

Abstract
Gene expression is regulated by orchestrated binding of regulatory proteins to promoters and other cis-regulatory DNA elements (CREs). Several plant databases have been developed for mapping promoters or DNA motifs associated with promoters. However, there is a lack of databases that allow investigation for all CREs. Here we present PlantDHS (http://plantdhs.org), a plant DNase I hypersensitive site (DHS) database that integrates histone modification, RNA sequencing, nucleosome positioning/occupancy, transcription factor binding sites, and genomic sequence within an easily navigated user interface. DHSs are indicative of all CREs, including promoters, enhancers, silencers, insulators and transcription factor binding sites; all of which play immense roles in global gene expression regulation. PlantDHS provides a platform to predict all CREs associated with individual genes from three model plant species, including Arabidopsis thaliana, Brachypodium distachyon and rice (Oryza sativa). PlantDHS is especially valuable in the detection of distant CREs that are located away from promoters.
INTRODUCTION
DNase I hypersensitive sites (DHSs) are genomic regions that exhibit hypersensitivity to cleavage by DNase I endonucleases. These specific sites are typically inferred as open chromatin, which is accessible to regulatory proteins and thus marks cis-regulatory enriched regions in eukaryotic genomes. Chromatin accessibility can be examined by the extent of DNase I digestion, which was discovered over 30 years ago (1). Formerly, this approach involved titration of DNase I followed by characterization of digested DNAs by Southern blot hybridization (2). Recent advances in massively parallel sequencing technologies have enabled genome-wide mapping of DHSs, which can be achieved by partial DNase I digestion followed by sequencing (DNase-seq). Genome-wide DHS mapping has laid the foundations for the assembly of comprehensive catalogs of regulatory DNA sequences (3,4). This method has been particularly useful in identifying accessible cis-regulatory DNA elements (CREs), including promoters and enhancers of actively transcribed genes (4,5).
Recently, DHS maps have been developed in plant species, such as Arabidopsis thaliana and rice, by using this high throughput method (6–9). However, information about such DHSs is often trapped in the supplementary materials of a publication or is only accessible through the NCBI GEO database. To address this problem, we developed PlantDHS, a web interface/application of plant DHSs. In PlantDHS, we have introduced unified DHS IDs for plant species comprising several tissues. In addition, we have also integrated histone modification, RNA-seq, nucleosome positioning/occupancy, and transcription factor (TF) binding site data. Moreover, by applying modern web infrastructure that allows user browsing and searching, DHS-related information can be readily visualized.
DATABASE ARCHITECTURE
The PlantDHS incorporates A. thaliana, B. distachyon and rice genomes, three model plant species, with DHSs, histone modification data sets developed from chromatin immunoprecipitation followed by sequencing (ChIP-seq), nucleosome positioning and occupancy, RNA-seq, and transcription factor binding site data. All the DHSs and DHS scores were identified using an in-house developed software, Popera (https://github.com/forrestzhang/Popera). First, Popera identifies the DHSs by applying the kernel density estimation algorithm, which is similar to the algorithm defined by F-seq (10). Second, all the DHSs identified from various tissues and developmental stages were merged to create a unified DHS file; DHSs were then assigned unique ID tags. Lastly, normalized scores of unified DHSs were calculated for each unique tissue and developmental stage.
For RNA-seq data analysis, we used TopHat2 (11) and Cufflinks (12) for mapping RNA-seq reads and calculating gene expression levels (Fragment Per Kilobase of exon per million fragments Mapped, (FPKM)), respectively. Histone modification ChIP-seq data were mapped using Bowtie (13). We calculated the normalized ChIP-seq read counts within DHSs and ±300 bp flanking the DHSs. The DHS, gene expression and histone modification data sets were integrated using SQLAlchemy, which is an object-relational mapper (ORM) for the Python programming language. This allowed us to build a virtual object database and import the data to a MySQL database server.
