

Abstract
The genome-wide transcriptome profiling of cancerous and normal tissue samples can provide insights into the molecular mechanisms of cancer initiation and progression. RNA Sequencing (RNA-Seq) is a revolutionary tool that has been used extensively in cancer research. However, no existing RNA-Seq database provides all of the following features: (i) large-scale and comprehensive data archives and analyses, including coding-transcript profiling, long non-coding RNA (lncRNA) profiling and coexpression networks; (ii) phenotype-oriented data organization and searching and (iii) the visualization of expression profiles, differential expression and regulatory networks. We have constructed the first public database that meets these criteria, the Cancer RNA-Seq Nexus (CRN, http://syslab4.nchu.edu.tw/CRN). CRN has a user-friendly web interface designed to facilitate cancer research and personalized medicine. It is an open resource for intuitive data exploration, providing coding-transcript/lncRNA expression profiles to support researchers generating new hypotheses in cancer research and personalized medicine.
INTRODUCTION
Over the past two decades, gene expression data generated by microarray technology has been widely used to study the causes and therapies of cancers. Research on the human transcriptome has produced a large amount of expression data at the gene level. Many gene-expression databases have been developed for cancer research, such as Oncomine (public version) (1), NextBio (2) and GCOD (3). However, a given gene is often spliced into multiple transcript isoforms, which in turn are translated into different proteins. More than 90% of human genes undergo alternative splicing (4). Therefore, the gene-level expression data generated by microarray platforms are insufficient to understand the involvement of specific proteins in cancer. RNA-Seq (5) is a revolutionary tool that can be used to study alternative splicing and to quantify gene/isoform expression levels across a genome. RNA-Seq has been widely used in many cancer studies.
Several major data portals contain cancer RNA-Seq data, such as the NCBI Gene Expression Omnibus (GEO) (6) and the Sequence Read Archive (SRA) (7). However, these portals mainly serve as raw data archives. They do not provide the full utility of RNA-Seq data for biologists, because highly developed bioinformatics skills are required to set up and manipulate data pipelines, tune parameters and control quality during data processing, analysis and visualization. The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) database stores genomics data (including RNA-Seq data) for a variety of cancer types, but it only contains data generated by TCGA consortium. The RNA-Seq Atlas (8) attempts to provide easy access to RNA-Seq expression profiles. However, it only has one RNA-Seq data set and 11 samples. Recently, we have constructed a database for isoform–isoform interactions using 19 RNA-Seq data sets (9), and performed high-resolution functional annotation of the human transcriptome using 29 RNA-Seq data sets (10). Nevertheless, these studies have not utilized long non-coding RNA (lncRNA) expression profiles, which we believe is complementary to the coding-transcript expression data in cancer research.
This paper presents the Cancer RNA-Seq Nexus (CRN), the first public database providing phenotype-specific coding-transcript/lncRNA expression profiles and mRNA–lncRNA coexpression networks in cancer cells. CRN includes cancer RNA-Seq data sets in the TCGA, SRA and GEO databases. Figure 1 shows the framework that we used to construct the database. It resulted in 54 human cancer RNA-Seq data sets, including 326 phenotype-specific subsets and 11 030 samples. Each subset is a group of RNA-Seq samples associated with a specific phenotype or genotype, e.g. breast cancer stage II, ER+ breast cancer or Her2+ breast cancer. This specificity facilitates research into personalized medicine. CRN provides a user-friendly interface to efficiently organize and visualize coding-transcript/lncRNA expression profiles. It also permits the visualization and analysis of mRNA–lncRNA coexpression networks for any pair of phenotype-specific subsets. CRN is freely available at http://syslab4.nchu.edu.tw/CRN.
Figure 1.

Framework for constructing the CRN database. Cancer RNA-Seq data sets were collected from NCBI GEO, SRA and TCGA, and then all samples were classified into the phenotype-specific subsets. For the GEO data sets, Bowtie2 and eXpress software were used to calculate isoform expressions using GENCODE v21 as a reference. For the TCGA data sets, we converted the expression values (tau values) of the TCGA Level 3 RNA-Seq version 2 data sets to TPM (transcripts per million). To identify phenotype-specific differentially expressed protein-coding transcripts and lncRNAs in each data set, we performed log2 scale t-tests with Benjamini–Hochberg adjustment between each pair of subsets with no overlapping samples and from the same data set. For each subset pair, we selected coding transcripts and lncRNAs with high expression variance, then calculated the correlations of expression profiles between selected coding transcripts and lncRNAs to construct an mRNA–lncRNA coexpression network.
