

Significance
Cells encode several effective antiviral proteins, yet these are sometimes not expressed in infected cells. Using a synthetic transcriptional activator based on the bacterial RNA-guided DNA binding protein Cas9, we demonstrate the efficient induction of two antiviral effectors, APOBEC3G (A3G) and APOBEC3B (A3B), in human cells that normally express neither protein. A3G is susceptible to degradation by the HIV-1 Vif protein, whereas A3B is resistant to Vif. As a result, only the induced A3B inhibited wild-type HIV-1 infectivity. However, both induced factors blocked the replication of a Vif-deficient HIV-1 mutant. These data demonstrate that Cas9-derived transcription factors can effectively induce human genes that regulate virus replication, thus setting the scene for their use in genomic screens to identify such factors.
Keywords: APOBEC3 proteins, restriction factors, CRISPR/Cas, synthetic transcription factors, HIV-1
Abstract
Whereas several mammalian proteins can restrict the replication of HIV-1 and other viruses, these are often not expressed in relevant target cells. A potential method to inhibit viral replication might therefore be to use synthetic transcription factors to induce restriction factor expression. In particular, mutants of the RNA-guided DNA binding protein Cas9 that have lost their DNA cleavage activity could be used to recruit transcription activation domains to specific promoters. However, initial experiments revealed only weak activation unless multiple promoter-specific single guide RNAs (sgRNAs) were used. Recently, the recruitment of multiple transcription activation domains by a single sgRNA, modified to contain MS2-derived stem loops that recruit fusion proteins consisting of the MS2 coat protein linked to transcription activation domains, was reported to induce otherwise silent cellular genes. Here, we demonstrate that such “synergistic activation mediators” can induce the expression of two restriction factors, APOBEC3G (A3G) and APOBEC3B (A3B), in human cells that normally lack these proteins. We observed modest activation of endogenous A3G or A3B expression using single sgRNAs but high expression when two sgRNAs were used. Whereas the induced A3G and A3B proteins both blocked infection by an HIV-1 variant lacking a functional vif gene by inducing extensive dC-to-dU editing, only the induced A3B protein inhibited wild-type HIV-1. These data demonstrate that Cas9-derived transcriptional activators have the potential to be used for screens for endogenous genes that affect virus replication and raise the possibility that synthetic transcription factors might prove clinically useful if efficient delivery mechanisms could be developed.
The clustered, regularly interspaced, short palindromic repeats (CRISPR)-associated (Cas) system initially evolved in bacteria as an adaptive immune response that can block infection by DNA bacteriophages or the uptake of plasmids (1, 2). The effector arm of type II CRISPR/Cas systems relies on a single protein, called Cas9, that is guided to specific DNA targets by two small RNA molecules called the transactivating CRISPR RNA (tracrRNA) and CRISPR RNA (crRNA) (3). These two RNAs can be fused to generate a single guide RNA (sgRNA), which contains the sequences required for Cas9 binding as well as a short sequence of ∼20 nt that is complementary to the target DNA (4, 5). Once bound to the target DNA, Cas9 introduces a double-stranded DNA break that, in bacteria, results in loss of the target DNA (1, 2). Importantly, this ∼20-nt sequence can be readily changed and combinations of Cas9 bound to artificial sgRNAs have also proven very effective at cleaving specific DNA targets in eukaryotic cells, including human cells (3). These cleavages can result in gene inactivation, due to repair by error prone nonhomologous end joining, or can be used to introduce specific sequence changes into the human genome, if an appropriate DNA sequence is provided in trans to promote homologous recombination.
Whereas the CRISPR/Cas system therefore initially evolved, and is also primarily used, as a tool for introducing specific sequence changes into the genome, Cas9 also has the potential to be used as a vehicle to deliver functional modules to specific sites on the genome. In particular, mutational inactivation of the two independent DNA cleavage modules in Cas9 gives rise to a protein called dCas9 that will bind with high specificity to a genomic DNA target that is complementary to the sgRNA used but is no longer capable of cleaving the bound DNA (6, 7). One potential application of the dCas9 protein would be to specifically activate individual human genes by fusing dCas9 to a potent transactivation domain, such as that found on VP64 (8–11). However, initial efforts to activate specific genes using this approach proved relatively ineffective unless sgRNAs targeting multiple sites within a given promoter were used, thus indicating that efficient activation of a given promoter would require the development of improved methods to recruit multiple, ideally synergistic, transcription activation domains to a promoter.
To address this problem, Konermann et al. (12) devised a method to recruit multiple proteins to a DNA sequence using a single sgRNA. They replaced the two terminal loops present in the sgRNA, which do not contribute to either Cas9 or DNA binding, with two minimal hairpins that selectively bind the dimeric MS2 bacteriophage coat protein. The MS2 coat protein was then expressed in the form of a fusion protein consisting of MS2 linked to two potent transcription activation domains derived from the NF-κB subunit p65 and from heat shock factor 1 (HSF1). A combination of plasmids expressing a dCas9/VP64 fusion protein, the MS2-p65-HSF1 fusion protein, and the sgRNA(MS2) single guide RNA fusion is therefore predicted to recruit nine diverse transcription activation domains to each sgRNA target DNA and, indeed, Konermann et al. (12) reported potent activation of several human genes when this complex, which they term a “synergistic activation mediator” (SAM), was recruited to DNA targets located within 200 base pairs (bp) of the transcription start site.
From the virological perspective, these newly developed molecular tools have the potential to open up a number of avenues of research. For example, dCas9-based programmable SAMs could be used to globally investigate the effects of individual human gene products on viral replication or latency. As proof of principle, we here present data showing that the recruitment of SAMs to either the apolipoprotein B mRNA-editing enzyme catalytic polypeptide (APOBEC) APOBEC3B (A3B) or APOBEC3G (A3G) promoter in human cells that normally do not express either restriction factor (13, 14) results in a dramatic and specific induction in either A3B or A3G expression. Moreover, we observed that the cells expressing induced endogenous A3G or A3B became unable to produce infectious particles of a Vif-deficient (ΔVif) HIV-1 provirus and gave rise to a characteristic pattern of C-to-T mutations in the defective proviruses produced upon viral infection (15–17). As expected, wild-type HIV-1, expressing the Vif protein, induced the degradation of the induced A3G, but not A3B, protein and was able to overcome the induced restriction in cells expressing the former, but not the latter, protein (13, 18–21). These data demonstrate a simple method for inducing the expression of endogenous human proteins that can either facilitate or hinder viral replication. Importantly, this dCas9-based method of transcriptional activation of endogenous genes via their normal promoter element is potentially superior to simply transfecting cells with cDNA expression plasmids as it allows all potential isoforms of a given protein to be expressed simultaneously. Moreover, this method is adaptable at a genome scale to search for factors that affect virus replication (12).
