

Significance
One missing or wrong nucleotide out of six billion in a human genome can cause a genetic disease. Detecting such a point mutation in a single human germ cell has been a daunting challenge in in vitro fertilization, yet one cannot afford to make any mistakes in selecting a viable embryo for transfer. Mutated allele revealed by sequencing with aneuploidy and linkage analyses (MARSALA) combines next-generation sequencing and single-cell whole-genome amplification methodologies, allowing embryo diagnosis with a single-molecule precision, significantly reducing false-positive or false-negative errors. MARSALA can benefit couples who desire to avoid transmitting their genetic diseases to their offspring.
Keywords: monogenic diseases, chromosome abnormality, IVF, PGD, MALBAC
Abstract
In vitro fertilization (IVF), preimplantation genetic diagnosis (PGD), and preimplantation genetic screening (PGS) help patients to select embryos free of monogenic diseases and aneuploidy (chromosome abnormality). Next-generation sequencing (NGS) methods, while experiencing a rapid cost reduction, have improved the precision of PGD/PGS. However, the precision of PGD has been limited by the false-positive and false-negative single-nucleotide variations (SNVs), which are not acceptable in IVF and can be circumvented by linkage analyses, such as short tandem repeats or karyomapping. It is noteworthy that existing methods of detecting SNV/copy number variation (CNV) and linkage analysis often require separate procedures for the same embryo. Here we report an NGS-based PGD/PGS procedure that can simultaneously detect a single-gene disorder and aneuploidy and is capable of linkage analysis in a cost-effective way. This method, called “mutated allele revealed by sequencing with aneuploidy and linkage analyses” (MARSALA), involves multiple annealing and looping-based amplification cycles (MALBAC) for single-cell whole-genome amplification. Aneuploidy is determined by CNVs, whereas SNVs associated with the monogenic diseases are detected by PCR amplification of the MALBAC product. The false-positive and -negative SNVs are avoided by an NGS-based linkage analysis. Two healthy babies, free of the monogenic diseases of their parents, were born after such embryo selection. The monogenic diseases originated from a single base mutation on the autosome and the X-chromosome of the disease-carrying father and mother, respectively.
There are about 7,000 known monogenic diseases; the genes for more than half of these diseases have been identified (1). Most can cause death, disability, or congenital malformation, bringing heavy burdens to both the affected families and to society’s health care system. In addition, chromosome abnormality, i.e., copy number variation (CNV) at particular chromosome locations, is a major cause of miscarriages and genetic disorders such as Down syndrome. The probability of aneuploidy rises drastically with maternal age, resulting in a decreased live-birth rate as women age.
Preimplantation genetic diagnosis (PGD) or preimplantation genetic screening (PGS) allows the selection of embryos free of single-gene disorders or aneuploidy (2), respectively. Previous PGD techniques include FISH to detect chromosome abnormalities (3) and Sanger sequencing after PCR to detect specific point mutations (4), but the two types of abnormality could not be detected at the same time. Genome-wide aneuploidy screening, such as comparative genomic hybridization (CGH) array (5, 6), SNP array (7), multiplex quantitative PCR (8), and next-generation sequencing (NGS), have been used to select embryos free of aneuploidy and are increasingly applied in the PGS field (5, 9). NGS offers many advantages, including reduced costs, increased precision, and higher base resolution (9–11). Recently NGS also has been used to detect monogenic diseases or de novo mutations (12, 13).
Because of the small amount of genetic material, genomic analyses of a single or a few cells require whole-genome amplification (WGA). There are three commercially available WGA methods, which recently have been compared (14): degenerate oligomer primer PCR (DOP-PCR) (15), multiple displacement amplification (MDA) (16), and multiple annealing and looping-based amplification cycles (MALBAC) (17). Unfortunately, any WGA method exhibits false-positive and/or false-negative single-nucleotide variations (SNVs), which could lead to wrong selection of embryos for in vitro fertilization (IVF). Eliminating these errors is a major challenge for PGD. Linkage analysis has become a standard method of circumventing this problem (18, 19) by detecting short tandem repeats (STR) or by karyomapping with an SNP array (20–24) to determine the disease allele.
Several groups have reported the combined use of two or three of these methods in the same embryo to increase precision in the selection of embryos (25–27). Konstantinidis et al. (23) reported a combined procedure of detecting chromosomal abnormality and linkage analysis using the karyomapping method. These reports proved that single-gene disorders and chromosome abnormalities can be detected simultaneously. However, separate procedures had to be used for each embryo (27). Moreover, the targeted mutations still could not be observed directly in these non-NGS methods (23).
Here we present, for the first time to our knowledge, an integrated NGS-based PGD/PGS procedure with simultaneous and direct detection of disease-causing mutations, chromosome abnormalities, and linkage analyses; this procedure has led to the live births of two baby girls free of their parents’ monogenic diseases.
Results and Discussion
Mutated Allele Revealed by Sequencing with Aneuploidy and Linkage Analyses.
We describe a novel NGS-based method, mutated allele revealed by sequencing with aneuploidy and linkage analyses (MARSALA), in which PGD, PGS, and linkage analyses of individual embryos can be carried out in a single procedure. As shown in Fig. 1, a few trophectoderm cells are biopsied from a blastocyst on day 5 and are amplified by WGA with MALBAC. Then the WGA product is reamplified by PCR primers at the targeted SNVs, and the product is mixed with the WGA product. This mixture is sequenced by NGS at a low sequencing depth (0.1–2×), which is sufficient for measuring CNVs accurately and for call-targeted SNVs in the same run by virtue of the preamplification (>1,000×).
Fig. 1.

Workflow of MARSALA. See text for a description of the workflow.
To circumvent the false-positive and false-negative SNV errors and to increase the certainty of allele identification, MARSALA relies on a large number of SNP markers for the disease allele, rather than on one uncertain marker; this approach is similar in principle to those of STR and karyomapping analyses and can be used if the genetic disorder comes from either parent. Alternatively, if the genetic disorder comes primarily from the female, we also can analyze the two polar bodies of each zygote on day 1 to deduce whether the embryo is viable for transfer (10, 28).
By NGS analyses of SNVs adjacent to a targeted mutation in a single cell (or in a few cells), MASALA is able to avoid false-positive and false-negative errors. Hence MARSALA allows simultaneous detection of both the disease-causing point mutation and the chromosome abnormality via a single low-depth NGS sequencing procedure.
Family History and Blastocyst Biopsy.