MATERIALS AND METHODS
RNA-seq data sets
We collected a total of 54 human cancer data sets and 11 030 samples from major cancer RNA-Seq data portals, including the Gene Expression Omnibus (GEO) (6), Sequence Read Archive (SRA) (7) and the Cancer Genome Atlas (TCGA http://cancergenome.nih.gov/). The final database consists of 28 TCGA data sets and 26 GEO/SRA data sets.
GEO/SRA data sets
We obtained annotation data for the RNA-Seq data sets from GEO, and downloaded RNA-Seq reads from SRA. Each data set consists of several subsets, where each subset is a group of RNA-Seq samples associated with a specific phenotype trait or cancer condition. For example, the subsets can be identified with a specific stage of cancer, a disease state, a cell line, a type of tissue or a genotype. We manually created these subsets from the raw data and annotations, assigning samples based on their textual descriptions. The GEO/SRA data yielded a total of 142 subsets and 1337 samples. To obtain expression profiles for both coding transcripts and lncRNAs, we downloaded the entire human transcriptome with Ensembl ID, including 93 139 protein-coding and 26 414 lncRNA transcripts, from GENCODE (Release 21, GRCh38) (11) as a reference. Bowtie (12), version 1.1.1, was used to build the reference index and align the RNA-Seq reads to the reference transcriptome. To ensure enough depth of sequencing coverage, we only selected samples with at least 1 million reads and 15% of reads mapped to the reference transcriptome (13). After this step, isoform abundances in each sample were quantified using eXpress (14), version 1.5.1, with the expression unit ‘fragments per kilobase of transcript per million mapped reads’ (FPKM) (15).
TCGA data sets
We collected all Level 3 RNA-Seq version 2 data sets and clinical information from the TCGA portal (http://cancergenome.nih.gov/). We defined subsets for all patient samples based on TCGA clinical information, e.g. primary tumor, metastatic, recurrent tumor, adjacent normal tissue or the pathologic tumor stage. Since the number of adjacent normal samples for each tumor stage is small, we pull all the adjacent normal samples from various tumor stages into a normal subset for each TCGA data set. This analysis resulted in a total of 239 subsets and 10,270 samples. The TCGA expression data contain 67 119 transcripts with UCSC KnownGene IDs that are given gene symbol assignments according to the UCSC KnownGene annotation (16). Since GENCODE provides annotation of protein-coding transcripts and lncRNAs, we classified these KnownGene IDs into 63 775 protein-coding transcripts and 3344 lncRNAs using the mapping of GENCODE and KnownGene IDs. We multiplied the expression values (tau values, calculated by RSEM) (17) of the TCGA Level 3 RNA-Seq version 2 data sets by 106 to obtain transcripts per million (TPM).
Phenotype-specific differentially expressed transcripts (DETs)
To identify phenotype-specific differentially expressed (DE) protein-coding transcripts and DE lncRNAs, within each data set we selected all subsets with at least three samples, then converted the expression value to log2 scale with added pseudo-counts and performed a t-test between every pair of subsets with no samples in common. We used Benjamini–Hochberg adjustment (18,19) to calculate the adjusted P-value. There were 601 subset pairs with differentially expressed transcripts (adjusted P-value < 0.01): 444 ‘cancer versus cancer’ and 157 ‘cancer versus normal’ subset pairs.
Construction of mRNA–lncRNA coexpression networks
To investigate the phenotype-specific regulation of mRNAs and lncRNAs, which may reveal the potential biological functions of lncRNAs, we constructed mRNA–lncRNA coexpression networks. For a given subset pair, we selected coding transcripts and lncRNAs with high expression variance (standard deviation > 0.3 and (standard deviation / mean) > 0.5), then calculated correlations between every pair of selected mRNA and lncRNA expression profiles. The samples of the subset pair were used for the correlation analysis. To assess the significance of these connections, we transferred Pearson's correlation coefficient r to a P-value using the following method: given a sample size n, we calculated the t-value (20):
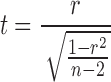 |
The P-value was calculated using this t-value for the t-distribution with n−2 degrees of freedom. The significant correlations between protein-coding transcripts and lncRNAs (P-value < 1E-6) were collected for network construction. In the web interface, Cytoscape (21) was used to demonstrate the mRNA–lncRNA coexpression networks. In the web interface, users can input a gene symbol and an lncRNA name, then select a correlation threshold.