The PlantDHS utilizes the Web Server Gateway Interface (WSGI), which facilitates an interaction between the server and the application, and is the most preferred interface for python-based web programming. For the construction of PlantDHS, we used HTML5 (http://www.w3.org/TR/html5/), CSS (http://www.w3.org/Style/CSS/), JavaScript, jQuery (https://jquery.com) and HighCharts (http://www.highcharts.com). The client side of the web application/interface is delivered via the apache2 HTTP server (http://httpd.apache.org) to provide modern web browsers on any platform, such as Mac OS, Windows, iOS, Android, Linux, etc. The server side was constructed using Flask (http://flask.pocoo.org), SQLAlchemy (http://www.sqlalchemy.org), MySQL (https://www.mysql.com) and JBrowse (14) on Ubuntu 14.04 LTS server (http://www.ubuntu.com).
CORE FUNCTIONALITY
The PlantDHS includes a total of five web pages within the navigational bar: Home, JBrowse, Genome, Download and Help (Figure 1A). The web application infrastructure provides a user-friendly interface that expedites exploring, visualizing and browsing DHSs information with relative ease. The Home page provides a quick search menu for immediate DHS browsing, basic help information and JBrowse quick links for species selection. JBrowse provides fast navigation, zooming and track selection functions for layering DHSs, TF binding sites and nucleosome positioning/occupancy features of the genome. The Genome page includes information regarding available data sets and a search menu for information about a specific species. Users can download the DHS data sets from the Download page. We supply GFF files, which contain the positional information of the DHSs. The CSV files contain DHS scores in different tissues and development states. The Help page contains links to the data sources, several demo/tutorial videos and information regarding website maintenance and version releases.
Figure 1.
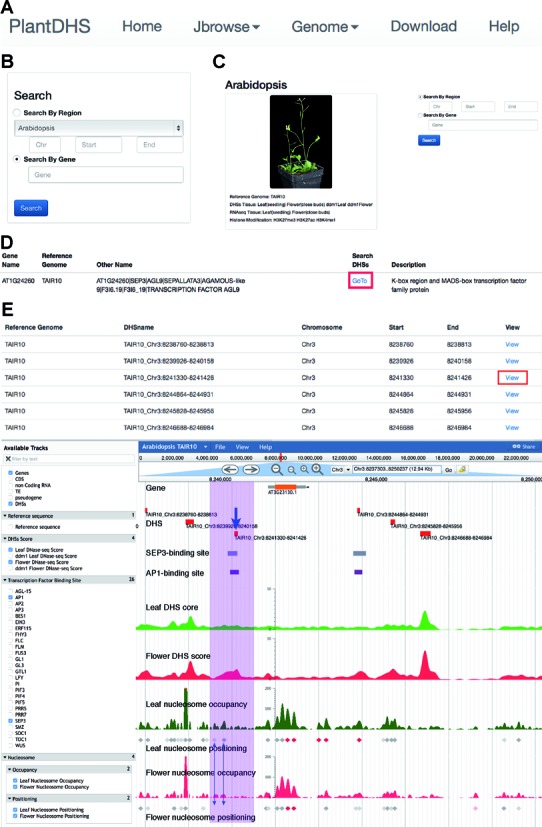
Structure of PlantDHS database. (A) Navigation bar of PlantDHS home page. (B) Quick search box for finding DHSs for a region or gene of interest. (C) Species-specific search engine (Genome Tab). (D) DHS search results and links to JBrowse pages (red box). This page shows the gene name, reference genome, gene name synonyms, if there are DHSs associated with the gene or region of interest, and a short description of the gene. (E) Details of region and/or gene of interest. Top panel represents the table of DHSs in the intersected region, and more specifically the coordinates of DHSs. The ‘View’ links take the user to the histone modification and RNA-seq information for each DHS, which is displayed in Figure 2. Bottom panel is the JBrowser view of the region of interest. The purple box highlights a DHS (blue arrow), a SEP3-binding site, a AP1-binding site, and two positioned nucleosomes that present in leaf tissue but miss in flower tissue (blue double arrows), which are all located upstream of the SUPERMAN gene (AT3G23130).