Our first case involves a couple of maternal age 34 y and paternal age 32 y. The husband has a family history of an autosomal dominant disorder and has suffered from hereditary multiple exostoses (HME), which is characterized by multiple bony spurs or lumps in the bones, from an early age. He previously underwent a series of operations to remove the exostoses. More recently, an exostosis developed on his spinal canal. His father, two paternal aunts, and one of his cousins are HME patients with the same symptoms (Fig. 2A). Genetic diagnosis of the husband showed a frame-shift point mutation c.233delC at the exostosin glycosyltransferase 2 (EXT2) gene, already known to cause this disease (29).
Fig. 2.

MARSALA analyses of blastocyst sequencing for case 1 in which the husband was affected by HME. (A) Pedigrees of the couple’s family with HME. Filled symbols represent affected individuals; open symbols represent unaffected individuals. Circles and squares indicate females and males, respectively. The arrow indicates the affected parent. Individuals indicated by asterisks were verified by Sanger sequencing. (B) Results of sequencing the targeted mutation site in the EXT2 gene of the husband (H), the wife (W), and their 18 embryos (E1–E18). Embryos E05, 06, 09, 10, 11, 12, and 16 had the disease-carrying mutated allele (red) and were excluded from transfer. The fraction of reads of the covered region shown in green is consistent with the reference genome. (C) CNVs of the 18 embryos at the low sequencing depth (0.1×) of NGS. Significant chromosomal abnormality was identified in embryos E06 (monosomy 18, deletion in part of chromosome 6), E07 (monosomy 22), E08 (monosomy 2, 6), E09 (trisomy 12), E12 (monosomy 8), E13 (trisomy 8, 15, deletion or duplication in part of chromosomes 9, 10, and 18), E14 (deletion or duplication in part of chromosome 3), E16 (deletion in part of chromosome 4), and E18 (trisomy 22); hence these embryos were not suitable for transfer. (D) Schematic representation of MARSALA for disease-carrying alleles. We sequenced the amplified genomes from each embryo, from the couple, and from the husband’s father. The gene loci heterozygous for the husband (e.g., A/G) but homozygous for the husband’s father (e.g., A/A) and the wife (e.g., A/A) are shown within a range of 1 Mb upstream or downstream from the mutated site. These SNPs are sufficient for determining whether an embryo carries the disease or the normal allele from the husband (see below). Even if the SNV is not detected directly in the low-coverage sequencing, the disease-carrying allele still can be identified with certainty. The purple “X*” indicates the deletion of a single base in the mutated allele (frame-shift mutation). The purple “C” indicates the base associated with the monogenic disease in the wild-type allele. “A” and “G” indicate the SNVs linked to the mutated allele. (E) Results of linkage analyses of the 18 embryos. Ten SNV markers were selected to identify the disease-carrying allele in each embryo. These 10 markers identified seven embryos (E05, E06, E09, E10, E11, E12, and E16) as carrying the disease-associated allele with high certainty. ALT, the SNPs of alternative allele; CHROM, chromosome number; FH, husband’s father; H, husband; POS, genomic location; REF, the SNPs of reference allele; RSID, reference SNP cluster ID; W, wife. - represents the alleles that are not covered by single-cell low-depth sequencing.
This couple underwent IVF and PGD treatment. Thirty-two metaphase-II oocytes were collected and fertilized by intracytoplasmic sperm injection (ICSI). Eighteen embryos developed to the blastocyst stage, and a cluster of trophectoderm (TE) cells was biopsied for PGD. The DNA of all biopsies was amplified successfully by MALBAC.
In our second case, the wife carried an X-linked chromosome recessive heredity disorder. The couple (maternal age, 33 y; paternal age, 32 y) already had an affected son who suffers from X-linked hypohidrotic ectodermal dysplasia characterized by hair, sweat gland, and teeth abnormalities (30). Genetic diagnosis of the son showed a point mutation c.T1085G at the ectodysplasin A1 (EDA1) gene, inherited from his mother (Fig. 3A). The SNV on the EDA gene has been known to be associated with this disorder. Five embryos developed to the blastocyst stage on day 6, and their TE cells were biopsied also. The TE cells of embryo E05 was not biopsied or amplified successfully.
Fig. 3.

MARSALA analyses of blastocyst sequencing for case 2 in which the wife carried a disease causing SNV in the EDA1 gene. (A) Pedigrees of the couple’s family with the EDA1 point mutation. Filled symbols represent affected individuals; open symbols represent unaffected individuals. Circles and squares indicate females and males, respectively. The arrow indicates the affected proband. Individuals indicated by asterisks were verified by Sanger sequencing. (B) Result of sequencing the targeted mutation site in the EDA1 gene of the husband (H), wife (W), and affected child (C) and in the four embryos. Embryos E01 and E03 had the disease-carrying mutated allele (red) and were excluded from transfer. The fraction of reads of the covered region shown in green is consistent with the reference genome. (C) CNVs of the four embryos at low sequencing depth (0.1×) of NGS. Significant chromosomal abnormality was identified in embryo E03 (monosomy 4), which therefore was not suitable for transfer. (D) Schematic representation of MARSALA for disease-carrying alleles. We sequenced the amplified genomes from each embryo and the bulk genomes of the couple and their affected son. The gene loci heterozygous for the wife (e.g., T/G) are shown within the range of 3 Mb upstream or downstream from the disease-causing mutation site. These SNPs are sufficient for determining whether an embryo carries the mutated or the normal allele from the wife. Even if the SNV is not detected in the low-coverage sequencing, the disease-carrying allele still can be identified with certainty. The purple “G*” indicates the mutated allele (a single base substitution). The purple “C” indicates the wild-type allele. “A,” “G,” and “T” indicate the SNVs linked to the disease-associated allele. (E) Result of linkage analyses of the blastocyst-stage embryos. Ten SNV markers were selected to identify the disease-carrying allele in each embryo. These 10 markers identified two embryos (E01 and E03) as carrying the disease-associated allele with high certainty. ALT, the SNPs of alternative allele; CHROM, chromosome number; H, husband; POS, genomic location; REF, the SNPs of reference allele; RSID, reference SNP cluster ID; S, affected son; W, wife. - represents the alleles that are not covered by single-cell low-depth sequencing.
Simultaneous SNV and CNV Detection by MARSALA.