WEB INTERFACES
We developed a user-friendly web interface to present the CRN database, which integrates large-scale RNA-Seq data sets and cancer phenotype/genotype information. As shown in Figure 2A, the CRN database displays a hierarchical menu in the disease-dataset panel (upper left) to help users search and browse the different phenotype-specific subsets. When users select a cancer type or a cancer subset, the associated subset pairs are displayed in the bottom-left panel. After a user selects a subset pair, the web server shows a complete description of the data set/subsets, and permits analysis of the data through four tabs in the right panel:
DE coding transcripts. Given a subset pair, CRN visualizes the expression profiles of all differentially expressed (DE) protein-coding transcripts, sorted by the significance level (adjusted P-value) between two subsets. Users can filter DE transcripts based on their adjusted P-value threshold or up/down regulation. The interface displays the rank, the adjusted P-value, the average expression values, the transcript ID and the gene symbol for each transcript. The transcript ID represents Ensembl transcript ID for GEO/SRA data sets and it represents UCSC KnownGene ID for TCGA data sets.
DE lncRNAs. Given a subset pair, CRN visualizes the expression profiles of DE lncRNAs sorted by the significance level between two subsets. Users can filter DE lncRNAs based on an adjusted P-value threshold or up/down regulation. CRN displays the rank, the adjusted P-value, the expression values of two subsets, the lncRNA ID and the lncRNA name.
mRNA–lncRNA coexpression network. Given a subset pair, CRN visualizes an mRNA–lncRNA co-expression network, displaying coding transcripts and lncRNAs with significant correlations between coding transcripts and lncRNAs (illustrated in Figure 2B). Users can input a gene symbol and an lncRNA name, then select a correlation threshold. They can also choose how many of the most significant connections to show (N = 10, 15, 20 or 25) for the given gene or the given lncRNA.
Search for coding transcripts and lncRNAs. Given a gene symbol, CRN provides a search function on the expression profiles of all transcripts associated with the given gene, allowing users to investigate the differential expressions of its various isoforms. An auto-complete function provides suggestions for gene or transcript symbols as the user types, quickly searching and displaying partially matched terms. The auto-complete function not only helps users search efficiently, but also make a quick filtering (illustrated in Figure 2C).
Figure 2.

Screenshots of the web interface of CRN. (A) The CRN web interface provides three major panels: (1) Disease-dataset panel (upper left). The hierarchical menu illustrates which subsets are associated with which cancer types. A subset is a group of RNA-seq samples associated with a specific phenotype or genotype, e.g. breast cancer stage II, ER+ breast cancer or Her2+ breast cancer. (2) Subset-pair panel (bottom left). There are two types of subset pairs: ‘Cancer versus Cancer’ and ‘Cancer versus Normal’. When users select a subset in the first panel, CRN shows all associated subset pairs in this panel. (3) Profile panel (right). After clicking a subset pair in the second panel, CRN displays a detailed description of the data set and subsets. In this panel, there are four analysis tabs: (i) DE coding transcripts visualizes the expression profiles of differentially expressed (DE) protein-coding transcripts. (ii) DE lncRNAs visualizes the expression profiles of DE lncRNAs. (iii) mRNA–lncRNA coexpression network The user can search for a specific gene and lncRNA in this tab, then visualize the most significant negative and positive correlations between coding transcripts and lncRNAs as a network (B). (iv) Search Users can search for gene/transcript names and transcript IDs. This panel provides an auto-complete function which displays partially matched terms as the user types, filtering the list to provide better and better matches (C). After selecting a gene or transcript, the panel shows the expression profiles.
EXAMPLE APPLICATIONS
To demonstrate the biological applications of CRN, we used the genes TP63, CCAT1 and MYC/PVT1 as examples to show the functionality of differentially expressed coding transcripts, differentially expressed lncRNAs and the mRNA–lncRNA coexpression network.