A major function of PlantDHS is mapping putative CREs that are likely binding sites of unknown regulatory proteins. PlantDHS allows for browsing and visualization of histone modification, gene expression and nucleosome positioning around possible regulatory factor binding sites. To begin investigating, users will start by entering a gene ID or by specifying genomic coordinates for the species of interest (Figure 1B and C). A successful gene ID search will yield a page displaying the gene name, reference genome, gene name synonyms and a description of the gene (Figure 1D). To retrieve DHS information for the target gene or region from this page, the user can click the ‘GoTo’ button under ‘Search DHSs’ (red box in Figure 1D). This action will display detailed DHSs information on JBrowse for target gene or region (Figure 1E); specifically, at the top of this page, the user can find a list of DHSs considered CREs associated with unknown regulators (Figure 1E). The bottom of this page shows the JBrowse window, which contains all listed DHSs (Figure 1E). Within the JBrowse page, users can integrate DHS genomic coordinates with DHS scores, transcription factor binding sites identified by ChIP-seq or ChIP-chip, and nucleosome positioning/occupancy information. Utilizing the ‘Available Tracks’ menu on the left hand side, the user can select multiple layers of available data to add or subtract to the JBrowse window.
For plotting DHS-related data such as DHS scores, histone modification and expression levels of the DHS-associated genes, users can either click ‘DHSs’ in the JBrowse window or the ‘View’ button in the DHSs list (red box in Figure 1E). There are three panels in the DHS-related information plot view page. The DHS score panel (Figure 2A) represents the mean and max score of the DHS in different tissues. The histone modification panel (Figure 2B) displays normalized scores for various histone modifications within the DHSs; where ‘flanking’ represents the normalized score of the histone modification ±300 bp flanking the DHS peak. The gene expression panel (Figure 2C) included RNA-seq-based expression data for all genes located within ±10 kb of the DHS's midpoint. By looking at these three plot panels, users can easily visualize DHS scores along with gene expression and histone modification data for different tissues or developmental stages simultaneously.
Figure 2.
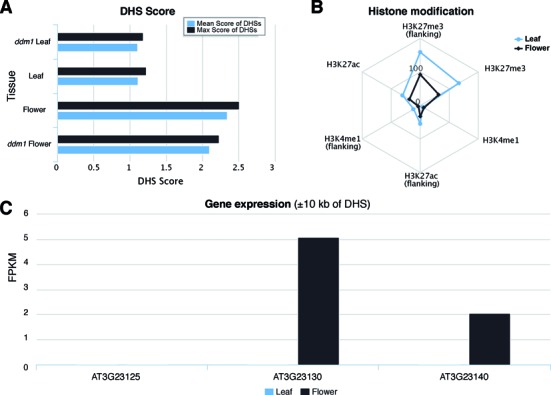
Histone modification, RNA-seq and DHS score information page. (A) Mean and max DHS scores in different tissues and developmental stages associated with a single DHS. (B) Normalized histone modification scores within and around (flanking) the DHS. (C) Expression levels of SUPERMAN (AT3G23130) and two nearby genes (within 10 kb of DHS) in flower and leaf tissues. All three genes are not expressed in leaf tissue.
EXAMPLE
To further demonstrate the utility of our database, we examined the collaborative effect of these data sets for the well-characterized floral regulator gene, SUPERMAN, in A. thaliana. A DHS (TAIR_chr3:8241330–8241426, indicated by a blue arrow in Figure 1E) upstream of the transcription start site (TSS) of SUPERMAN lies congruently with ChIP-predicted binding sites of floral MADS box transcription factors AP1 and SEP3. The DNase-seq (DHS) read depth at this position in the floral tissue was significantly higher compared to the leaf sample, suggesting that this particular DHS is specific to floral tissues (Figures 1E and 2A). In addition, there is a depletion of nucleosome occupancy at this site in the flower tissue compared with the leaf tissue, indicative of open chromatin and thereby increasing accessibility to transcription factors, and subsequent downstream consequences. Of the three genes within 10 kb surrounding TAIR10_Chr3:8241330–8241426, SUPERMAN is the most expressed gene in flower tissue, whereas none of the three genes are expressed in the leaf tissue (Figure 2C). Interestingly, the other florally expressed gene downstream of SUPERMAN is the hormone-induced developmental tissue regulator, UPRIGHT ROSETTE (15). Furthermore, there is an enrichment of the repressive H3K27me3 histone modification in leaf tissue, compared with flower tissue (Figure 2B). Several additional DHSs can be detected at both upstream and downstream of the SUPERMAN gene. Only one of these additional DHSs overlapped with predicted binding sites of AP1 and SEP3 (Figure 1E). These DHSs are potential CREs that may regulate SUPERMAN and/or its neighboring genes. Taken together, these results corroborate the TF binding site data and reveal the complex regulation for the SUPERMAN gene.