Results
To determine whether recruitment of dCas9-based programmable SAMs (12) to the promoter element of an antiviral restriction factor would result in a level of expression that would convert a normally permissive cell (i.e., able to replicate HIV-1ΔVif) into a nonpermissive cell, we focused on two members of the APOBEC3 family of restriction factors. A3G is normally expressed in primary T cells and macrophages and potently restricts the replication of HIV-1 ΔVif (19–21). In the absence of Vif, A3G is packaged into newly produced virions where it edits dC residues to dU residues on the newly synthesized minus DNA strand produced during reverse transcription, resulting in the production of fewer DNA proviruses and extensive mutagenesis of those that are produced (16, 17). However, in the presence of Vif, A3G is degraded and HIV-1 infectivity is unaffected (19–23). Like A3G, A3B is also an effective inhibitor of HIV-1 replication and acts via a very similar mechanism, though the sequence context of edited dC residues differs between A3G and A3B, being 5′-CC*-3′ in the former case and 5′-TC*-3′ in the latter (13, 18). However, A3B differs from A3G in that it is not expressed in primary T cells or macrophages (24, 25) and A3B expression is, in fact, normally restricted to pluripotent cell types, such as embryonic stem cells, where it functions as an effective inhibitor of retrotransposon mobility (26, 27). Importantly, perhaps because A3B is not normally expressed in the cells that are infected by HIV-1 in vivo, A3B is also refractory to inhibition by Vif and therefore inhibits wild-type HIV-1 and HIV-1ΔVif equivalently (13, 18).
To identify sgRNA target sequences that would permit efficient activation of the endogenous A3G promoter, but not the A3B promoter, by a dCas9-derived SAM, and vice versa, we aligned the A3G and A3B promoter regions and selected two sgRNA sequences specific for each promoter sequence for analysis (Fig. 1). Binding of the Streptococcus pyogenes (Spy) Cas9 protein to a given DNA sequence is mediated by an invariant sequence, called the protospacer adjacent motif (PAM), which is directly bound by the Spy Cas9 protein and has the sequence 5′-XGG-3′ (3). In addition, binding requires essentially complete complementarity to the “seed” region of the sgRNA, which is the 3′ ∼13 nt of the target complementary region of the sgRNA (28). We therefore inspected the A3G and A3B promoter sequences (29, 30) for targets within the −200 to −1 promoter sequence (where +1 is the transcription start site) for potential Spy Cas9 binding sites that were present in one promoter but potentially mutationally inactivated in the second promoter. In the case of the A3G promoter, we identified two potential target sites for Spy Cas9, located at −91 to −69 and at −46 to −24, that were lacking in the A3B promoter due to loss of the PAM sequence (Fig. 1). In the case of the A3B promoter, we noted two target sites, between −161 and −139 and between −124 to −102, which bore either six or two point mutations in the sgRNA seed in the same region of the A3G promoter (Fig. 1). To test these for activity and specificity, we cloned the relevant segments of the A3G or A3B promoter upstream of a minimal TATA box linked to the firefly luciferase (FLuc) indicator gene and then cotransfected each of the indicators, together with plasmids expressing the cognate sgRNA(MS2) guide, as well as the dCas9-VP64 and the MS2-p65-HSF1 fusion proteins (12), into HeLa cells. As shown in Fig. 2A, both A3G promoter-specific guides, called sgG1 and sgG2, activated the A3G promoter by >1,000-fold but did not affect the A3B promoter. Similarly, the A3B-specific sgRNA sgB1 activated the A3B promoter by >100-fold but did not affect the A3G promoter (Fig. 2B). Finally, the A3B-specific sgB2 guide RNA activated the A3B promoter by ∼800-fold but also had a weak but readily detectable activity on the analogous A3G promoter fragment, resulting in an ∼10-fold activation of this promoter (Fig. 2C). We note that the A3G promoter retains the PAM used by sgB2 that is present in the A3B promoter and that the sgB2 sgRNA has only two seed sequence mismatches to the A3G promoter sequence (Fig. 1), thus perhaps explaining this weak residual activity.
Fig. 1.

Alignment of genomic human A3B and A3G promoter sequences used. Regions targeted by each sgRNA tested are shown. The coordinates given are based on the reported major transcription start sites (29, 30) for each gene. Identical bases are boxed and the PAM sequences indicated. Note that both A3G-specific PAMs are on the sense strand (5′-XGG-3′), whereas both A3B-specific PAMs are on the antisense strand (5′-CCX-3′).
Fig. 2.

Specific activation of FLuc expression in HeLa cells by sgRNAs targeting A3B or A3G promoter sequences. The A3G- and A3B-derived promoter sequences targeted in these assays are shown in Fig. 1. (A) A3G sgG1 and sgG2 activate FLuc expression from a reporter plasmid containing the targeted region of the A3G promoter (A3G rep1) but do not activate a reporter containing the homologous region from the A3B promoter (A3B rep1). (B) A3B sgB1 specifically activates the targeted region of the A3B promoter (A3B rep2) and does not activate via the homologous region of the A3G promoter (A3G rep2). (C) A3B sgB2 strongly activates the targeted region of the A3B promoter (A3B rep3) and modestly activates the homologous region of the A3G promoter (A3G rep3). Data shown represent the average of three independent experiments with SD indicated. Note that the A3G rep1 indicator construct contains both targeted regions of the A3G promoter, whereas the two regions analyzed in the A3B promoter are present in two distinct indicator plasmids.
We next asked whether any of these sgRNAs would be able to induce expression of the endogenous A3B or A3G gene in a human cell line, 293T, that normally expresses neither protein (13, 14). Although the paper initially describing the SAM methodology reported no evidence of synergy when multiple sgRNAs were used (12), we also wished to ask if we could see detectable synergy between sgRNAs in our system, given previous reports demonstrating strong synergy when a simpler system using only a dCas9-activation domain protein as an inducer of transcription was tested (8–11). For this purpose, we cotransfected 293T cells with expression plasmids encoding dCas9/VP64 and MS2-p65-HSF1 as well as plasmids encoding either one or both sgRNAs specific for the A3B or A3G promoters and then examined the pattern of APOBEC3 protein expressing by Western blot using a rabbit polyclonal antiserum that recognizes both A3G and A3B (Fig. 3 and Fig. S1).
Fig. 3.

Activation of the endogenous A3B and A3G genes in human cells. The 293T cells were cotransfected with pdCAS-VP64 and pMS2-p65-HSF1 plus either pGU6(Neg), pGsgB1, pGsgB2, pGsgB1+pGsgB2, pGsgG1, pGsgG2, or pGsgG1+pGsgG2. At 96 h posttransfection, cells were lysed and analyzed for A3B and A3G protein expression. Induced expression was detected using a rabbit polyclonal antibody that recognizes both A3B and A3G. The combination of two sgRNAs (sgB1+sgB2 in the case of A3B or sgG1+sgG2 in the case of A3G) resulted in the synergistic activation of each endogenous gene. This was also seen with sgG1 and sgG2 when an indicator construct was tested (Fig. S2). Data shown are representative of three independent experiments.