The mutated region of a target gene was reamplified with a pair of specific primers, and then the PCR products were mixed with the WGA product for NGS. In this way, the existence of the point mutation and aneuploidy can be detected in one NGS run, as shown in Fig. 2. Fig. 2 B and C show that the region of interest can be sequenced to ultra-high coverage (>1,000×) while still maintaining accurate CNV measurement throughout the whole genome. In doing so, a significant portion of the allele dropout caused by allele amplification imbalance can be overcome. This method is applicable to almost all monogenic diseases with known mutations, even when multiple sites are mutated in the alleles from one or both parents.
The NGS data for the first case with a 0.1× genome coverage for each embryo are shown in Table S1. The number of the total reads covering the disease mutation site among the 18 TE samples varied from 4,421 to 134,373 (Table S1). Seven embryos were identified as being carriers of the mutated SNV (Fig. 2B). The CNV data of all chromosomes for each embryo are presented in Fig. 2C; nine of them have aneuploidy.
Table S1.
High-throughput MARSALA sequencing data for case 1
| Sample | Raw data | Clean data | Mapped ratio, % | Coverage ratio, % | Coverage depth, × | PCR coverage, number of reads (range) | Mutation coverage, number of reads | |||
| Reads, M | Bases, G | Reads, M | Bases, G | Ratio, % | ||||||
| Husband | 2.37 | 0.36 | 2.36 | 0.33 | 99.8 | 90.8 | 7.2 | 0.12 | 93,565 (89,880–179,880) | 45,626 |
| Wife | 2.26 | 0.34 | 2.26 | 0.32 | 99.8 | 93.4 | 6.9 | 0.11 | 90,747 (87,057–174,597) | 88,343 |
| Embryo 1 | 57.03 | 5.76 | 47.18 | 4.14 | 71.8 | 74.5 | 44.0 | 1.08 | 78,724 (67,338–84,378) | 78,986 |
| Embryo 2 | 1.95 | 0.30 | 1.78 | 0.24 | 80.7 | 86.7 | 4.9 | 0.08 | 4,988 (4,631–9,510) | 4,807 |
| Embryo 3 | 1.88 | 0.28 | 1.71 | 0.23 | 79.4 | 87.9 | 4.1 | 0.07 | 138,376 (132,250–267,006) | 134,373 |
| Embryo 4 | 2.37 | 0.36 | 2.19 | 0.29 | 81.7 | 88.2 | 5.2 | 0.10 | 98,989 (94,862–190,982) | 96,087 |
| Embryo 5 | 61.95 | 6.26 | 51.54 | 4.53 | 72.3 | 80.6 | 48.6 | 1.28 | 101,268 (89,705–108,092) | 53,175 |
| Embryo 6 | 2.17 | 0.33 | 2.05 | 0.28 | 85.4 | 88.4 | 4.8 | 0.09 | 113,555 (108,469–217,346) | 68,570 |
| Embryo 7 | 69.80 | 7.05 | 56.76 | 5.04 | 71.5 | 80.0 | 44.7 | 1.41 | 73,399 (63,270–78,426) | 73,907 |
| Embryo 8 | 1.92 | 0.29 | 1.78 | 0.24 | 81.2 | 86.4 | 4.3 | 0.08 | 85,301 (81,715–164,500) | 126,922 |
| Embryo 9 | 2.00 | 0.30 | 1.84 | 0.25 | 81.1 | 82.3 | 4.5 | 0.08 | 112,544 (107,858–214,737) | 103,694 |
| Embryo 10 | 1.54 | 0.23 | 1.48 | 0.22 | 82.4 | 87.9 | 4.0 | 0.06 | 74,050 (70,783–141,802) | 60,681 |
| Embryo 11 | 64.19 | 6.48 | 54.35 | 4.81 | 74.3 | 79.1 | 48.0 | 1.33 | 76,183 (70,044–80,863) | 11,244 |
| Embryo 12 | 62.81 | 6.34 | 50.81 | 4.51 | 71.1 | 74.8 | 39.5 | 1.18 | 86,191 (75,850–91,089) | 70,031 |
| Embryo 13 | 53.78 | 5.43 | 46.37 | 4.12 | 75.8 | 83.1 | 45.8 | 1.20 | 95,731 (81,121–102,536) | 96,351 |
| Embryo 14 | 2.13 | 0.32 | 1.89 | 0.25 | 76.0 | 85.5 | 5.0 | 0.08 | 72,325 (68,956–139,359) | 70,067 |
| Embryo 15 | 63.30 | 6.39 | 54.85 | 4.95 | 77.4 | 85.0 | 48.9 | 1.47 | 93,407 (80,412–99,998) | 94,364 |
| Embryo 16 | 2.18 | 0.33 | 1.82 | 0.23 | 69.6 | 87.7 | 5.0 | 0.08 | 11,348 (10,444–20,997) | 4,421 |
| Embryo 17 | 2.35 | 0.36 | 2.09 | 0.27 | 76.6 | 83.9 | 5.3 | 0.09 | 101,793 (97,353–195,010) | 98,741 |
| Embryo 18 | 1.63 | 0.25 | 1.47 | 0.19 | 78.3 | 87.2 | 4.0 | 0.06 | 67,031 (61,240–128,853) | 64,964 |
For the second case, the number of the total reads covering the disease mutation among the polar body and TE samples varied from 316,944 to 753,555 (Table S2). Two embryos, E01 and E03, were identified as having the mutated SNV (Fig. 3B). Embryo E03 also was found to have chromosome abnormality (Fig. 3C).
Table S2.