Differentially expressed coding transcripts
To demonstrate the biological function of DE protein-coding transcripts in CRN, we use the TP63 gene as an example. TP63 has multiple transcript isoforms as a result of alternative promoter usage and alternative splicing. Its transcript isoforms fall into two groups: (i) the upstream promoter of the gene generates TAp63 isoforms containing the N-terminal transactivation (TA) domain, and (ii) an alternative internal promoter generates ΔNp63 isoforms lacking the TA domain (22). In each group, additional variants (e.g. α, β and γ) with various C-terminal tails are generated through alternative splicing at the 3′ end. It has been suggested that ΔNp63 isoforms are highly specific for squamous cells: they are overexpressed in squamous cell carcinoma (SCC) (23–25), while TAp63 expression is either lost or extremely low in squamous cells and SCC (25,26). Therefore, ΔNp63 may be used to precisely distinguish between lung adenocarcinoma and lung SCC, allowing for novel targeted therapies (27,28). As shown in Figure 3, the database confirms that all ΔNp63 isoforms are significantly overexpressed with respect to normal tissue in lung SCC, while the TAp63 isoforms are not. On the other hand, ΔNp63 isoforms are not differentially expressed between cancer and normal subsets in lung adenocarcinoma.
Figure 3.

The differential expressions of two TP63 major isoform groups ‘TAp63 and ΔNp63’ in lung squamous cell carcinoma (A, B) and lung adenocarcinoma (C, D). Based on the adjusted P-values calculated by CRN, ΔNp63 isoforms are significantly overexpressed with respect to normal tissue in lung squamous cell carcinoma, but not in lung adenocarcinoma. In contrast, TAp63 has low expression in all four subset pairs. In the adjusted P-value column, the absent values indicate insignificance (adjusted P-value > 0.01) in differential expression analysis.
Table 1 shows the average transcript expressions of the TAp63α and ΔNp63α isoforms (those with the longest 3′-terminus) in lung SCC and lung adenocarcinoma. The expression of ΔNp63α is extremely high (Average TPM = 109.61) and significantly overexpressed (adjusted P-value = 8.12E-68) in lung SCC, compared with normal samples (Average TPM = 1.205). The expression of ΔNp63α is very low (Average TPM = 1.229 in cancer, 0.807 in normal) and has no significant differential expression (adjusted P-value = 3.00E-1) in lung adenocarcinoma. The expression of TAp63α is extremely low in both lung SCC and lung adenocarcinoma (Average TPM = 0.239–0.833), consistent with the previous reports (23–25).
Table 1. The transcript expressions of TAp63α and ΔNp63α in lung squamous cell carcinoma and lung adenocarcinoma.
| Subset pair | Isoform ID | Isoform name | Average expression in cancer | Average expression in normal | Adjusted P-value |
|---|---|---|---|---|---|
| Lung squamous cell carcinoma versus adjacent normal | uc003fry.2 | TAp63α | 0.833 | 0.456 | 1.22E-01 |
| Lung squamous cell carcinoma versus adjacent normal | uc003fsc.2 | ΔNp63α | 109.610 | 1.205 | 8.12E-68 |
| Lung adenocarcinoma versus adjacent normal | uc003fry.2 | TAp63α | 0.646 | 0.239 | 1.35E-01 |
| Lung adenocarcinoma versus adjacent normal | uc003fsc.2 | ΔNp63α | 1.229 | 0.807 | 3.00E-01 |
Differentially expressed lncRNAs
The Colon Cancer Associated Transcript-1 (CCAT1) lncRNA is significantly up-regulated in colon and colorectal cancer compared with the normal human tissues (29,30). Recently, CCAT1 has also been found to be highly up-regulated in gastric carcinoma, compared with adjacent normal gastric tissues (31,32). Our data analysis confirms these findings. Specifically, there are seven colon adenocarcinoma subset pairs of the type ‘Cancer versus Normal’ in CRN. In all these pairs, CCAT1 is significantly overexpressed (adjusted P-values 9.48E-10–1.35E-50) in cancer subsets (average TPM = 4.53–9.89) with respect to normal subsets (average TPM = 0.318) (Table 2A). Additionally, there are eight stomach adenocarcinoma subset pairs categorized as ‘Cancer versus Normal’ in CRN, and CCAT1 is significantly overexpressed (adjusted P-values 5.02E-3–4.77E-8) in seven cancer subsets (average TPM = 4.43–10.17) with respect to normal subsets (average TPM = 1.575) (Table 2B). Interestingly, the expression values of CCAT1 are extremely similar between colon adenocarcinoma and stomach adenocarcinoma, consistent with the previous reports.
