

Abstract
Purpose of review
The sodium glucose cotransporter SGLT2 reabsorbs most of the glucose filtered by the kidneys. SGLT2 inhibitors reduce glucose reabsorption thereby lowering blood glucose levels and have been approved as new anti-hyperglycemic drugs. While the therapeutic strategy is very promising, many questions remain.
Recent findings
Using validated antibodies SGLT2 expression was localized to the brush border of the early proximal tubule in human kidney and was found upregulated in genetic murine models of type 1 and 2 diabetes. SGLT2 may functionally interact with the Na/H exchanger NHE3 in the proximal tubule. SGLT1-mediated reabsorption explains the fractional glucose reabsorption of 40–50% during SGLT2 inhibition. SGLT2 is expressed on pancreatic alpha cells where its inhibition induces glucagon secretion. SGLT2 inhibition lowers GFR in hyperfiltering diabetic patients consistent with the tubular hypothesis of diabetic hyperfiltration. New data indicate a potential of SGLT2 inhibition for renal medullary hypoxia and ketoacidosis, but also for blood glucose effect-dependent and independent nephroprotective actions, renal gluconeogenesis inhibition, reduction in cardiovascular mortality, and cancer therapy.
Summary
The findings expand and refine our understanding of SGLT2 and its inhibition, have relevance for clinical practice, and will help interpret ongoing clinical trials on the long-term safety and cardiovascular effects of SGLT2 inhibitors.
Keywords: diabetic kidney disease, glycemic control, glucose transport, SGLT2 inhibition
Introduction
Diabetes mellitus is a growing health concern worldwide [1]. Normalizing hyperglycemia is vital to slowing the progression of the disease process and preventing the devastating secondary consequences including cardiovascular disease and nephropathy. Inhibitors of sodium glucose cotransporter SGLT2 have recently been approved as new anti-hyperglycemic drugs in type 2 diabetes mellitus (T2DM). Studies are under way in type 1 diabetes (T1DM) using SGLT2 inhibitors as add-on to insulin therapy. These compounds inhibit the reabsorption of glucose in the kidney, a process that is enhanced in diabetes, thereby spilling glucose into the urine and lowering blood glucose levels (Figure 1&2). In contrast to many of the current anti-diabetic therapies, SGLT2 inhibitors can lower body weight, work independent of endogenous insulin, and have little risk of hypoglycemia (see [2–5] for review).
Figure 1.
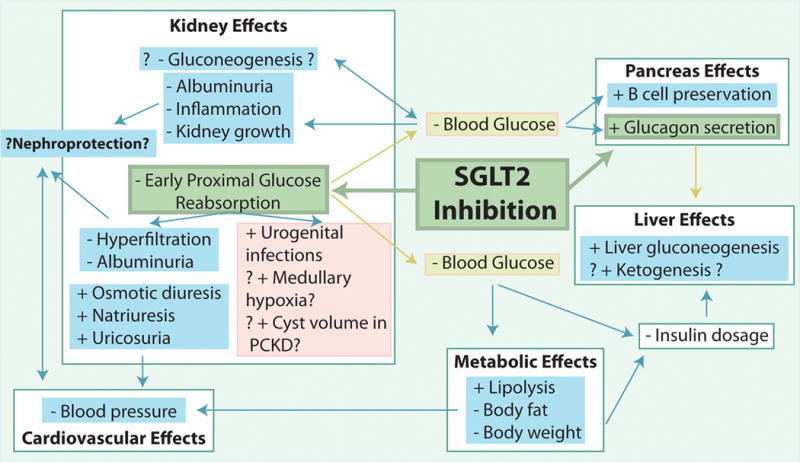
Direct and indirect effects of SGLT2 inhibition on the kidney and other organs, and the potential for beneficial cardiovascular effects and nephroprotection as well as unwanted side effects. PCKD, polycystic kidney disease.
Figure 2.