High-throughput MARSALA sequencing data for case 2
| Sample | Raw data | Clean data | Mapped ratio, % | Coverage ratio, % | Coverage depth, × | PCR coverage, number of reads (range) | Mutation coverage, number of reads | |||
| Reads, M | Bases, G | Reads, M | Bases, G | Ratio, % | ||||||
| Husband | 41.89 | 4.23 | 41.02 | 3.73 | 88.13 | 81.71 | 31.55 | 0.96 | 1,985,807 (1,018,825–3,244,252) | 2,164,021 |
| Wife | 44.58 | 4.5 | 43.68 | 3.98 | 88.43 | 90.26 | 34.89 | 1.13 | 814,906 (386,658–1,281,107) | 899,346 |
| Affected child | 52.68 | 5.32 | 51.64 | 4.7 | 88.33 | 89.17 | 37.72 | 1.3 | 733,151 (385,406–1,116,409) | 810,748 |
| Embryo 1 | 53.37 | 5.39 | 50.96 | 4.39 | 81.53 | 89.93 | 29.64 | 1.27 | 451,500 (217,331–748,674) | 492,943 |
| Embryo 2 | 45.6 | 4.61 | 44.46 | 3.88 | 84.29 | 92.12 | 30.83 | 1.15 | 389,042 (193,080–639,323) | 424,858 |
| Embryo 3 | 40.9 | 4.13 | 38.72 | 3.34 | 80.77 | 88.79 | 22.1 | 0.95 | 288,556 (141,402–481,496) | 316,944 |
| Embryo 4 | 47.3 | 4.78 | 44.8 | 3.87 | 81.01 | 86.33 | 24.15 | 1.05 | 399,507 (173,666–644,369) | 441,110 |
| PB1 E1 | 51.02 | 5.15 | 49 | 4.25 | 82.51 | 91.44 | 27.96 | 1.25 | 556,207 (274,427–915,859) | 607,222 |
| PB1 E2 | 56.65 | 5.72 | 55.34 | 4.84 | 84.67 | 92.85 | 32.87 | 1.45 | 461,956 (248,913–739,343) | 504,376 |
| PB1 E3 | 44.53 | 4.5 | 43.48 | 3.83 | 85.13 | 92.76 | 28.94 | 1.15 | 410,505 (213,272–665,295) | 448,109 |
| PB1 E4 | 44.54 | 4.5 | 42.26 | 3.68 | 81.8 | 86.88 | 26.58 | 1 | 460,226 (205,293–735,709) | 508,065 |
| PB1 E5 | 55.98 | 5.65 | 54.56 | 4.8 | 84.86 | 92.79 | 31.98 | 1.44 | 584,035 (293,514–956,384) | 637,222 |
| PB2 E1 | 46.99 | 4.75 | 45.37 | 4 | 84.28 | 87.31 | 27.26 | 1.09 | 485,146 (198,801–792,917) | 535,596 |
| PB2 E2 | 44.52 | 4.5 | 42.64 | 3.75 | 83.43 | 87.62 | 24.55 | 1.04 | 546,635 (208,545–907,638 ) | 603,549 |
| PB2 E3 | 47.03 | 4.75 | 45.28 | 4.03 | 84.75 | 87.11 | 25.76 | 1.11 | 682,078 (266,726–1,128,088) | 753,555 |
| PB2 E4 | 51.04 | 5.15 | 49.56 | 4.37 | 84.71 | 91.97 | 28.49 | 1.3 | 442,102 (213,730–731,660) | 483,077 |
| PB2 E5 | 47.39 | 4.79 | 45.78 | 4.05 | 84.59 | 88.62 | 26.62 | 1.13 | 590,904 (261,178–948,621) | 652,306 |
Linkage Analyses by MARSALA.
In the first case, we sequenced the blood samples of the couple and of the husband’s disease-carrying father. We called all SNPs with sequencing depth >10× within 1 Mb of the mutated site (X/C) and focused on the heterozygous SNPs of the husband (for example, the first row, A/G in Fig. 2D), homozygous SNPs of the wife (A/A), and homozygous SNPs of the husband’s disease-carrying father (A/A). We know the husband’s allele with base G must come from his (normal) mother, because his disease-carrying father is A/A homozygous; hence, the husband’s disease allele must be the allele with the base A. We used a similar strategy to deduce the inherited allele of the embryos under screening. For example, at the first SNP position (Fig. 2E, first row), embryo E05 is identified as homozygous A/A; thus one of its alleles (A) must be derived from the husband’s disease-carrying father (A), and therefore embryo E05 should not be transferred.
The MARSALA results at the 10 selected genomic positions are listed in Fig. 2E and are highly concordant in all 18 embryos, as is consistent with the PCR results of the MALBAC products (Fig. 2B). Multiple SNPs close to the target mutation can increase the accuracy of identifying the mutated allele significantly, even if the direct mutation calling is unsuccessful. Accordingly, we applied this strategy to all 18 embryos; seven were identified as affected, and the remaining 11 were identified as unaffected (Table S3). Of the embryos that were neither affected nor aneuploid, embryo E04 was selected for transfer.
Table S3.
Summary of PGS/PGD results in case 1 and case 2
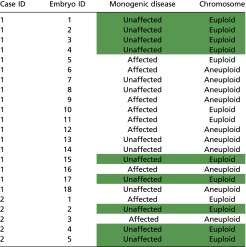 |
Normal embryos with no disease-associated allele or aneuploidy are shown in green background and were established as being viable for embryo transfer.
For the second case, to carry out the linkage analysis of each embryo, we sequenced the genomes of the blood samples of the wife, husband, and disease-carrying son. We called all SNPs of the three individuals with a sequencing depth >10× within 3 Mb of the EDA1 gene and focused on the heterozygous SNPs in the X chromosome of the wife and the SNPs in the X chromosomes of the affected son and unaffected husband (Fig. 3D). As in case 1, we can deduce whether the disease-carrying allele from the mother is present in each embryo (Fig. 3D). The MARSALA results at the 10 selected genomic positions, listed in Fig. 3E, are highly concordant in all four embryos and are consistent with the PCR results of the MALBAC-amplified DNA (Fig. 3B).
The results for all embryos in case 2 are summarized in Table S3. After excluding all affected and aneuploid embryos, we selected embryo E02 for transfer.
When using MALBAC for single-cell WGA, we were able to achieve linkage analyses with only 2× sequencing depth for each embryo as well as a 2× depth for the parents and a disease-carrying relative. This low sequencing depth is adequate because the sequence-dependent bias of MALBAC is highly reproducible from cell to cell, allowing some genomic regions to be covered at ∼10× depth at the 2× average depth. In contrast, the 2× sequencing depth is not enough for MARSALA using MDA for WGA.
Comparison of PGD/PGS Results of Blastocyst and Polar Body Biopsies.
We previously have demonstrated the proof of principle that when a single-gene disorder and aneuploidy are maternal, it is possible to determine whether an embryo is viable for transfer by sequencing the polar bodies, PB1 and PB2 (10). Polar body biopsy is less invasive than blastocyst biopsy and can offer more time for diagnosis when fresh embryo transfer is desired, but the cost of its genetic analysis is higher. Because the monogenic disease in the second case is of maternal origin, we performed MARSALA in a polar body biopsy to compare that with the blastocyst biopsy.