DATA SOURCE
TAIR10 (https://www.arabidopsis.org) (16), TIGR7 (http://rice.plantbiology.msu.edu) (17) and MIPS1.2 (http://www.brachypodium.org) (18) were used as the reference genomes of A. thaliana, rice and B. distachyon, respectively. We included a total of seven DHS libraries: Arabidopsis (Col-0) leaf, Arabidopsis (Col-0) flower, Arabidopsis (Col-0) ddm1 mutant (deficient in DNA methylation1) leaf, ddm1 mutant flower (6), rice (Nipponbare) seedling tissue, rice (Nipponbare) callus (7) and B. distachyon (BD21) seedling tissue. RNA-seq libraries included: Arabidopsis leaf, Arabidopsis flower (6), rice seedlings, rice callus (19) and B. distachyon leaf (20). Histone modification data sets contain: H3K27me3, H3K27ac and H3K4me1 from Arabidopsis leaf and flower tissues; and H3K36me3, H3K4me3, H3K9me2, H4K12ac, H3K9ac, H3K4me2 and H3K27me3 from rice leaf tissue (7,21). Histone modification was calculated in two distinct regions: (i) ±300 bp flanking the full DHS and (ii) exclusively within the DHS. The nucleosome positioning data includes: rice leaf, Arabidopsis leaf and Arabidopsis flower (22,23). Arabidopsis transcription factor data sets: AGL-15 (24), AP1 (25), AP3 (26), BES1 (27), EIN3 (28), ERF115 (29), FHY3 (30), FLC (31), FLM (32), FUS3 (33), GL1 (34), GL3 (34), GTL1 (35), LFY (36), PI (26), PIF3 (37), PIF4 (38), PIF5 (39), PRR5 (40), PRR7 (41), SEP3 (42), SMZ (43), SOC1 (44), TOC1 (45) and WUS (46). All Arabidopsis transcription factor binding site information was downloaded from http://bioinformatics.psb.ugent.be/cig_data/RegNet/ (47).
DISCUSSION AND FUTURE DIRECTIONS
Identification of CREs in plants has been mainly dependent on bioinformatic and computational predictions (48). Several algorithms and bioinformatic tools have been developed to identify CREs in plants. Most of these tools were established either exclusively based on analysis of DNA sequences from the upstream regions of genes (49,50), or based on identification of co-expressed genes in different tissues or/and under the same biotic or abiotic stress, followed by sequence/motif analysis of the presumed upstream regulatory regions of the co-expressed genes (48). These tools and the established databases have been valuable to the plant research community. However, since these prediction tools have mainly focused on promoter regions, the vast majority of other types of CREs, including enhancers, are missed in these predictions.
DNase I hypersensitivity is a universal mark for all active CREs (51). For example, more than 90% of the SEP3-binding and AP1-binding sites detected by ChIP-seq were covered by DHSs (6). Thus, DHSs provide corroborative information of TF-binding sites predicted based on the classical ChIP-seq method. PlantDHS provides a platform that allows predicting of all potential CREs associated with specific plant genes. The position of the promoter of a plant gene can be readily predicted based on various tools and databases developed by the plant research community. By contrast, plant enhancers have proved to be difficult to identify, which is due to the fact that enhancers can be located at various positions relative to a specific gene. DHSs located outside of the promoter regions are putative enhancers. We recently examined the function of several intergenic DHSs in A. thaliana using the β-glucuronidase gene reporter. Enhancer function was found to be associated with of more than 70% of these candidates (52). This result confirmed the power of mapping CREs using DHSs.
We plan to maintain and improve the PlantDHS by adding additional DHS data sets, including those from additional plant species and from model plant species grown under various stress conditions. Epigenomic data sets will also be added in the database. We are currently developing a genome-wide enhancer map in A. thaliana based largely on the DHS information (52). The enhancer information will be integrated into PlantDHS, which will be one of our near future goals.