Table 2. The lncRNA expression of CCAT1 in colon adenocarcinoma and stomach adenocarcinoma.
| Average expression in cancer | Average expression in normal | Adjusted P-value | |
|---|---|---|---|
| A. Colon adenocarcinoma subset pair | |||
| Colon adenocarcinoma—Stage I versus Normal (adjacent normal) | 8.584 | 0.318 | 1.67E-34 |
| Colon adenocarcinoma—Stage II versus Normal (adjacent normal) | 9.887 | 0.318 | 7.99E-13 |
| Colon adenocarcinoma—Stage IIA versus Normal (adjacent normal) | 6.926 | 0.318 | 1.35E-50 |
| Colon adenocarcinoma—Stage III versus Normal (adjacent normal) | 4.527 | 0.318 | 9.48E-10 |
| Colon adenocarcinoma—Stage IIIB versus Normal (adjacent normal) | 8.182 | 0.318 | 8.00E-23 |
| Colon adenocarcinoma—Stage IIIC versus Normal (adjacent normal) | 9.078 | 0.318 | 4.12E-13 |
| Colon adenocarcinoma—Stage IV versus Normal (adjacent normal) | 7.583 | 0.318 | 4.17E-29 |
| B. Stomach adenocarcinoma Subset Pair | |||
| Stomach adenocarcinoma—Stage IA versus Normal (adjacent normal) | 4.986 | 1.575 | 9.48E-04 |
| Stomach adenocarcinoma—Stage IB versus Normal (adjacent normal) | 5.815 | 1.575 | 5.90E-04 |
| Stomach adenocarcinoma—Stage II versus Normal (adjacent normal) | 4.430 | 1.575 | 5.02E-03 |
| Stomach adenocarcinoma—Stage IIA versus Normal (adjacent normal) | 5.615 | 1.575 | 1.79E-02 |
| Stomach adenocarcinoma—Stage IIB versus Normal (adjacent normal) | 5.809 | 1.575 | 7.91E-06 |
| Stomach adenocarcinoma—Stage IIIA versus Normal (adjacent normal) | 7.738 | 1.575 | 4.77E-08 |
| Stomach adenocarcinoma—Stage IIIB versus Normal (adjacent normal) | 7.857 | 1.575 | 2.56E-05 |
| Stomach adenocarcinoma—Stage IV versus Normal (adjacent normal) | 10.166 | 1.575 | 3.50E-06 |
mRNA–lncRNA coexpression network
The overexpression of PVT1 lncRNA is induced by MYC, and is involved in the suppression of apoptosis (33,34). PVT1 lncRNA and MYC protein expressions are correlated in primary human tumors, and the copy number of PVT1 was increased in more than 98% of cancers that also showed an increase of MYC copies (35). Figure 4 shows the mRNA–lncRNA coexpression network centered on PVT1 and MYC. The gene MYC has two isoforms, uc003ysh.1 and uc003ysi.2, and both of them are significantly overexpressed (adjusted P-values 1.25E-14 and 4.56E-17, respectively) in kidney renal clear cell carcinoma Stage I (TPM = 1.60 and 77.47, respectively) with respect to the adjacent normal subset (TPM = 0.60 and 28.20, respectively). PVT1 lncRNA also has two isoforms, uc003ysl.2 and uc010mdp.1, and both of them are significantly overexpressed (adjusted P-values < 1E-10) in the cancer subset (TPM = 1.33 and 0.55, respectively) with respect to adjacent normal tissues (TPM = 0.03 and 0.03, respectively). As shown in Figure 4, there is a significant positive correlation between MYC isoform uc003ysi.2 and PVT1 isoform uc010mdp.1.
Figure 4.