Each pair of PBs (PB1 and PB2) from the five embryos was biopsied for single-cell sequencing by NGS, allowing us to determine the SNVs and aneuploidy of each female pronucleus (10, 28). If the haploid PB2 and diploid PB1 together have a total of two copies of the disease-causing SNV, then the corresponding female pronucleus is unaffected. Conversely, detecting one copy of the targeted mutation in PB2 but none in PB1 of embryo E03 (Fig. 4A) indicated that E03 is affected.
Fig. 4.

MARSALA analyses of polar body sequencing for case 2. (A) Result of sequencing the targeted mutation site in the EDA1 gene using polar body biopsies of two embryos, E03 and E05. The deduction from E03 polar body SNVs is consistent with the affected SNV identified in the blastocyst biopsy of E30. However, the result of the polar body biopsy in E05 indicates that there is no affected SNV in this embryo; this information could not be obtained from the E05 blastocyst, because the WGA of the E05 biopsy failed. (B) CNV patterns of PB1 and PB2 of embryo E03 at the low sequencing depth (0.1×) of NGS, from which the CNV of the female pronucleus is deduced to be normal. In contrast, the directly measured CNV pattern of the E03 blastocyst biopsy showed an abnormal chromosome 4 (compare Figs. 4B and 3C). This result can be explained by the aneuploidy of the sperm, consistent with chromosome 4 of this blastocyst exhibiting only the maternal rather than the paternal SNVs. (C) Schematic representation of MARSALA for linkage analysis to identify the existence of disease-carrying alleles in the female pronucleus to avoid false-positive and false-negative SNVs in determining the presence of the disease allele in the female pronucleus.
The results obtained from the analyses of PB1 and PB2 are consistent with those obtained from the blastocyst biopsy, with the exception that the blastocyst WGA of embryo E05 contained errors. In this case, polar body biopsy provided an accurate alternative for determining the status of the female pronucleus. The deduced CNV profile of the female pronucleus of embryo E05 in Fig. 4B shows that this embryo is normal.
We note that polar body analysis can detect only maternal disorders, not paternal disorders or errors in embryo development. Surprisingly, the blastocyst biopsy analysis identified chromosomal abnormalities in embryo E03 (Fig. 3C). The copy number of Chr4 was one instead of two. However, the results of polar body biopsy sequencing showed a normal copy number of Chr4 in this same embryo (Fig. 4B). We hypothesized that this discrepancy is caused by the aneuploidy of the sperm cell, with ∼5% probability (31). Indeed this notion was verified by Chr4 of this blastocyst exhibiting only the maternal rather than the paternal SNPs (Table S4).
Table S4.
SNP genotypes of chromosome 4 in mother, father, and embryo 3
| SNP ID | Position | Mother | Father | Embryo 3* |
| rs11931543 | chr4: 527,176 | C | G | C |
| rs11938226 | chr4: 527,419 | C | T | C |
| rs11931692 | chr4: 527,534 | A | G | A |
| rs13149933 | chr4: 528,289 | C | A | C |
| rs61334630 | chr4: 528,403 | C | T | C |
| rs3888807 | chr4: 641,421 | T | C | T |
| rs77049343 | chr4: 1,194,586 | A | T | A |
| rs56401136 | chr4: 1,194,630 | T | C | T |
| rs2293633 | chr4: 1,291,640 | C | T | C |
| rs73075548 | chr4: 1,334,260 | C | G | C |
| rs13130121 | chr4: 1,653,858 | T | A | T |
| rs10031530 | chr4: 3,468,031 | T | C | T |
| rs4364330 | chr4: 6,135,908 | G | C | G |
| rs79680227 | chr4: 7,368,200 | C | T | C |
| rs79755463 | chr4: 7,374,122 | T | C | T |
| rs4044946 | chr4: 7,374,179 | A | G | A |
| rs3965270 | chr4: 7,374,203 | T | C | T |
| rs4689109 | chr4: 7,374,220 | A | G | A |
| rs7691124 | chr4: 7,418,330 | C | T | C |
| rs4696711 | chr4: 8,654,761 | G | C | G |
| rs11724246 | chr4: 8,658,448 | C | T | C |
| rs35730276 | chr4: 8,658,642 | A | G | A |
| rs145027448 | chr4: 8,791,132 | T | G | T |
| rs4245955 | chr4: 8,912,354 | T | C | T |
| rs933212 | chr4: 8,912,813 | G | C | G |
| rs4697968 | chr4: 10,198,628 | C | T | C |
| rs6448124 | chr4: 10,603,360 | G | T | G |
| rs10022255 | chr4: 12,976,777 | T | C | T |
| rs1547578 | chr4: 24,888,984 | T | G | T |
| rs1030351 | chr4: 37,771,023 | T | A | T |
| rs28464959 | chr4: 38,324,347 | G | C | G |
| rs68115994 | chr4: 38,750,200 | G | T | G |
| rs73234989 | chr4: 38,754,888 | G | A | G |
| rs10938524 | chr4: 48,183,646 | T | C | T |
| rs61795729 | chr4: 49,290,033 | A | G | A |
| rs11133288 | chr4: 54,630,659 | C | T | C |
| rs2899015 | chr4: 55,747,697 | T | C | T |
| rs6811163 | chr4: 56,027,577 | A | T | A |
Mother and embryo have the same genotypes in all selected SNPs, indicating that the chromosome 4 in embryo 3 was from the mother and the chromosome 4 from the father was missing.
In principle, linkage analysis also can be carried out with the genomes of the wife and her disease-carrying son to avoid false positives and false negatives in SNV detection (Fig. 4C).
Confirmation by Sanger Sequencing, CGH Array, and STR Analyses.
To validate our MARSALA method further, we subjected our targeted PCR fragment from the MALBAC products of the 18 trophectoderm biopsies to Sanger sequencing using the primers of EXT2 c.233delC listed in Table S5. Six of the 18 embryos (E05, E06, E10, E11, E12, and E16) carrying the paternal mutation were confirmed by Sanger sequencing (Figs. S1A and S2). Notably, the high-throughput sequencing results using MARSALA were more accurate than Sanger sequencing because no deletion on EXT2 c.233 was found in embryo E09 by the latter method, likely because of allelic dropout (Fig. S2).
Table S5.
Primers for the detection of point mutations in EXT2, EDA1, and STR linkage analysis
| Forward primer | Reverse primer | Size, bp |
| GGGAGTATAATGAACTGCTCATGG | GGCTGTATGAGTGTGAGATACCTA | 175 |
| 6FAM-GGACAGATGCTTCCAGAAAA | AGATTATGCATGTGTAAAGAGCC | 224–245 |
| 6FAM-AAATCCCTCTGGCTGC | AGTTCTTGAGCCCACCC | 166–206 |
| CACTGACTTTGCCAGCTATGA | GTGTGCTTGCTCATGTTGATG | 147 |
Fig. S1.