Fig. S1.
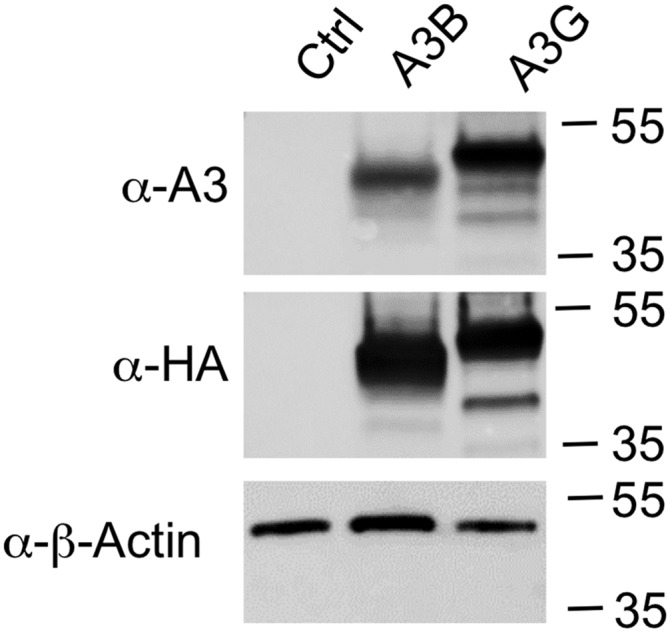
Recognition of A3B and A3G by a single APOBEC3 antibody. The 293T cells were transfected with 1 µg of pK, pK-A3B, or pK-A3G plasmids (27). Cells were lysed and analyzed by Western blot. Blots were probed with a mouse monoclonal antibody specific for the included HA epitope tag (901501, Covance) as well as a rabbit polyclonal antibody raised against APOBEC3G (10201, NIH AIDS Reagent Program) and a mouse monoclonal antibody specific for β-actin (sc-47778, Santa Cruz). These data confirm that the A3G antiserum also recognizes A3B and that A3B migrates faster than A3G during gel electrophoresis.
As expected, 293T cells expressing both fusion proteins but no sgRNA (Neg) did not give a detectable signal at the size predicted for either A3G or A3B (both are predicted to be ∼46 kDa in size) although we did note the presence of a small protein, of ∼25 kDa in size, that was recognized by this antiserum in all tested cultures and therefore serves as useful loading control. When 293T cells were cotransfected with a plasmid expressing sgB1, a weak signal, running somewhat slower than the 35-kDa size marker, was detectable (Fig. 3, lane 2). This signal likely represents the A3B protein, which migrates at a smaller size than expected and somewhat faster than A3G, even though both proteins are expected to have a similar molecular mass (Fig. S1) (13, 27). Use of sgB2 produced a slightly stronger signal at the same size but also a band migrating slightly more slowly (Fig. 3, lane 3). Given that sgB2 can also apparently bind to the A3G promoter (Fig. 2C), we considered that this might be induced A3G protein expression. Interestingly, when both sgRNA(MS2) guides specific for the A3B promoter were used together, we observed strong expression of the predicted A3B band and the same low level of the slower migrating, potentially A3G signal seen when sgB2 was used alone (compare lanes 3 and 4 in Fig. 3).
Use of the sgRNA(MS2) guides specific for the A3G promoter also revealed clear synergy. Both single sgRNA(MS2) guides sgG1 and sgG2 gave rise to single bands at the size expected for A3G and their simultaneous expression revealed a much stronger signal than when either sgRNA(MS2) was used alone (Fig. 3, compare lanes 5, 6, and 7). No signal at the size expected for A3B was detected, consistent with the lack of functional binding of either sgG1 or sgG2 to the A3B promoter reported in Fig. 2A. Consistent with the potential of two sgRNA(MS2) guides specific for a single promoter, in this case the A3G promoter, to show synergistic activity, we also observed enhanced activity when both A3G-specific sgRNAs were tested by cotransfection using the A3G rep1 indicator plasmid described in Fig. 2A (Fig. S2).
Fig. S2.

Synergy of A3G guides. The 300,000 HeLa cells were transfected in a 12-well dish with a total of 333 ng of a pGsg-derived plasmid (pG, pGsgG1, pGsgG2, or pGsgG1+pGsgG2), 333 ng pMS2-p65-HSF1, 333 ng pdCAS-VP64, and 25 ng of the pFLucA3G/rep1 reporter. Cells were lysed and analyzed for induced FLuc expression 24 h posttransfection using the Luciferase Assay System (E1501, Promega). These data confirm the guide RNA synergy seen in Fig. 3.
We next asked if induction of either APOBEC3 protein would inhibit HIV-1 replication. For this purpose, we cotransfected 293T cells as described in Fig. 3 but also added a vector expressing an HIV-1 provirus containing the FLuc indicator gene in place of nef (HIV-1WT) or a derivative in which the vif gene had been mutationally inactivated (HIV-1ΔVif) as well as a plasmid expressing the vesicular stomatitis virus (VSV) glycoprotein (pHIT/G) to allow infection of cells lacking CD4 (13, 14). We harvested the supernatant media at 104 h posttransfection and used these to infect naïve 293T cells. As shown in Fig. 4A, induction of either the endogenous A3G or A3B gene resulted in a profound inhibition of HIV-ΔVif infectivity, with both A3G and A3B reducing the expression of the virus-encoded FLuc gene by ∼10-fold. In the case of HIV-1WT, the high level of inhibition seen upon A3B induction was unaffected, as expected (13, 18). However, in the case of A3G, inhibition of the HIV-1WT vector was decreased from ∼90% to only ∼40% inhibition (Fig. 4A).
Fig. 4.

