and ‘Bivalent Enhancer’) from the 15-state model were used to annotate the chromatin state of a variant's surrounding region. Purely repressed states in the 15-state model were not included.
lncRNAs and miRNA target sites
Regulatory elements that cover or overlap with analyzed variants are identified as variant-related elements. In addition to the regulatory elements that are included in rSNPBase (CpG islands, chromatin-interactive regions, TF-binding regions and mature miRNAs), lncRNAs and miRNA target sites were also introduced into the variants’ annotations. lncRNA information was drawn from the LNCipedia database (13); experimentally supported lncRNA target genes were obtained from the LncRNA2Target database (16). Considering the important roles that microRNA target site polymorphisms play in human diseases (21), miRNA target sites in the 3′ UTRs of experimentally supported miRNA target genes were also included for comparison with variants. miRNA target genes were obtained from the miR2Disease (22) and miRTarBase (23) databases, and matched miRNA binding sites were scanned using TargetScan (24,25) and miRnada (26). Detailed information about the utilized regulatory elements is shown in Supplementary Table S1 and http://rv.psych.ac.cn/datacontent.do.
TF binding sites and TF matrixes
In rSNPBase, experimentally supported TF-binding regions (∼150 bp) that had been generated by the ENCODE project were used to annotate variants. Because exact TF binding sites are often smaller than 15 bp, a more detailed annotation is necessary for functional analysis and experimental design. Using predicted genome-wide TFBS maps from UCSC TFBS conserved (Z score greater than 2.33) (20), JASPAR (27) and ENCODE-motif (28), the potential binding sites of matched TF families inside TF-binding regions were identified and compared with variants. Corresponding TF-binding matrixes from TRANSFAC (29), JASPAR (27) and ENCODE-motif (28) were also included in rVarBase.
More extended information
As in rSNPBase, an extended information analysis was performed on all rVarBase-hosted variants. In addition to the LD-proxies of rSNPs, extended SNPs that located in rCNVs were also added. eQTL information from more data sources, including the RTeQTL database (30), BrainEAC (31), the skin eQTL database (32) and the GTEx Portal (17,33), was added to provide eQTL labels. Variants’ associated diseases/traits were integrated from the database of GWAS catalog (34) and the database of CNVD (35). Detailed information about the reference data that were used in the extended analysis is shown in Supplementary Table S2 and http://rv.psych.ac.cn/datacontent.do.
Web interface
The web interface was refined to make data acquisition more convenient. The input format of queried variants may be as a dbSNP ID (for a known SNP) or as a genome position with zero-based coordinates (for all types of variants). In addition to ‘Variant search’ and ‘Gene search’, a new search module, ‘Element search’, was added to facilitate searches based on TFs/miRNAs/lncRNAs of interest. As shown in Figure 2A, variants in experimentally supported binding regions or predicted TFBSs, variants in mature miRNA or predicted miRNA-binding sites and variants in lncRNAs may be queried by entering the element name and the target gene name. An FTP site (ftp://rv.psych.ac.cn/pub/rv/) was added to facilitate the download of the whole database.
Figure 2.

New search module of rVarBase and an example of data retrieving process.
DATABASE USAGE
The rVarBase was developed to bridge genetic studies with functional researches. This database can provide potential functional interpretation in terms of gene expression regulation for results of genetic studies. rVarBase can also assist researchers in filtering candidate variants by genes of interest or regulatory mechanisms. Furthermore, for queried variants, rVarBase provides detailed regulatory information, which is practical for the design of experiments that explore biological function. Because rVarBase can perform regulatory feature analysis on novel variants, it can be utilized not only with disease-associated SNPs that are generated by traditional genetic association studies, but also with more other types of genetic data.
We provide a demonstration dataset as an example to show the database usage with novel variants. This dataset includes nine novel non-coding SNVs that are associated with tumors and were identified by Nils et al. (36) in 2014. Detailed chromosomal locations of the nine SNVs can be seen in Supplementary Table S3 and http://rv.psych.ac.cn/tutorial.do. As shown in Figure 2B, these variants can be quickly entered into the model ‘Variant search’ with their chromosomal locations (hg19 genome coordinates). The regulatory features of and extended information about the queried variants are summarized in the ‘Search Results’. One of the nine variants (located at chr5:1295243–1295244) has been included in NCBI dbSNP database with the ID ‘rs35550267’. All of the nine novel SNVs have regulatory features. They are located in active chromatin regions and inside TF-binding regions and chromatin-interactive regions; two genes are potentially regulated by the regulatory elements in which they are located. These regulatory variants are appropriate candidates for further validation studies and functional researches. Detailed information about each regulatory variant, such as the genomic locations of their overlapping active chromatin regions or regulatory elements, specific tissue types, target genes and related regulatory modes, are shown on the ‘Variant report’ page. Since all variants are overlapped with TF-binding regions, additional information about matched TFBS and TF-binding motif is also provided in this page. These detailed reports, as practical reference data, may directly support experimental design in functional research.