Confirmation of MARSALA results in case 1 with Sanger sequencing, STR linkage analysis, and CGH array. The DNA templates for these analyses were also from the products of MALBAC amplification from the same biopsy. (A) Sanger sequencing results of two representative embryos, E04 and E06, showed that embryo E06 carried the mutated allele whereas embryo E04 was normal, free of the mutated allele. Sanger sequencing results of the other 16 embryos are shown in Fig. S2. These results were all consistent with MARSALA, except for embryo E09, likely because of the lower sensitivity of the Sanger sequencing method. (B) Array CGH verification of chromosome abnormalities in some embryos. The data showed that embryo E12 was monosomy 8, E14 had deletion or duplication in part of chromosome 3, and E18 was trisomy 22; these findings are consistent with our low-depth sequencing results. (C) STR linkage analysis showed that embryos E05, E06, E09, E10, E11, E12, and E16 had the mutant allele; this finding is consistent with our MARSALA results.
Fig. S2.

Sanger sequencing results of the remaining 16 embryos for detecting paternal frameshift c.del233C in the EXT2 gene.
DNA from the same MALBAC products of embryos E02, E03, E04, E12, E14, E17, and E18 were analyzed by CGH array. The results obtained were exactly the same as the results obtained from the low-coverage MALBAC-NGS (Fig. S1B).
The two D11S1993 alleles from the father were distinguished by two STRs of different sizes. A 229-bp STR was linked with the normal EXT2 allele, and a 235-bp STR was linked with the mutated allele associated with HME. As for the D11S4103 allele, the 195-bp STR was linked with the normal allele, and the 197-bp STR was linked with the mutated allele. Thus, based on STR genotyping, seven embryos (E05, E06, E09, E10, E11, E12, and E16) were identified as carrying the mutated allele of EXT2; these findings were consistent with our NGS results (Fig. S1C).
Fetus Validation.
Ultrasound examination on day 30 after embryo transfer revealed a single intrauterine gestational sac with normal fetal heart beat for the two IVF cases reported here. In the first case, to verify the absence of the mutated EXT2 allele and chromosomal normality, a prenatal diagnosis was conducted with Sanger sequencing, SNP array, and karyotype analysis using amniotic fluid cells at 17-wk gestation. For the second case, to verify the absence of the mutated EDA1 allele as well as chromosomal normality, a prenatal diagnosis was conducted with Sanger sequencing, SNP array, and karyotype analysis using amniotic fluid cells at 19-wk gestation. A baby girl from the case 1 couple was born on September 19, 2014. A baby girl from the case 2 couple was born on November 30, 2014. Both babies were confirmed to be healthy by physical examination by a senior pediatrician. The umbilical cord blood of each baby was collected for further validation, and the babies were again confirmed to be free of the EXT2 paternal mutation and EDA1 maternal mutation, respectively (Fig. S3).
Fig. S3.

Validations of the fetus and newborn baby for case 1. (A) Sanger sequencing of the amniotic fluid cells and umbilical cord blood confirmed that the baby was free of the mutated allele of the EXT2 gene. (B and C) Karyotype analysis (B) and SNP array (C) for the detection of aneuploidy showed that the newborn did not exhibit chromosome abnormalities.
Cost of MARSALA.
Identifying a point mutation normally requires a 30× sequencing depth, which is costly. However, the detection of aneuploidy requires only 0.1× sequencing depth data (0.3G data), at a current cost of ∼$30 on the Illumina HiSEq. 2500 platform. By adding specific primers for the targeted genes, MARSALA avoids the need for high sequencing depths and allows simultaneous detection of aneuploidy and specific point mutations at a 0.1× sequencing depth, thereby substantially decreasing the cost. In our experience, the linkage analyses using MALBAC requires only a 2× sequencing depth. Overall, therefore, MARSALA is cost effective.
Limitations of MARSALA.
MARSALA is not capable of detecting de novo mutations. To use MARSALA, prior knowledge of a single gene associated with the disorder from either parent is needed. In fact, the genome sequences of the parents, which are available from carrier testing, are prerequisites for MARSALA. The use of specific primers for MARSALA might be considered a limitation. Although not costly, they could be time consuming. However, the primer sequences can be determined even before the PGD procedure based on the parents’ genomes and therefore should not add extra time to the overall procedure.
We note that the primed amplification of MALBAC products can be omitted because it serves only as an additional validation for the detectable SNVs, which could not be read directly in the previous non-NGS–based STR analyses and karyomapping.
In the absence of an affected relative of the parents, an affected embryo identified by direct calling of the target SNV can be used as an equivalent of a proband for linkage analyses. Alternatively, sequencing a few cells of a sperm or PB2 can be used in order to phase the genome of the husband or wife, respectively (10, 31).
Conclusions
First and foremost, MARSALA allows the simultaneous direct observation of aneuploidy, targeted mutation sites, and their linked SNPs. Compared with previous methods of detecting point mutations such as FISH and PCR, MARSALA significantly reduces the false-positive and -negative SNVs by virtue of linkage analyses. NGS-based MARSALA offers more accurate CNV measurements than previous methods of detecting aneuploidy, such as array CGH and SNP array. Unlike previous linkage analyses such as STR and karyomapping methods, MARSALA provides direct visualization of the mutation sites, avoiding misidentification resulting from loss of linkage because of homologous recombination. MARSALA has improved the precision of PGD and PGS markedly and has streamlined the PGD/PGS procedure.
Second, MARSALA is cost effective compared with microarray-based methods. In principle, MARSALA can be carried out with any WGA methods. We chose MALBAC because it has the lowest allelic dropout (false-negative rate for SNV) and the highest precision for CNV detection (14). A 2× sequencing depth is sufficient when MALBAC is used for linkage analyses.
We have demonstrated that normal embryos can be selected by a one-step NGS procedure, MARSALA, to circumvent both monogenic diseases and chromosomal abnormalities with high precision. MARSALA works for both autosome and X-linked diseases and for male and female carriers. The first two neonates resulting from such embryo selection for a male and a female, patient each carrying a monogenic disease, were born successfully.
Materials and Methods
Preimplantation verification by standard methods and prenatal and postnatal confirmation are discussed in SI Materials and Methods.
Blastocyst Biopsy.