The coexpression network of the gene MYC and lncRNA PVT1 in the subset pair of kidney renal clear cell carcinoma: Stage I V.S. Adjacent normal. There is a significant positive correlation between MYC isoform uc003ysi.2 and PVT1 isoform uc010mdp.1. MYC has two isoforms, uc003ysh.1 and uc003ysi.2, and both of them are significantly overexpressed in the subset of kidney renal clear cell carcinoma: Stage I versus adjacent normal (adjusted P-values 1.25E-14 and 4.56E-17, respectively). PVT1 also has two isoforms, uc003ysl.2 and uc010mdp.1, and both of them are significantly overexpressed (adjusted P-values < 1E-10) in the cancer samples where MYC is also overexpressed.
DISCUSSION
We processed massive data sets collected from NCBI GEO and TCGA in order to obtain complete transcriptome profiles, extract the differentially expressed protein-coding transcripts and lncRNAs involved in various cancer types and infer the lncRNA regulatory network. Although CRN was developed with the goal of collecting, processing, analyzing and visualizing all publically available cancer RNA-seq data, it is still limited by the characteristics of these data. Firstly, different subsets from the same RNA-Seq data set have different sizes, which can influence the power of statistical analysis. Secondly, the cancer RNA-Seq data were generated using different experiment designs, sequencing platforms, cDNA library protocols, genetic backgrounds (cell lines or patients) and so on. It is a great challenge to integrate such diverse data resources. We have therefore applied the strategy of only comparing subsets from the same data set. Thirdly, some clinical information has not yet been integrated into CRN yet, such as treatments, pharmaceutical therapies and survival data.
As more and more cancer RNA-Seq data sets accumulate in the TCGA and NCBI GEO databases, we will update our CRN database to ensure that it can empower biologists with comprehensive cancer transcriptome knowledge.
FUNDING
National Institutes of Health (NIH) [NHLBI MAPGEN U01HL108634, NIGMS R01GM105431 to X.J.Z.]; Taiwan Ministry of Science and Technology [MOST 103-2320-B-005-004, MOST 104-2621-M-005-005-MY3, MOST 104-2320-B-005-005 to C.C.L.]; Taichung Veterans General Hospital [TCVGH-NCHU1047608 to C.C.L.]. Funding for open access charge: Taiwan Ministry of Science and Technology [MOST 104-2621-M-005-005-MY3].
Conflict of interest statement. None declared.
REFERENCES
- 1.Rhodes D.R., Kalyana-Sundaram S., Mahavisno V., Varambally R., Yu J., Briggs B.B., Barrette T.R., Anstet M.J., Kincead-Beal C., Kulkarni P., et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kupershmidt I., Su Q.J., Grewal A., Sundaresh S., Halperin I., Flynn J., Shekar M., Wang H., Park J., Cui W., et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One. 2010;5:e13066. doi: 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu F., White J.A., Antonescu C., Gusenleitner D., Quackenbush J. GCOD—GeneChip Oncology Database. BMC Bioinformatics. 2011;12:46. doi: 10.1186/1471-2105-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler D.L., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvernin V., Church D.M., Dicuccio M., Edgar R., Federhen S., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krupp M., Marquardt J.U., Sahin U., Galle P.R., Castle J., Teufel A. RNA-Seq Atlas–a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics. 2012;28:1184–1185. doi: 10.1093/bioinformatics/bts084. [DOI] [PubMed] [Google Scholar]
- 9.Tseng Y.T., Li W., Chen C.H., Zhang S., Chen J.J., Zhou X., Liu C.C. IIIDB: a database for isoform-isoform interactions and isoform network modules. BMC Genomics. 2015;16(Suppl 2):S10. doi: 10.1186/1471-2164-16-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W., Kang S., Liu C.C., Zhang S., Shi Y., Liu Y., Zhou X.J. High-resolution functional annotation of human transcriptome: predicting isoform functions by a novel multiple instance-based label propagation method. Nucleic Acids Res. 2014;42:e39. doi: 10.1093/nar/gkt1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalek A.K., Satija R., Shuga J., Trombetta J.J., Gennert D., Lu D., Chen P., Gertner R.S., Gaublomme J.T., Yosef N., et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts A., Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benjamini Y., Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 1995;57:289–300. [Google Scholar]