Potent inhibition of HIV-1 infectivity by induced, endogenous A3B and A3G. (A) The 293T cells were cotransfected with expression plasmids encoding HIV-1WT or HIV-1ΔVif variants encoding the FLuc indicator gene in place of nef, plus pHIT/G, pdCAS-VP64, pMS2-p65-HSF1, and either pgU6 (Neg), pGsgB1+pGsgB2 (expressing sgRNAs specific for the A3B promoter), or pGsgG1+pGsgG2 (sgRNAs specific for the A3G promoter). Media were changed at 24 h and 96 h posttransfection. Eight hours after the second media change, the virus-containing supernatant media were collected, passed through a 0.45-µm filter and used to infect naïve 293T cells. After an additional 24 h, the infected cells were lysed and analyzed for FLuc expression. Induced A3G was able to efficiently inhibit HIV-1ΔVif infectivity but only modestly inhibited HIV-1WT. In contrast, induced, endogenous A3B protein was able to efficiently inhibit the infectivity of both HIV-1WT and HIV-1-ΔVif. Average of three independent experiments with SD are indicated. (B) The viral producer 293T cells from A were lysed in SDS loading buffer and the lysates analyzed by Western blot. Neither the A3B nor A3G protein could be detected in the control (CTRL) lanes 1 and 4, as expected. Robust expression of the endogenous A3B gene was detected in both HIV-1WT and HIV-1ΔVif expressing cells treated with the combination of the two sgRNAs specific for the A3B promoter (lanes 2 and 5). Induced A3G expression in cells transfected with the two sgRNAs specific for the A3G promoter was readily detected in the cells expressing HIV-1ΔVif (lane 6), whereas A3G expression in the cells expressing HIV-1WT was, as expected, almost entirely blocked by the HIV-1 Vif protein (lane 3). Expression of the viral p24 capsid protein and endogenous cellular β-actin was not significantly changed upon induction of the endogenous A3B or A3G proteins.
We also harvested and lysed the virus-producing 293T cells at the time of supernatant harvest and examined the expression of endogenous A3G and A3B. As shown in Fig. 4B, we detected high levels of A3B expression in both the HIV-1WT– and HIV-1ΔVif–expressing cultures (lanes 2 and 5). However, in the case of A3G, expression in the culture producing the Vif+ HIV-1WT was almost undetectable (lane 3), whereas high levels of A3G were seen in the culture expressing HIV-1ΔVif (lane 6). Importantly, we did not see any difference in the level of HIV-1 capsid (p24) expression or in the expression of endogenous β-actin (Fig. 4B).
If the inhibition in infectivity seen in Fig. 4A is due to the A3G or A3B cytidine deaminase editing the minus strand of nascent HIV-1 proviral intermediates, we should observe C-to-T mutations in the presence of A3B on both HIV-1WT– and HIV-1ΔVif–derived proviruses and also in the presence of A3G in the case of HIV-1ΔVif (13, 14, 18, 23). As shown in Fig. 5A, we indeed detected a high level of C-to-T mutation when we sequenced segments of the FLuc gene present in both the HIV-1WT and HIV-1ΔVif vector in the presence of A3B and also high levels of C-to-T mutations in the case of HIV-1ΔVif in the presence of A3G. We also noted an increase in the number of G-to-A mutations, which may have occurred due to editing during second strand proviral DNA synthesis.
Fig. 5.

Pattern of proviral mutagenesis induced by A3B and A3G. (A) Wells of infected 293T cells, obtained as described in Fig. 4A, were lysed and total DNA purified using a DNeasy kit (Qiagen). After DpnI digestion to remove possible residual transfected plasmid DNA, a region of the HIV-1 genome was amplified by PCR, cloned, and sequenced. Sequence data were then analyzed for the presence of the expected C-to-T substitutions induced by the APOBEC3 proteins. A3B induced a similar level of C-to-T mutations in the presence and absence of the viral Vif protein (HIV-1WT compared with HIV-1ΔVif). A3G induced a substantial number of C-to-T mutations in HIV-1ΔVif and also a small number of G-to-A mutations, which most likely result from deamination of cytidine residues during second strand DNA synthesis. A3G mutagenesis of HIV-1WT was not analyzed due to the observed weak inhibition of the virus by the induced A3G expression (Fig. 4A) and the almost complete loss of A3G expression in the presence of Vif (Fig. 4B). Neg control samples, obtained from cultures lacking a targeting sgRNA, were sequenced and revealed a small number of random mutations. (B) The sequence data from A were also analyzed to identify any consensus sequence around edited deoxycytidine residues. We observed different consensus nucleotides at the −1 position, required for mutagenesis by the induced, endogenous A3B protein (5′-TC*-3′) or A3G protein (5′-CC*-3′), and these mirror published data (13, 14) for these two proteins and confirm the specificity of the mutagenesis of HIV-1 proviruses observed upon A3B and A3G induction.
A key difference between A3G and A3B is that A3G preferentially edits dC residues in the sequence context 5′-CC*-3′, whereas A3B edits dC residues in the sequence context 5′-TC*-3′ (13, 14, 23). A shown in Fig. 5B and Fig. S3, we observed the expected sequence contexts for edited dC residues in the case of both of these distinct human cytidine deaminases.
Fig. S3.

Sequence specificity of A3G and A3B editing sites. Logos were generated using WEBLOGO (weblogo.berkeley.edu/) using the data shown in (A) Fig. 5B and (B) Fig. S4.
All of the data reported thus far were generated in 293T cells that, although highly permissive for HIV-1 replication, do not represent a physiologically relevant cell context. To extend these results to human T cells, we selected the CD4+ T-cell line CEM-SS, which expresses neither A3G nor A3B (22, 24). To express the dCas-VP64 and MS2-p65-HSF1 fusion proteins, as well as the two sgRNAs G1 and G2 specific for the endogenous A3G promoter, we performed sequential transductions with two lentiviral vectors, selecting initially for blasticidin resistance conferred by the lentiviral dCas9-VP64 expression vector. Single cell clones were screened for high dCas9-VP64 expression and then transduced with MS2-p65-HSF1, or with a derivative of the MS2-p65-HSF1 lentiviral vector modified to express both A3G promoter-specific sgRNAs. The transduced cells were then selected for hygromycin resistance and screened for A3G expression by Western blot. As may be observed, the polyclonal transduced CEM-SS cells expressing the dCas9-VP64 and MS2-p65-HSF1 fusion proteins as well as the two A3G sgRNAs gave rise to readily detectable levels of A3G expression, whereas control CEM-SS cells expressing both fusion proteins but no sgRNA remained negative (Fig. S4B).
Fig. S4.

Infection of a polyclonal CEM-SS cell line expressing endogenous A3G. (A) A total of 2 × 106 293T cells were transfected with 10 µg of HIV-1-NLuc-WT or HIV-1-NLuc-ΔVif. Virus containing supernatants were passed through a 0.45-µm filter and the virus was then used to infect the polyclonal cell lines CEM-SS(A3G) or CEM-SS(CTRL) at a concentration of 105 cells/mL in a total volume of 5 mL. Cells were visually monitored for cell proliferation. After 2 wk the CEM-SS(CTRL) cells infected with either HIV-1-NLuc-WT or HIV-1-NLuc-ΔVif were determined to be nonviable. The CEM-SS(A3G) cells infected with HIV-1-NLuc-WT also contained few, if any, viable cells. However, the CEM-SS(A3G) cells infected with HIV-1-NLuc-ΔVif were actively growing. After an additional week in culture, total DNA was isolated from CEM-SS(A3G) cells infected with HIV-1-NLuc-ΔVif using a DNeasy kit (Qiagen). A region of HIV-1 proviral DNA was PCR amplified and cloned, sequenced, and analyzed for A3G-induced C-to-T mutations in the (−) strand HIV DNA. (B) Expression of endogenous A3G in polyclonal CEM-SS(A3G) cells was confirmed by Western blot, as described above.