We adapted the widely used approach of embryo biopsy at the blastocyst stage (10), collecting a few TE cells from each hatching blastocyst on day 5 or day 6. All embryos were obtained from patients who chose to be subjected to an IVF procedure and voluntarily gave their consent for providing the samples for these studies. This study was carefully reviewed and approved by the Reproductive Study Ethics Committee at Peking University Third Hospital (research license 2014SZ001) and the Harvard University Committee on the Use of Human Subjects.
Single-Cell WGA with MALBAC.
WGA of lysed cells was performed using the MALBAC method (17) following the standard protocol provided by the commercial MALBAC amplification kit (Yikon Genomics Inc.). Briefly, the cell was lysed by heating (3 h at 50 °C and 10 min at 80 °C) in 5 μL of lysis buffer. Then 30 μL of freshly prepared preamplification mix was added to each tube and was incubated at 94 °C for 3 min. Then DNA was amplified using eight cycles of 40 s at 20 °C, 40 s at 30 °C, 30 s at 40 °C, 30 s at 50 °C, 30 s at 60 °C, 4 min at 70 °C, 20 s at 95 °C, and 10 s at 58 °C and was placed on ice immediately. We then added 30 μL of the amplification reaction mix to each tube and incubated at 94 °C for 30 s followed by 17 cycles of 20 s at 94 °C, 30 s at 58 °C, and 3 min at 72 °C. For blood samples, we extracted the gDNA by using the QIAamp DNA Blood Mini Kit (Qiagen Inc.) and amplified 1 ng gDNA using the MALBAC kit (Yikon Genomics Inc.). For comparison with the MDA method, we used REPLI-g Single Cell kit (Qiagen, Inc.) to amplify the two embryos in case 1. According to a recent review, MALBAC offers the highest accuracy for CNV detection and the lowest allele dropout rate, therefore producing fewer false negatives (14).
MARSALA Simultaneously Detects Aneuploidy and a Targeted Mutation.
Whole-genome sequencing of MALBAC products can identify chromosome abnormalities efficiently at low sequencing depths (<0.1×). However, precise detection of inherited point mutations would require a higher sequencing depth (>10×), which significantly increases the cost. We developed a simple and inexpensive method to detect single mutations by amplifying the MALBAC products using PCR primers specific to the mutated allele (Table S5). To do so, we incubated 8 ng of the MALBAC product from a single cell, using PCR primers specific to the mutated allele, at 98 °C for 30 s, 32 cycles of 15 s at 98 °C, 30 s at 60 °C, and 30 s at 72 °C, and an additional 2 min at 72 °C. This PCR product was mixed with the MALBAC product (1–5% of MALBAC product) and was used to construct a sequencing library using the NEBNext Ultra DNA library Prep kit (New England Biolabs, Inc.). The sequencing was done on the Illumina HiSEq. 2500 platform. Following this procedure, targeted point mutations and aneuploidy can be detected simultaneously in one NGS run with only a 0.1× sequencing depth, although 2× was used for linkage analysis (see below).
Linkage Analyses with MARSALA.
To identify homozygous SNP positions adjacent to the target heterozygous point mutation of the patient, we sequenced the genomes of the parents, as well as the genome from a relative carrying the monogenic disease allele. Then, from the sequences of each embryo (at blastocyst stage or the first and second polar bodies), the SNP readouts (heterozygous or homozygous) at these adjacent positions allowed the identification of the disease-carrying allele in the embryo (Figs. 2D, 3D, and 4C). We noted that this method worked well and reproducibly with only a 2× sequencing depth for each MALBAC-amplified DNA sample involved, reducing the high cost of multiple sequencing rounds. Potential background noise decreased to <5% in samples that had two rounds of MALBAC (Fig. 3B).
Comparison of Blastocyst and Polar Body Biopsies.
In our second case, which involves a mutation in the EDA1 gene, the monogenic disease is maternal. For this female patient, in addition to analyzing biopsied cells at the blastocyst stage, we also biopsied the first (PB1) and second (PB2) polar bodies at 1 and 9 h after ICSI (10) to compare these results with those obtained from the blastocyst biopsies. The biopsied PBs were transferred to the lysis buffer as described in Hou et al. (10) for WGA and NGS, followed by MARSALA analyses using the genomes of the couple and their affected son.
SI Materials and Methods
Preimplantation Verification by Standard Methods.
Detection of targeted point mutations by Sanger sequencing, aneuploidy by CGH array or low-depth NGS, and linkage analyses by STR or karyomapping have become standard methods for IVF evaluation. To compare our novel one-step NGS-based MARSALA method with these standard methods, we designed specific PCR primers for the two cases of patients carrying genetic disorders. The PCR products were subjected to Sanger sequencing (Table S5). For CNV detection, the DNA from the purified MALBAC amplification product was processed for CGH array analysis on 24sure-plus chips (BlueGenome). Two STR markers (D11S1993 and D11S4103) within ∼1 Mb of the EXT2 gene and two STR markers (DXS8111 and DXS8107) within ∼1 Mb of the EDA1 gene (Table S5) were used to confirm the linkage analysis results of the mutant alleles.
Prenatal and Postnatal Confirmation.
In all cases, the amniotic fluid cells were divided into three equal parts, one for the detection of mutation sites, another for karyotype analysis, and the third for SNP array screening. Each sample was compared with the results obtained before implantation.
Acknowledgments
We thank Dr. P. Purcell (Harvard University) and Dr. C. Racowsky (Brigham & Women’s Hospital) for critical reading and editing of the manuscript, the staff at the Department of Obstetrics and Gynecology in Peking University Third Hospital for assistance during the patient’s pregnancy, and the staff of the BIOPIC sequencing facility at Peking University, particularly Y. Zhang, for assistance. This work was supported by National Natural Science of China Grants 31230047, 31322037, 31440063, and 31522034 (to J.Q., F.T., and L.Y.), National High Technology Research and Development Program Grant 2015AA020407 (to L.Y. and L.H.), Beijing Municipal Science and Technology Commission Grants Z131100005213006 and D151100002415000 (to J.Q., F.T., and X.S.X.), a Peking University Grant for Translational Research (to X.S.X., J.Q., and F.T.), and National Basic Research Program of China Grants 2011CB944500 and 2012CB966704 (to J.Q. and F.T.).
Footnotes
Conflict of interest statement: S.L. and X.S.X. are cofounders and shareholders of Yikon Genomics.