- 19.Liu C.C., Lin C.C., Chen W.S., Chen H.Y., Chang P.C., Chen J.J., Yang P.C. CRSD: a comprehensive web server for composite regulatory signature discovery. Nucleic Acids Res. 2006;34:W571–W577. doi: 10.1093/nar/gkl279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruan Q., Dutta D., Schwalbach M.S., Steele J.A., Fuhrman J.A., Sun F. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics. 2006;22:2532–2538. doi: 10.1093/bioinformatics/btl417. [DOI] [PubMed] [Google Scholar]
- 21.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mangiulli M., Valletti A., Caratozzolo M.F., Tullo A., Sbisa E., Pesole G., D'Erchia A.M. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37:6092–6104. doi: 10.1093/nar/gkp674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westfall M.D., Pietenpol J.A. p63: Molecular complexity in development and cancer. Carcinogenesis. 2004;25:857–864. doi: 10.1093/carcin/bgh148. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara R., Kawano S., Kiyosue T., Goto Y., Hirano M., Jinno T., Toyoshima T., Kitamura R., Oobu K., Nakamura S. Increased DeltaNp63 expression is predictive of malignant transformation in oral epithelial dysplasia and poor prognosis in oral squamous cell carcinoma. Int. J. Oncol. 2011;39:1391–1399. doi: 10.3892/ijo.2011.1151. [DOI] [PubMed] [Google Scholar]
- 25.Rocco J.W., Ellisen L.W. p63 and p73: life and death in squamous cell carcinoma. Cell Cycle. 2006;5:936–940. doi: 10.4161/cc.5.9.2716. [DOI] [PubMed] [Google Scholar]
- 26.Barbieri C.E., Tang L.J., Brown K.A., Pietenpol J.A. Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res. 2006;66:7589–7597. doi: 10.1158/0008-5472.CAN-06-2020. [DOI] [PubMed] [Google Scholar]
- 27.Bishop J.A., Teruya-Feldstein J., Westra W.H., Pelosi G., Travis W.D., Rekhtman N. p40 (DeltaNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod. Pathol. 2012;25:405–415. doi: 10.1038/modpathol.2011.173. [DOI] [PubMed] [Google Scholar]
- 28.Nobre A.R., Albergaria A., Schmitt F. p40: a p63 isoform useful for lung cancer diagnosis—a review of the physiological and pathological role of p63. Acta Cytol. 2013;57:1–8. doi: 10.1159/000345245. [DOI] [PubMed] [Google Scholar]
- 29.Nissan A., Stojadinovic A., Mitrani-Rosenbaum S., Halle D., Grinbaum R., Roistacher M., Bochem A., Dayanc B.E., Ritter G., Gomceli I., et al. Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. Int. J. Cancer. 2012;130:1598–1606. doi: 10.1002/ijc.26170. [DOI] [PubMed] [Google Scholar]
- 30.Alaiyan B., Ilyayev N., Stojadinovic A., Izadjoo M., Roistacher M., Pavlov V., Tzivin V., Halle D., Pan H., Trink B., et al. Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer. 2013;13:196. doi: 10.1186/1471-2407-13-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang F., Xue X., Bi J., Zheng L., Zhi K., Gu Y., Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J. Cancer Res. Clin. Oncol. 2013;139:437–445. doi: 10.1007/s00432-012-1324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizrahi I., Mazeh H., Grinbaum R., Beglaibter N., Wilschanski M., Pavlov V., Adileh M., Stojadinovic A., Avital I., Gure A.O., et al. Colon Cancer Associated Transcript-1 (CCAT1) Expression in Adenocarcinoma of the Stomach. J. Cancer. 2015;6:105–110. doi: 10.7150/jca.10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riquelme E., Suraokar M.B., Rodriguez J., Mino B., Lin H.Y., Rice D.C., Tsao A., Wistuba II. Frequent coamplification and cooperation between C-MYC and PVT1 oncogenes promote malignant pleural mesothelioma. J. Thorac. Oncol. 2014;9:998–1007. doi: 10.1097/JTO.0000000000000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chinen Y., Sakamoto N., Nagoshi H., Taki T., Maegawa S., Tatekawa S., Tsukamoto T., Mizutani S., Shimura Y., Yamamoto-Sugitani M., et al. 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J. Hematol. Oncol. 2014;7:68. doi: 10.1186/s13045-014-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng Y.Y., Moriarity B.S., Gong W., Akiyama R., Tiwari A., Kawakami H., Ronning P., Reuland B., Guenther K., Beadnell T.C., et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]