CONCLUSION AND FUTURE PLAN
Here, we upgraded the rSNPBase database, which provides reliable regulatory annotation of human SNPs, to the rVarBase database, which now provides more comprehensive regulatory annotation for multiple types of human variants. The updates include the regulatory annotations of short and structural variants with reference to up-to-date epigenetic advancements. The updated rVarBase supports the functional analysis of known and novel variants and will thus assist users in exploring data from new types of research, such as novel results from next-generation sequencing. Integrative, tissue/cell-based chromatin-state data were introduced to annotate the variants; these data will be helpful to users in gathering more biologically meaningful information. New types of regulatory elements, more detailed annotation, additional extended information and a new search module in the updated database will further aid researchers in future functional analyses of genetic studies and will provide more comprehensive reference data for candidate variant selection and for the experimental design of subsequent genetic and functional research.
rVarBase will be continuously updated with newly reported human genetic and epigenetic data. In addition to continuously adding newly reported variants in dbSNP and dbVar, new annotation dimensions and new types of regulatory elements will be considered and followed. For example, the method for lncRNA target site prediction (37) is appeared and developed; we hope to add the corresponding data in the future, when the method is mature and validated. The integration of multi-dimensional regulatory features is also being considered.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Key Laboratory of Mental Health, Institute of Psychology; Chinese Academy of Sciences; the CAS/SAFEA International Partnership Program for Creative Research Teams [Y2CX131003]; Knowledge Innovation Program of the Chinese Academy of Sciences [KSCX2-EW-J-8]; National Natural Science Foundation of China [81201046]. Funding for open access charge: Key Laboratory of Mental Health,Institute of Psychology; Chinese Academy of Sciences; the CAS/SAFEA International Partnership Program for Creative Research Teams [Y2CX131003]; Knowledge Innovation Program of the Chinese Academy of Sciences [KSCX2-EW-J-8]; National Natural Science Foundation of China [81201046].
FUNDING
Conflict of interest statement. None declared.
REFERENCES
- 1.Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albert F.W., Kruglyak L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- 3.Weischenfeldt J., Symmons O., Spitz F., Korbel J.O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 2013;14:125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 4.Haider S.A., Faisal M. Human aging in the post-GWAS era: further insights reveal potential regulatory variants. Biogerontology. 2015;16:529–541. doi: 10.1007/s10522-015-9575-y. [DOI] [PubMed] [Google Scholar]
- 5.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward L.D., Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li M.J., Wang L.Y., Xia Z., Sham P.C., Wang J. GWAS3D: Detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res. 2013;41:W150–W158. doi: 10.1093/nar/gkt456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritchie G.R., Dunham I., Zeggini E., Flicek P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo L., Du Y., Chang S., Zhang K., Wang J. rSNPBase: a database for curated regulatory SNPs. Nucleic Acids Res. 2014;42:D1033–D1039. doi: 10.1093/nar/gkt1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shalem O., Sanjana N.E., Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung D., Jung I., Rajagopal N., Schmitt A., Selvaraj S., Lee A.Y., Yen C.A., Lin S., Lin Y., Qiu Y., et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–354. doi: 10.1038/nature14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volders P.J., Verheggen K., Menschaert G., Vandepoele K., Martens L., Vandesompele J., Mestdagh P. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 2015;43:4363–4364. doi: 10.1093/nar/gkv295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quek X.C., Thomson D.W., Maag J.L., Bartonicek N., Signal B., Clark M.B., Gloss B.S., Dinger M.E. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015;43:D168–D173. doi: 10.1093/nar/gku988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volders P.J., Verheggen K., Menschaert G., Vandepoele K., Martens L., Vandesompele J., Mestdagh P. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 2015;43:D174–D180. doi: 10.1093/nar/gku1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Q., Wang J., Wu X., Ma R., Zhang T., Jin S., Han Z., Tan R., Peng J., Liu G., et al. LncRNA2Target: a database for differentially expressed genes after lncRNA knockdown or overexpression. Nucleic Acids Res. 2015;43:D193–D196. doi: 10.1093/nar/gku1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lappalainen I., Lopez J., Skipper L., Hefferon T., Spalding J.D., Garner J., Chen C., Maguire M., Corbett M., Zhou G., et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41:D936–D941. doi: 10.1093/nar/gks1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sethupathy P., Collins F.S. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24:489–497. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu S.D., Lin F.M., Wu W.Y., Liang C., Huang W.C., Chan W.L., Tsai W.T., Chen G.Z., Lee C.J., Chiu C.M., et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 25.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betel D., Wilson M., Gabow A., Marks D.S., Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kheradpour P., Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42:2976–2987. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wingender E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Briefings Bioinformatics. 2008;9:326–332. doi: 10.1093/bib/bbn016. [DOI] [PubMed] [Google Scholar]
- 30.Ma B., Huang J., Liang L. RTeQTL: Real-Time Online Engine for Expression Quantitative Trait Loci Analyses. Database: J. Biological Databases Curation. 2014;2014:bau066. doi: 10.1093/database/bau066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramasamy A., Trabzuni D., Guelfi S., Varghese V., Smith C., Walker R., De T., Coin L., de Silva R., Cookson M.R., et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 2014;17:1418–1428. doi: 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding J., Gudjonsson J.E., Liang L., Stuart P.E., Li Y., Chen W., Weichenthal M., Ellinghaus E., Franke A., Cookson W., et al. Gene expression in skin and lymphoblastoid cells: refined statistical method reveals extensive overlap in cis-eQTL signals. Am. J. Hum. Genet. 2010;87:779–789. doi: 10.1016/j.ajhg.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu F., Xu Y., Li K., Li Z., Liu Y., DuanMu H., Zhang S., Li Z., Chang Z., Zhou Y., et al. CNVD: text mining-based copy number variation in disease database. Hum. Mutat. 2012;33:E2375–E2381. doi: 10.1002/humu.22163. [DOI] [PubMed] [Google Scholar]
- 36.Fredriksson N.J., Ny L., Nilsson J.A., Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014;46:1258–1263. doi: 10.1038/ng.3141. [DOI] [PubMed] [Google Scholar]
- 37.He S., Zhang H., Liu H., Zhu H. LongTarget: a tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics. 2015;31:178–186. doi: 10.1093/bioinformatics/btu643. [DOI] [PubMed] [Google Scholar]