To examine the phenotypic consequences of this level of A3G expression, we infected control CEM-SS transductants or the polyclonal A3G+ CEM-SS culture with either HIV-1WT or HIV-ΔVif. After 3 wk in culture, both the control and A3G+ CEM-SS cultures infected with HIV-1WT contained almost no viable cells, as did the CEM-SS culture infected with the HIV-ΔVif virus. However, the transduced, A3G+ CEM-SS culture infected with HIV-ΔVif virus continued to grow actively. To confirm that this culture had indeed been infected by HIV-1ΔVif and that the infecting virions had encountered A3G activity, we again used PCR to recover HIV-1 proviral sequences from this culture that were then subjected to DNA sequencing. In fact, these samples again contained numerous C-to-T mutations and the edited C residues were again found in the 5′-CC*-3′ sequence context characteristic of A3G editing (Figs. S3B and S4A).
To further analyze the ability of the induced, endogenous A3G protein to protect CEM-SS T cells from HIV-1 pathogenesis, we subcloned individual cells from the A3G+ CEM-SS culture expressing the dCas9-VP64 and MS2-p65-HSF1 fusion protein, as well as both A3G-specific sgRNAs, and identified clones expressing higher levels (A3G-H) or lower levels (A3G-L) of A3G, as shown by Western in Fig. 6A. Of note, the A3G-H CEM-SS cells express slightly higher levels of A3G than seen in the A3G+ T-cell line CEM, whereas the level of A3G expression in the A3G-L CEM-SS clone is somewhat below that seen in CEM (Fig. S5). We next transfected the A3G+ CEM-SS cultures, or a control CEM-SS culture, with a replication-competent HIV-1ΔVif–based vector encoding the Nano luciferase (NLuc) indicator gene. All three cultures were transfected with equal efficiency, as determined by measurement of NLuc expression (Fig. 6B). The virus-containing supernatant was then harvested, filtered, and used to infect naïve CEM-SS cells. As may be observed in Fig. 6C, high-level expression of the endogenous A3G protein almost totally blocked the infectivity of progeny HIV-1 virions, whereas low-level A3G production resulted in an intermediate, but still substantial, reduction in infectious virus production. Therefore, CRISPR/Cas-mediated induction of endogenous, cellular antiviral restriction factors can also exert the predicted phenotype in T cells.
Fig. 6.

Inhibition of HIV-1 infectivity by induced, endogenous A3G in CEM-SS. (A) Protein expression in CEM-SS cells engineered to express A3G. The CEM-SS Ctrl cell line is negative for A3G expression, whereas the clonal cell line CEM-SS (A3G-H) expresses a high level of induced A3G and CEM-SS (A3G-L) cells express a lower level of A3G. β-Actin provides a loading control. (B) A3G expression does not inhibit viral production in the HIV-1 producer cells. The CEM-SS cell lines (Ctrl, A3H-H and A3G-L) were transfected with the HIV-NLucΔVif proviral clone and an aliquot of cells lysed at 24 h posttransfection to verify equivalent transfection efficiency as assayed by NLuc expression. (C) Naïve CEM-SS cells were infected with virus collected at 48 h posttransfection and assayed for NLuc activity at 48 h postinfection from the cells in B. The infectivity of the virus produced in CEM-SS (A3G-H) cells was reduced by ∼95%, relative to control CEM-SS cells, whereas the HIV-1 virions produced in the CEM-SS (A3G-L) cells were reduced by ∼75%, consistent with the lower A3G expression seen in the latter. B and C show the average of three independent experiments with SD indicated.
Fig. S5.
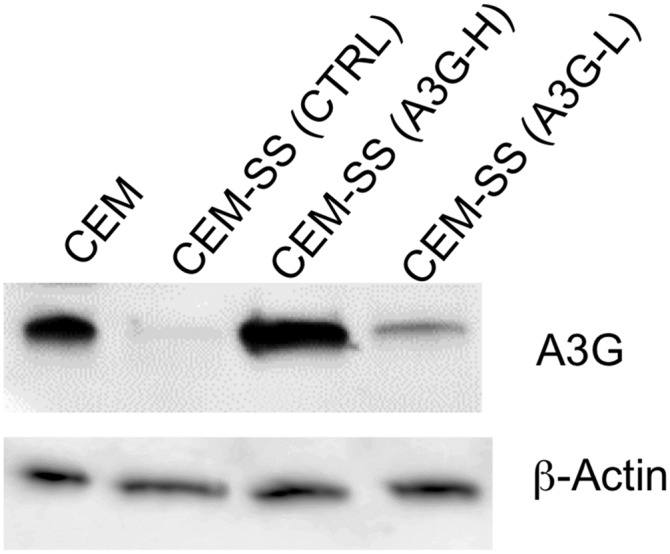
Induction of physiological levels of A3G expression in T cells by Cas9-targeted transcriptional activators. This Western blot for A3G expression was performed as described in Fig. 6 with the exception that a CEM cell culture was also analyzed in parallel. CEM, unlike the CEM-SS derivative, naturally express the endogenous protein and restrict HIV-1ΔVif replication (22).
Discussion
The identity of cellular factors that specifically promote or restrict the replication of particular viral species remains of considerable interest. Efforts to identify viral cofactors have generally relied on loss-of-function screens and a number of screens using small interfering RNAs (siRNAs) to down-regulate cellular messenger RNAs (mRNAs) have been reported for several viral species, including HIV-1 (31–33), influenza virus (34, 35), and dengue virus (36), although there has been significant variability in the identity of cellular cofactors for HIV-1 identified in different screens (37). More recently, CRISPR/Cas-based methodologies have also emerged as powerful tools to screen for loss-of-function phenotypes (38–40) and, as CRISPR/Cas results in complete ablation of a specific gene rather than partial knockdown, these screens may result in the more reproducible identification of cellular genes required for viral replication but dispensable for cellular viability in culture.
Whereas the identification of cellular cofactors for viral replication is of interest, the identification of novel restriction factors for specific viruses has attracted even more attention (16, 17). In the case of HIV-1, this effort has resulted in the identification of several factors capable of inhibiting primate lentivirus replication, including members of the APOBEC3 protein family, BST2/tetherin, TRIM5α, and SAMHD1 (16, 17). However, the identification of viral restriction factors remains a challenge, as wild-type viruses have often evolved mechanisms to overcome inhibition by specific restriction factors, including inhibition of A3G by HIV-1 Vif and of tetherin by HIV-1 Vpu. Moreover, many restriction factors are not constitutively expressed but are instead induced upon viral infection, including numerous IFN-stimulated genes (ISGs). Whereas the identification of restriction factors therefore remains challenging, screens for factors whose knockdown using siRNAs enhances viral replication have had some success (41).