FUNDING
National Science Foundation [MCB-1412948 to J.J.]. Funding for open access charge: National Science Foundation [MCB-1412948 to J.J.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Wu C., Wong Y.C., Elgin S.C. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell. 1979;16:807–814. doi: 10.1016/0092-8674(79)90096-5. [DOI] [PubMed] [Google Scholar]
- 2.Kodama Y., Nagaya S., Shinmyo A., Kato K. Mapping and characterization of DNase I hypersensitive sites in Arabidopsis chromatin. Plant Cell Physiol. 2007;48:459–470. doi: 10.1093/pcp/pcm017. [DOI] [PubMed] [Google Scholar]
- 3.Crawford G.E., Davis S., Scacheri P.C., Renaud G., Halawi M.J., Erdos M.R., Green R., Meltzer P.S., Wolfsberg T.G., Collins F.S. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W.L., Zhang T., Wu Y., Jiang J.M. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell. 2012;24:2719–2731. doi: 10.1105/tpc.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W.L., Wu Y., Schnable J.C., Zeng Z., Freeling M., Crawford G.E., Jiang J.M. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012;22:151–162. doi: 10.1101/gr.131342.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pajoro A., Madrigal P., Muino J.M., Matus J.T., Jin J., Mecchia M.A., Debernardi J.M., Palatnik J.F., Balazadeh S., Arif M., et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 2014;15:R41. doi: 10.1186/gb-2014-15-3-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan A.M., Arsovski A.A., Lempe J., Bubb K.L., Weirauch M.T., Sabo P.J., Sandstrom R., Thurman R.E., Neph S., Reynolds A.P., et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014;8:2015–2030. doi: 10.1016/j.celrep.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Boyle A.P., Guinney J., Crawford G.E., Furey T.S. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y., Yang Y., Yuan Z., Muller J.L., Yu C., Xu Y., Shao X., Li X., Decker E.L., Reski R., et al. Overexpression of the Arabidopsis gene UPRIGHT ROSETTE reveals a homeostatic control for indole-3-acetic acid. Plant Physiol. 2010;153:1311–1320. doi: 10.1104/pp.110.154021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamesch P., Berardini T.Z., Li D., Swarbreck D., Wilks C., Sasidharan R., Muller R., Dreher K., Alexander D.L., Garcia-Hernandez M., et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–D1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawahara Y., de la Bastide M., Hamilton J.P., Kanamori H., McCombie W.R., Ouyang S., Schwartz D.C., Tanaka T., Wu J.Z., Zhou S.G., et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y., Kikuchi S., Yan H., Zhang W., Rosenbaum H., Iniguez A.L., Jiang J.M. Euchromatic subdomains in rice centromeres are associated with genes and transcription. Plant Cell. 2011;23:4054–4064. doi: 10.1105/tpc.111.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davidson R.M., Gowda M., Moghe G., Lin H., Vaillancourt B., Shiu S.H., Jiang N., Robin Buell C. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012;71:492–502. doi: 10.1111/j.1365-313X.2012.05005.x. [DOI] [PubMed] [Google Scholar]
- 21.He G.M., Zhu X.P., Elling A.A., Chen L.B., Wang X.F., Guo L., Liang M.Z., He H., Zhang H.Y., Chen F.F., et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y.F., Zhang W.L., Jiang J.M. Genome-wide nucleosome positioning is orchestrated by genomic regions associated with DNase I hypersensitivity in rice. PLoS Genet. 2014;10:e1004378. doi: 10.1371/journal.pgen.1004378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T., Zhang W.L., Jiang J.M. Genome-wide nucleosome occupancy and positioning and their impact on gene expression and evolution in plants. Plant Physiol. 2015;168:1406–1416. doi: 10.1104/pp.15.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y.M., Ren N., Wang H., Stromberg A.J., Perry S.E. Global identification of targets of the Arabidopsis MADS domain protein AGAMOUS-Like15. Plant Cell. 2009;21:2563–2577. doi: 10.1105/tpc.109.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann K., Wellmer F., Muino J.M., Ferrier T., Wuest S.E., Kumar V., Serrano-Mislata A., Madueno F., Krajewski P., Meyerowitz E.M., et al. Orchestration of floral initiation by APETALA1. Science. 2010;328:85–89. doi: 10.1126/science.1185244. [DOI] [PubMed] [Google Scholar]