Data deposition: The sequencing data reported in this paper is deposited in the NCBI Sequence Read Archive database (accession no. SRP067387).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1523297113/-/DCSupplemental.
References
- 1.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat Rev Genet. 2013;14(10):681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 2.Yan L, et al. Advances in preimplantation genetic diagnosis/screening. Sci China Life Sci. 2014;57(7):665–671. doi: 10.1007/s11427-014-4683-5. [DOI] [PubMed] [Google Scholar]
- 3.Rubio C, et al. Use of array comparative genomic hybridization (array-CGH) for embryo assessment: Clinical results. Fertil Steril. 2013;99(4):1044–1048. doi: 10.1016/j.fertnstert.2013.01.094. [DOI] [PubMed] [Google Scholar]
- 4.Handyside AH, Kontogianni EH, Hardy K, Winston RM. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 1990;344(6268):768–770. doi: 10.1038/344768a0. [DOI] [PubMed] [Google Scholar]
- 5.Yin X, et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol Reprod. 2013;88(3):69. doi: 10.1095/biolreprod.112.106211. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, et al. Validation of copy number variation sequencing for detecting chromosome imbalances in human preimplantation embryos. Biol Reprod. 2014;91(2):37. doi: 10.1095/biolreprod.114.120576. [DOI] [PubMed] [Google Scholar]
- 7.Tobler KJ, et al. Two different microarray technologies for preimplantation genetic diagnosis and screening, due to reciprocal translocation imbalances, demonstrate equivalent euploidy and clinical pregnancy rates. J Assist Reprod Genet. 2014;31(7):843–850. doi: 10.1007/s10815-014-0230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treff NR, et al. Development and validation of an accurate quantitative real-time polymerase chain reaction-based assay for human blastocyst comprehensive chromosomal aneuploidy screening. Fertil Steril. 2012;97(4):819–824. doi: 10.1016/j.fertnstert.2012.01.115. [DOI] [PubMed] [Google Scholar]
- 9.Wells D, et al. Clinical utilisation of a rapid low-pass whole genome sequencing technique for the diagnosis of aneuploidy in human embryos prior to implantation. J Med Genet. 2014;51(8):553–562. doi: 10.1136/jmedgenet-2014-102497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou Y, et al. Genome analyses of single human oocytes. Cell. 2013;155(7):1492–1506. doi: 10.1016/j.cell.2013.11.040. [DOI] [PubMed] [Google Scholar]
- 11.Fiorentino F. Molecular genetic analysis of single cells. Semin Reprod Med. 2012;30(4):267–282. doi: 10.1055/s-0032-1313906. [DOI] [PubMed] [Google Scholar]
- 12.Treff NR, et al. Evaluation of targeted next-generation sequencing-based preimplantation genetic diagnosis of monogenic disease. Fertil Steril. 2013;99(5):1377–1384.e6. doi: 10.1016/j.fertnstert.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 13.Peters BA, et al. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res. 2015;25(3):426–434. doi: 10.1101/gr.181255.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang L, Ma F, Chapman A, Lu S, Xie XS. Single-cell whole-genome amplification and sequencing: Methodology and applications. Annu Rev Genomics Hum Genet. 2015;16:79–102. doi: 10.1146/annurev-genom-090413-025352. [DOI] [PubMed] [Google Scholar]
- 15.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99(8):5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dreesen J, et al. Evaluation of PCR-based preimplantation genetic diagnosis applied to monogenic diseases: A collaborative ESHRE PGD consortium study. Eur J Hum Genet. 2014;22(8):1012–1018. doi: 10.1038/ejhg.2013.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilton L, Thornhill A, Traeger-Synodinos J, Sermon KD, Harper JC. The causes of misdiagnosis and adverse outcomes in PGD. Hum Reprod. 2009;24(5):1221–1228. doi: 10.1093/humrep/den488. [DOI] [PubMed] [Google Scholar]
- 20.Thornhill AR, et al. Karyomapping-a comprehensive means of simultaneous monogenic and cytogenetic PGD: Comparison with standard approaches in real time for Marfan syndrome. J Assist Reprod Genet. 2015;32(3):347–356. doi: 10.1007/s10815-014-0405-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Natesan SA, et al. Live birth after PGD with confirmation by a comprehensive approach (karyomapping) for simultaneous detection of monogenic and chromosomal disorders. Reprod Biomed Online. 2014;29(5):600–605. doi: 10.1016/j.rbmo.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Natesan SA, et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet Med. 2014;16(11):838–845. doi: 10.1038/gim.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konstantinidis M, et al. Live births following Karyomapping of human blastocysts: Experience from clinical application of the method. Reprod Biomed Online. 2015;31(3):394–403. doi: 10.1016/j.rbmo.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Handyside AH. Live births following karyomapping - a “key” milestone in the development of preimplantation genetic diagnosis. Reprod Biomed Online. 2015;31(3):307–308. doi: 10.1016/j.rbmo.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Shen J, et al. Successful PGD for late infantile neuronal ceroid lipofuscinosis achieved by combined chromosome and TPP1 gene analysis. Reprod Biomed Online. 2013;27(2):176–183. doi: 10.1016/j.rbmo.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Daina G, et al. First successful double-factor PGD for Lynch syndrome: Monogenic analysis and comprehensive aneuploidy screening. Clin Genet. 2013;84(1):70–73. doi: 10.1111/cge.12025. [DOI] [PubMed] [Google Scholar]
- 27.Rechitsky S, et al. First systematic experience of preimplantation genetic diagnosis for single-gene disorders, and/or preimplantation human leukocyte antigen typing, combined with 24-chromosome aneuploidy testing. Fertil Steril. 2015;103(2):503–512. doi: 10.1016/j.fertnstert.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Kuliev A, Rechitsky S. Polar body-based preimplantation genetic diagnosis for Mendelian disorders. Mol Hum Reprod. 2011;17(5):275–285. doi: 10.1093/molehr/gar012. [DOI] [PubMed] [Google Scholar]
- 29.Seki H, et al. Mutation frequencies of EXT1 and EXT2 in 43 Japanese families with hereditary multiple exostoses. Am J Med Genet. 2001;99(1):59–62. doi: 10.1002/1096-8628(20010215)99:1<59::aid-ajmg1115>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 30.Monreal AW, Zonana J, Ferguson B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet. 1998;63(2):380–389. doi: 10.1086/301984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu S, et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science. 2012;338(6114):1627–1630. doi: 10.1126/science.1229112. [DOI] [PMC free article] [PubMed] [Google Scholar]