Conversely, a gain-of-function approach that allows the selective and efficient activation of promoters in cultured cells using a cleavage defective Cas9 (dCas9) derivative and a promoter-specific sgRNA would represent a promising tool to screen for cellular factors that can restrict viral replication. However, initial efforts to activate otherwise silent genes using fusion proteins consisting of dCas9 linked to transcription activation domains proved ineffective unless multiple sgRNAs specific for the promoter in question were expressed simultaneously (8–11), which would clearly render screens much more difficult. We were therefore intrigued by a recent report (12) describing a SAM consisting of not only a dCas/VP64 fusion but also a modified sgRNA scaffold containing two MS2 coat protein binding sites, each of which could bind two copies of a fusion protein consisting of MS2 coat protein linked to two transcription activation domains derived from the NF-κB p65 subunit and HSF1. SAMs were reported to efficiently activate cellular genes when guided to a single binding site located <200 bp 5′ to the transcription start site. The use of a single sgRNA per promoter would clearly facilitate gain-of-function screens for cellular genes that can regulate virus replication.
Here, we report proof-of-principle experiments to validate the ability of SAMs to induce the expression of phenotypically relevant levels of two known viral restriction factors, A3G and A3B (13, 16–18). We were able to confirm that each of these endogenous human gene products could indeed be induced by the use of sgRNAs specific for either the A3G or A3B promoter (Figs. 1–3). In particular, two sgRNAs that functioned via PAMs present in the A3G promoter but lacking in the A3B promoter (sgG1 and sgG2) proved highly selective for activation of A3G expression and this was also true for an A3B-specific sgRNA (sgG1) that targeted a sequence that, in the A3G promoter, retained a functional PAM but had six “seed” mutations. In contrast, an A3B promoter-specific sgRNA that retained a PAM in the A3G promoter and featured only two seed target mutations (sgB2, Fig. 1) was not totally specific for A3B, though activation for the A3B promoter was still ∼50-fold higher as determined by indicator assays (Fig. 2C). Nevertheless, the sgB1 guide did appear to induce modest levels of the endogenous A3G protein expression when expressed in human cells (Fig. 3).
Whereas the data reported in this manuscript were focused on the activation of endogenous A3G or A3B expression, we did not select these genes because of their potential as actual treatments for HIV-1 but because they provide a convenient and specific inhibitory phenotype. In particular, we note that wild-type HIV-1 is resistant to restriction by A3G, due to the viral Vif protein (Fig. 4B) (19–21), whereas overexpression of A3B has been associated with mutagenesis of the human genome and transformation (42–44). However, this does not affect the conclusion that activation of endogenous restriction factors using CRISPR/Cas could be both informative and potentially useful.
Whereas all of the sgRNAs tested activated the expression of their cognate endogenous promoter and gene product when tested in human cells, we observed higher levels of expression of both A3G and A3B when two sgRNAs specific for the cognate promoter were tested simultaneously (Fig. 3 and Fig. S2). This result differs from data reported by the inventors of this technology (12), who reported that they did not see enhanced gene expression from endogenous promoters when multiple sgRNAs specific for that promoter were tested together. It is unclear why we instead observed a significant degree of synergy (Fig. 3) but this result does raise the possibility that screens using the SAM methodology would require two sgRNAs to assure robust target gene activation, although it is certainly possible that the lower level of expression induced by the use of a single sgRNA (Fig. 3) would suffice to give a phenotypic read-out in some cases. On the other hand, if two sgRNAs are indeed required, then this would have the advantage of increasing the DNA target specificity from ∼15 bp to ∼30 bp, which would assure that only the targeted promoter is fully activated. The potentially high specificity of this activation methodology is supported not only by reporter analysis (Fig. 2) but also by Western blot (Figs. 3 and 4), which showed that sgRNAs lacking a PAM target in the promoter or lacking extensive seed homology fail to activate very similar nontarget promoters. The fact that A3G, but not A3B, expression is activated in the presence of both the sgG1 and sgG2 guide RNA is further demonstrated by the almost complete degradation of the induced A3G protein in the presence of Vif, which does not have the ability to inhibit A3B expression (Fig. 4B) (13) as well as by the sequence context of the DNA editing observed, which was the expected 5′-CC*-3′ in the presence of induced endogenous A3G and 5′-TC*-3′ in the presence of induced A3B (Fig. 5 and Fig. S3) (13, 18). We note, however, that two base mismatches in the seed target in the A3G promoter in the sgB2 guide RNA did still permit a modest activation of the A3G promoter as measured by reporter assay (Fig. 2C) and also by Western blot of the induced A3G and A3B proteins (Fig. 3). These data therefore suggest that full discrimination between promoters in screens using this technology may indeed require the enhanced specificity afforded by the use of two sgRNAs.
Whereas the focus of this work has been on the induction of cellular antiviral restriction factors, using the endogenous A3G and A3B genes as exemplars, we note that this same approach could also be used to induce latent viruses, such as latent HIV-1 proviruses present in resting memory T-cells, while simultaneously treating patients with antiretroviral drugs. This so-called “kick and kill” strategy has been the focus of considerable research focusing on drugs that have the potential to induce latent HIV-1 proviruses by, for example, inducing chromatin modifications or inducing cellular signaling pathways (45, 46). However, this approach has so far not proven able to activate a substantial percentage of latent proviruses even when drugs that induce high levels of T-cell activation, that would likely induce unacceptable side effects in vivo, were tested. The use of synthetic, Cas9-based transcription factors specific for the HIV-1 LTR U3 promoter might offer an alternative approach to proviral activation that would be far more specific. However, it is not obvious at present how these activators could be efficiently and specifically delivered to latently infected T cells. Nevertheless, if this technical issue can be resolved, then Cas9-based synthetic transcription factors may emerge as a clinically useful tool in the treatment of viral and other disease states.
Materials and Methods
Molecular Clones.
The previously described lentiviral vectors encoding the MS2-p65-HSF1 (61426) and dCAS-VP64 (61425) fusion proteins and the sgRNA(MS2) (61427) chimeric RNA (12) were obtained from Addgene (courtesy of Feng Zhang, Broad Institute, Cambridge, MA). The gene inserts from MS2-p65-HSF and dCAS-VP64 were amplified by PCR and cloned into pcDNA3 to create pMS2-p65-HSF1, and pdCAS-VP64. Oligonucleotides encoding sgRNAs, designed to be specific for either the A3B or A3G promoter regions (29, 30), were ligated into sgRNA(MS2). The U6 promoter cassettes from sgRNA(MS2), with either no sgRNA or containing the inserted promoter-specific sgRNA, were amplified by PCR and then cloned into pGEM3Zf(+) as EcoRI/HindIII fragments to create pGU6, pGsgB1, pGsgB2, pGsgG1, and pGsgG2.