- 26.Wuest S.E., O'Maoileidigh D.S., Rae L., Kwasniewska K., Raganelli A., Hanczaryk K., Lohan A.J., Loftus B., Graciet E., Wellmer F. Molecular basis for the specification of floral organs by APETALA3 and PISTILLATA. Proc. Natl. Acad. Sci. U.S.A. 2012;109:13452–13457. doi: 10.1073/pnas.1207075109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X.F., Li L., Zola J., Aluru M., Ye H.X., Foudree A., Guo H.Q., Anderson S., Aluru S., Liu P., et al. A brassinosteroid transcriptional network revealed by genome-wide identification of BESI target genes in Arabidopsis thaliana. Plant J. 2011;65:634–646. doi: 10.1111/j.1365-313X.2010.04449.x. [DOI] [PubMed] [Google Scholar]
- 28.Chang K.N., Zhong S., Weirauch M.T., Hon G., Pelizzola M., Li H., Huang S.S.C., Schmitz R.J., Urich M.A., Kuo D., et al. Temporal transcriptional response to ethylene gas drives growth hormone cross-regulation in Arabidopsis. Elife. 2013;2:e00675. doi: 10.7554/eLife.00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heyman J., Cools T., Vandenbussche F., Heyndrickx K.S., Van Leene J., Vercauteren I., Vanderauwera S., Vandepoele K., De Jaeger G., Van der Straeten D., et al. ERF115 controls root quiescent center cell division and stem cell replenishment. Science. 2013;342:860–863. doi: 10.1126/science.1240667. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang X., Li J., Li G., Li B., Chen B., Shen H., Huang X., Mo X., Wan X., Lin R., et al. Genome-wide binding site analysis of FAR-RED ELONGATED HYPOCOTYL3 reveals its novel function in Arabidopsis development. Plant Cell. 2011;23:2514–2535. doi: 10.1105/tpc.111.085126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng W.W., Ying H., Helliwell C.A., Taylor J.M., Peacock W.J., Dennis E.S. Flowering Locus C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6680–6685. doi: 10.1073/pnas.1103175108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pose D., Verhage L., Ott F., Yant L., Mathieu J., Angenent G.C., Immink R.G.H., Schmid M. Temperature-dependent regulation of flowering by antagonistic FLM variants. Nature. 2013;503:414–417. doi: 10.1038/nature12633. [DOI] [PubMed] [Google Scholar]
- 33.Wang F.F., Perry S.E. Identification of Direct Targets of FUSCA3, a Key Regulator of Arabidopsis Seed Development. Plant Physiol. 2013;161:1251–1264. doi: 10.1104/pp.112.212282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morohashi K., Grotewold E. A systems approach reveals regulatory circuitry for Arabidopsis trichome initiation by the GL3 and GL1 selectors. Plos Genet. 2009;5:e1000396. doi: 10.1371/journal.pgen.1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breuer C., Morohashi K., Kawamura A., Takahashi N., Ishida T., Umeda M., Grotewold E., Sugimoto K. Transcriptional repression of the APC/C activator CCS52A1 promotes active termination of cell growth. EMBO J. 2012;31:4488–4501. doi: 10.1038/emboj.2012.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moyroud E., Minguet E.G., Ott F., Yant L., Pose D., Monniaux M., Blanchet S., Bastien O., Thevenon E., Weigel D., et al. Prediction of regulatory interactions from genome sequences using a biophysical model for the Arabidopsis LEAFY transcription factor. Plant Cell. 2011;23:1293–1306. doi: 10.1105/tpc.111.083329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Mayba O., Pfeiffer A., Shi H., Tepperman J.M., Speed T.P., Quail P.H. A quartet of PIF bHLH factors provides a transcriptionally centered signaling hub that regulates seedling morphogenesis through differential expression-patterning of shared target genes in Arabidopsis. Plos Genet. 2013;9:e1003244. doi: 10.1371/journal.pgen.1003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh E., Zhu J.Y., Wang Z.Y. Interaction between BZR1 and PIF4 integrates brassinosteroid and environmental responses. Nat. Cell Biol. 2012;14:802–809. doi: 10.1038/ncb2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornitschek P., Kohnen M.V., Lorrain S., Rougemont J., Ljung K., Lopez-Vidriero I., Franco-Zorrilla J.M., Solano R., Trevisan M., Pradervand S., et al. Phytochrome interacting factors 4 and 5 control seedling growth in changing light conditions by directly controlling auxin signaling. Plant J. 2012;71:699–711. doi: 10.1111/j.1365-313X.2012.05033.x. [DOI] [PubMed] [Google Scholar]
- 40.Nakamichi N., Kiba T., Kamioka M., Suzuki T., Yamashino T., Higashiyama T., Sakakibara H., Mizuno T. Transcriptional repressor PRR5 directly regulates clock-output pathways. Proc. Natl. Acad. Sci. U.S.A. 2012;109:17123–17128. doi: 10.1073/pnas.1205156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu T., Carlsson J., Takeuchi T., Newton L., Farre E.M. Direct regulation of abiotic responses by the Arabidopsis circadian clock component PRR7. Plant J. 2013;76:101–114. doi: 10.1111/tpj.12276. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann K., Muino J.M., Jauregui R., Airoldi C.A., Smaczniak C., Krajewski P., Angenent G.C. Target genes of the MADS transcription factor SEPALLATA3: Integration of developmental and hormonal pathways in the Arabidopsis flower. Plos Biol. 2009;7:854–875. doi: 10.1371/journal.pbio.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathieu J., Yant L.J., Murdter F., Kuttner F., Schmid M. Repression of flowering by the miR172 target SMZ. Plos Biol. 2009;7:e1000148. doi: 10.1371/journal.pbio.1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao Z., Shen L.S., Liu C., Liu L., Yan Y.Y., Yu H. Genome-wide identification of SOC1 and SVP targets during the floral transition in Arabidopsis. Plant J. 2012;70:549–561. doi: 10.1111/j.1365-313X.2012.04919.x. [DOI] [PubMed] [Google Scholar]
- 45.Huang W., Perez-Garcia P., Pokhilko A., Millar A.J., Antoshechkin I., Riechmann J.L., Mas P. Mapping the core of the Arabidopsis circadian clock defines the network structure of the oscillator. Science. 2012;336:75–79. doi: 10.1126/science.1219075. [DOI] [PubMed] [Google Scholar]
- 46.Busch W., Miotk A., Ariel F.D., Zhao Z., Forner J., Daum G., Suzaki T., Schuster C., Schultheiss S.J., Leibfried A., et al. Transcriptional control of a plant stem cell niche. Dev. Cell. 2010;18:849–861. doi: 10.1016/j.devcel.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 47.Heyndrickx K.S., Van de Velde J., Wang C., Weigel D., Vandepoele K. A functional and evolutionary perspective on transcription factor binding in Arabidopsis thaliana. Plant Cell. 2014;26:3894–3910. doi: 10.1105/tpc.114.130591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Priest H.D., Filichkin S.A., Mockler T.C. cis-Regulatory elements in plant cell signaling. Curr. Opin. Plant Biol. 2009;12:643–649. doi: 10.1016/j.pbi.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 49.Davuluri R.V., Sun H., Palaniswamy S.K., Matthews N., Molina C., Kurtz M., Grotewold E. AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics. 2003;4:25. doi: 10.1186/1471-2105-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yilmaz A., Mejia-Guerra M.K., Kurz K., Liang X.Y., Welch L., Grotewold E. AGRIS: the Arabidopsis gene regulatory Information server, an update. Nucleic Acids Res. 2011;39:D1118–D1122. doi: 10.1093/nar/gkq1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang J.M. The ‘dark matter’ in the plant genomes: non-coding and unannotated DNA sequences associated with open chromatin. Curr. Opin. Plant. Biol. 2015;24:17–23. doi: 10.1016/j.pbi.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 52.Zhu B., Zhang W.L., Zhang T., Liu B., Jiang J.M. Genome-wide prediction and validation of intergenic enhancers in Arabidopsis thaliana using open chromatin signature. Plant Cell. 2015 doi: 10.1105/tpc.15.00537. doi:10.1105/tpc.15.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]