The lentiviral dual A3G guide expression vector was generated by modifying MS2-p65-HSF1 (Addgene 61426) as follows: the A3G sgG2 guide cassette from pGsgG2 was amplified by PCR and inserted into the unique NheI site in MS2-p65-HSF1 to create MS2-p65-HSF1/sgG2. The A3G sgG1 guide cassette from pGsgG1 was also amplified by PCR and inserted into the unique EcoRI site in MS2-p65-HSF1/sgG2 to create the dual guide lentiviral expression vector MS2-p65-HSF1/sgG(1+2).
The FLuc-based indicator construct pGL4.10 (Promega) was digested with BglII and HindIII and complementary synthetic oligonucleotides containing a minimal TATA box were annealed and inserted. This modified pGL4.10+TATA was then digested with XhoI and BglII and oligonucleotides were annealed and inserted to generate the indicator constructs pFLucA3Brep1, pFLucA3Brep2, pFLucA3Brep3, pFLucA3Grep1, pFLucA3Grep2, and pFLucA3Grep3. Oligonucleotide sequences used to generate the sgRNA expression constructs and pFLuc reporter plasmids are listed in SI Materials and Methods.
The HIV-1 proviral FLuc expression vectors pNL-HXB-FLuc (HIV-1WT) and pNL-HXB-FLucΔVif (HIV-1ΔVif), as well as the VSV glycoprotein expression plasmid pHIT/G, have been previously described (13, 14). The gene for NLuc (Promega) was amplified by PCR and inserted between the unique NotI and XhoI sites of pNL-HXB-LucΔVif (HIV-1ΔVif) to generate pHIV-1-NLuc-ΔVif. The identical strategy was used to construct pHIV-1-NLucWT.
Cell Culture and Transfection.
HeLa and 293T cells were cultured in DMEM supplemented with 5% (vol/vol) FBS and gentamicin (Life Technologies). The human CD4+ T-cell line CEM-SS (776, NIH AIDS Reagent Program) was maintained in RPMI-1640 medium, supplemented with 10% (vol/vol) FBS and gentamicin. All HeLa and 293T transfections were performed using polyethylenimine (PEI). All CEM-SS transfections were performed using Lipofectamine LTX (ThermoFisher).
Generation of a CEM-SS Cell Line Expressing Endogenous A3G.
The 2 × 106 293T cells were transfected with the lentiviral vector dCas9-VP64 (15 µg), the lentiviral packaging plasmid ΔCR8.74 (15 µg) (Addgene 22036), and the VSV-G envelope expression plasmid pMD2.G (3 µg) (Addgene 12259) to generate lentiviral particles that were then used to transduce CEM-SS cells, which do not normally express APOBEC3 proteins (22). After blasticidin (10 µg/mL) selection, the transduced cells were cloned and a cell clone expressing a high level of dCas9/VP64 was identified. The CEM-SS/dCas9 clonal cell line was then transduced with virus generated in 293T cells using MS2-p65-HSF1-sgG(1+2) or MS2-p65-HSF1, ΔCR8.74, and pMD2.G. Transduced cells were then selected for both blasticidin (10 µg/mL) and hygromycin (25 µg/mL) resistance to generate the polyclonal cell line CEM-SS(A3G), which expresses dCas9/VP64, MS2-p65-HSF1, and A3G sgG(1+2) or the CEM-SS(CTRL) cell line, which expresses dCas9/VP64 and MS2-p65-HSF1 (but no sgRNAs). The CEM-SS high expressing A3G (A3G-H) and low expressing A3G (A3G-H) cell lines were obtained by single cell cloning of the polyclonal CEM-SS (A3G) cell line.
Specificity of A3B and A3G sgRNAs Demonstrated by Reporter Assay.
The 3 × 105 HeLa cells were transfected in a 12-well dish with 333 ng of a pGsg-derived plasmid, 333 ng pMS2-p65-HSF1, 333 ng pdCAS-VP64, and 25 ng of a pFLuc reporter. Cells were lysed and analyzed for induced FLuc expression 24 h posttransfection using the Luciferase Assay System (E1501, Promega), as previously described (13, 14).
Induction of Endogenous A3B and A3G Detected by Western Blot.
The 3 × 105 293T cells were transfected in a six-well dish with a total of 3 µg DNA: 1 µg of pGsg-derived plasmid(s), 1 µg pMS2-p65-HSF1, and 1 µg pdCAS-VP64. Media were changed 24 h posttransfection and cells were harvested after an additional 72 h (96 h posttransfection) and analyzed for induced A3B and A3G expression. Lysates were subjected to SDS gel electrophoresis and then transferred to a nitrocellulose membrane. The membranes were probed with a rabbit polyclonal α-A3G antiserum (AIDS reagent 10201), which recognizes both A3B and A3G (Fig. S1). The blots were washed and then incubated with an anti-rabbit HRP conjugated antibody. Chemiluminesence was used to visualize bound proteins using the WesternBright Sirius Western blotting detection kit (Advansta K-12043-D20) and the G:Box Imaging System and GeneSys software.
Inhibition and Mutagenesis of HIV-1 by Induced A3B and A3G in 293T Cells.
The 2.5 × 106 293T cells in a 10-cm dish were transfected with 2.5 µg of pMS2-p65-HSF1, 2.5 µg of pdCAS-VP64, 1.25 µg of each of two pGsg-derived plasmids, 500 ng of pHIT/G, and 5 µg of either the HIV-1WT– or HIV-1ΔVif–derived lentiviral FLuc-expression vector. Media were changed at 24 h and again at 96 h posttransfection. Virus-containing supernatants were collected after an additional 8 h and filtered through a 0.45-µm filter (Pall) before being used to infect duplicate wells each containing 500,000 naïve 293T cells. At 24 h postinfection, one well was lysed and analyzed for FLuc activity. At the same time, the second well was harvested and total DNA purified using a DNeasy Kit (Qiagen). The DNA was DpnI digested to remove any residual plasmid DNA and used as a template for PCR using primers specific to the FLuc gene inserted into HIV-1 in place of the nef gene. The purified PCR product was then digested, ligated into pcDNA, and single colonies were isolated and sequenced.
In addition, HIV-1 producer cells were lysed and assayed by Western blot as described above for induced A3B and A3G, the HIV-1 p24 capsid protein, and endogenous β-actin using a rabbit polyclonal α-A3G antibody (AIDS reagent 10201), a mouse anti-p24 antibody (AIDS reagent 3537) (47), and a mouse monoclonal anti–β-actin antibody (Santa Cruz, sc-47778).
Inhibition of HIV Replication in CEM-SS (A3G) Clonal Cell Lines.
The 106 CEM-SS CTRL, CEM-SS (A3G-L), or CEM-SS (A3G-H) cells were transfected with HIV-1-NLuc-ΔVif (2.5 µg) using Lipofectamine LTX (Thermo Fisher) using the manufacturer’s protocol. At 24 h posttransfection, an aliquot of cells from each transfection was harvested and assayed for NLuc activity to verify equivalent initial transfection of the cell lines. Virus-containing supernatants were collected at 48 h posttransfection, filtered, and then used to infect naïve, parental CEM-SS cells. At 48 h postinfection, cells were washed with PBS and assayed for NLuc activity (Promega).
SI Materials and Methods
Oligonucleotides Used to Generate sgB1, sgB2, sgG1, and sgG2.
A3B
sgB1-F CACCGCTGGCTCTGGCTCTGGCTC
sgB1-R AAACGAGCCAGAGCCAGAGCCAGC
sgB2-F CACCGCATTGGTGTGGGAGGCCCC
sgB2-R AAACGGGGCCTCCCACACCAATGC
A3G
sgG1-F CACCGGATCCGGGAGTCCCTGCGG
sgG1-R AAACCCGCAGGGACTCCCGGATCC
sgG2-F CACCGTGGAGAGGAGGCTCCAGCT
sgG2-R CACCGTGGAGAGGAGGCTCCAGCT.
Oligonucleotides Used to Generate GL4.10-Based Reporters.
Insertion of TATA box
TATA box-F GATCTAGGGTATATAATGGAAGCTCGACTTCCA
TATA box-R AGCTTGGAAGTCGAGCTTCCATTATATACCCTA.
A3G Promoter Sequences.
A3G rep1-F
TCGAGGATCCGGGAGTCCCTGCGGAGGCATCAGCCTGTCTGTCTTGATGGTGGAGAGGAGGCTCCAGCTGGGCGGGACCACCAGGGGAGG
A3G rep1-R
GATCCCTCCCCTGGTGGTCCCGCCCAGCTGGAGCCTCCTCTCCACCATCAAGACAGACAGGCTGATGCCTCCGCAGGGACTCCCGGATCC
A3G rep2-F
TCGAAGAGCGGCCTGTCTTTATCAGAGGTCCCTCTGCCAGGGGGAGGGCCCCAGAGAAAACCAGAAAGAGGGTGAGAGACA
A3G rep2-R
GATCTGTCTCTCACCCTCTTTCTGGTTTTCTCTGGGGCCCTCCCCCTGGCAGAGGGACCTCTGATAAAGACAGGCCGCTCT
A3G rep3-F
TCGATGAGGAAGATAAAGCGTCCCAGGGCCTCCTACACCAGCGCCTGAGCAGGAAGCGGA
A3G rep3-R
GATCTCCGCTTCCTGCTCAGGCGCTGGTGTAGGAGGCCCTGGGACGCTTTATCTTCCTCA.
A3B Promoter Sequences.
A3B rep1-F
TCGAGTGCTGCTCAAAGCTCTGGGCACACATGGGCTGGGGAGGGGGTGGTGGAGAGGGGGCTCCAGCCTGCCCAACCACCAGGCAACG
A3B rep1-R
GATCCGTTGCCTGGTGGTTGGGCAGGCTGGAGCCCCCTCTCCACCACCCCCTCCCCAGCCCATGTGTGCCCAGAGCTTTGAGCAGCAC
A3B rep2-F
TCGAAGAGCAGCCTGTCTTTATTGGGGTTCCTCTGCCAGCGGGAAGGGTCCGGGGAAAACCAGAGCCAGAGCCAGAGCCA
A3B rep2-R
GATCTGGCTCTGGCTCTGGCTCTGGTTTTCCCCGGACCCTTCCCGCTGGCAGAGGAACCCCAATAAAGACAGGCTGCTCT
A3B rep3-F
TCGAAGAGAAACATGAAGCACCCCGGGGCCTCCCACACCAATGCCTGAGCAGGAATGGGA
A3B rep3-R
GATCTCCCATTCCTGCTCAGGCATTGGTGTGGGAGGCCCCGGGGTGCTTCATGTTTCTCT.
Acknowledgments
This publication resulted in part from research supported by the Duke University Center for AIDS Research, an NIH funded program (P30-AI064518). The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: rabbit anti-APOBEC3G antibody from Dr. Jaisri Lingappa and HIV-1 p24 monoclonal antibody from Drs. Bruce Cheesbro and Kathy Wehrly.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.S.H. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1516305112/-/DCSupplemental.
References
- 1.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468(7320):67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 2.Barrangou R, Marraffini LA. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54(2):234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109(39):E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339(2):281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23(12):2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80(3):1067–1076. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris RS, Hultquist JF, Evans DT. The restriction factors of human immunodeficiency virus. J Biol Chem. 2012;287(49):40875–40883. doi: 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malim MH, Bieniasz PD. HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med. 2012;2(5):a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop KN, et al. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14(15):1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 19.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9(11):1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 20.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13(22):2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 21.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9(11):1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 22.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 23.Mangeat B, et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 24.Refsland EW, et al. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010;38(13):4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koning FA, et al. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83(18):9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogerd HP, et al. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006;103(23):8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34(1):89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muckenfuss H, et al. Sp1 and Sp3 regulate basal transcription of the human APOBEC3G gene. Nucleic Acids Res. 2007;35(11):3784–3796. doi: 10.1093/nar/gkm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori S, Takeuchi T, Ishii Y, Kukimoto I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem Biophys Res Commun. 2015;460(3):555–560. doi: 10.1016/j.bbrc.2015.03.068. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4(5):495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 33.König R, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.König R, et al. Human host factors required for influenza virus replication. Nature. 2010;463(7282):813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlas A, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463(7282):818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 36.Sessions OM, et al. Discovery of insect and human dengue virus host factors. Nature. 2009;458(7241):1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hao L, et al. Limited agreement of independent RNAi screens for virus-required host genes owes more to false-negative than false-positive factors. PLOS Comput Biol. 2013;9(9):e1003235. doi: 10.1371/journal.pcbi.1003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509(7501):487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 41.Benitez AA, et al. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza A virus. Cell Reports. 2015;11(11):1714–1726. doi: 10.1016/j.celrep.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45(9):977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns MB, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor BJ, et al. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siliciano JD, Siliciano RF. HIV-1 eradication strategies: Design and assessment. Curr Opin HIV AIDS. 2013;8(4):318–325. doi: 10.1097/COH.0b013e328361eaca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Archin NM, Margolis DM. Emerging strategies to deplete the HIV reservoir. Curr Opin Infect Dis. 2014;27(1):29–35. doi: 10.1097/QCO.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology. 1995;213(1):70–79. doi: 10.1006/viro.1995.1547. [DOI] [PubMed] [Google Scholar]