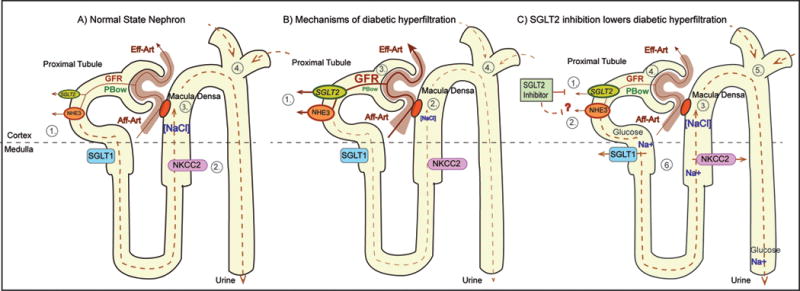
The tubular hypothesis of diabetic glomerular hyperfiltration: effect of SGLT2 inhibition. Panel A) 1. Glucose reabsorption occurs via SGLT2 in early proximal tubule (PT) and to a small degree via SGLT1 in late PT; Na+ is reabsorbed via NHE3. 2. NaCl is reabsorbed via NKCC2 in thick ascending limb (TAL). 3. Luminal NaCl concentration at macula densa ([NaCl]) inversely regulates GFR via tubuloglomerular feedback (TGF). 4. Fluid delivery to distal tubule (site of main tubular resistance) is proportional to hydrostatic pressure in Bowman space (PBow) which imposes inhibitory influence on GFR. Panel B) 1. Hyperglycemia and tubular growth increase Na+/ glucose reabsorption via SGLT2 and SGLT1 and increase Na+ reabsorption via NHE3. 2. & 3. This lowers [NaCl] at macula densa and increases GFR via TGF. 4. The hyperreabsorption also decreases fluid delivery to distal tubule which reduces PBow and increases GFR. Panel C) 1. & 2. SGLT2 inhibitor blocks Na+/ glucose reabsorption via SGLT2 and potentially inhibits NHE3. 3. & 4. This increases [NaCl] at the macula densa and lowers GFR via TGF. 5. Inhibition of hyperreabsorption increases fluid delivery to distal tubule which increases PBow and lowers GFR. 6. In addition, SGLT2 inhibition increases load to and Na+ reabsorption via SGLT1 and NKCC2 in outer medulla. This may enhance medullary hypoxia unless counterbalanced by GFR reduction.
While this new class of anti-diabetic compounds is very promising, many questions remain. In this review, we will discuss new insights on the biology of SGLT2 and the consequences of SGLT2 inhibition on the kidney and beyond.
SGLT2 and renal glucose reabsorption in normoglycemia
Under conditions of normal serum glucose levels and GFR about 160–180 grams of glucose are filtered each day and nearly completely reabsorbed by the kidneys. The sodium-glucose transporters SGLT2 and SGLT1 in the luminal membrane of the proximal tubule have been established as primary mechanisms of glucose reabsorption in the kidney [4–6] (Figure 2). Using validated antibodies and knockout mice as negative controls, the SGLT2 protein has been localized in the luminal membrane of the early proximal tubule (S1 and S2 segments) whereas the SGLT1 protein is expressed in the late proximal tubule (S3 segment) in rodents [7;8]. Recent studies used well-validated antibodies and confirmed this expression pattern in the human kidney, and found that, in contrast to rodents, renal SGLT2 expression was sex-independent [9].
Studies in knockout mice demonstrated that SGLT2 is responsible for all glucose reabsorption in the early proximal tubule, and for most kidney glucose reabsorption overall [7], whereas SGLT1 reabsorbs 3% of the filtered glucose in euglycemia [10]. Net renal glucose reabsorption is absent during pharmacologic SGLT2 inhibition in SGLT1−/− mice as well as in SGLT1/SGLT2 double knockout mice [11], indicating that SGLT2 and SGLT1 can explain all renal glucose reabsorption in euglycemia and that SGLT2 reabsorbs ~97% in euglycemia.
Diabetes increases renal SGLT2 expression and glucose reabsorption
In T1DM and T2DM, the kidneys increase their glucose transport maximum by ~20% to 600 gram/day. This could be secondary to proximal tubular growth and an increase in SGLT2 expression, however, no data using primary tissue samples and validated antibodies is available in humans, and previous studies in rodents provided conflicting results [5;12]. Using knockout mice as critical negative antibody controls, renal protein expression of SGLT2 was shown to be increased by 40–80% in genetic models of T2DM db/db mice and T1DM Akita mice [13;14] (Figure 2).
Renal SGLT1 protein expression was increased in T2DM ob/ob mice, while SGLT1 mRNA was reduced [15]. Other studies indicated that renal SGLT1 protein expression is suppressed whenever the late proximal tubule is exposed to high glucose levels, as observed in normoglycemic SGLT2−/− mice [7] as well as in non-diabetic mice given a SGLT2 inhibitor or in T1DM Akita mice [14]. Despite this downregulation, overall glucose transport through SGLT1 was greatly increased in those settings due to the increased glucose load to the late proximal tubule [7;11] (Figure 2). We speculated that down-regulation of SGLT1 is a protective mechanism to attenuate glucotoxicity in the vulnerable S3 segments within the outer medulla [4]. Upregulation of SGLT2 and downregulation of SGLT1 in diabetes are both expected to enhance the glucosuric and thereby the blood glucose lowering effect of SGLT2 inhibition.
Molecular regulation of SGLT2 expression and activity
Studies in mice have shown that pharmacological SGLT2 inhibition itself also increases renal SGLT2 protein expression, i.e. under conditions when the physiologic uptake of glucose into the early proximal tubule is inhibited [14]. The mechanisms involved in SGLT2 upregulation under these conditions and in response to diabetes (see above) remain poorly understood. Studies in human embryonic kidney (HEK-293 T) cells indicated that insulin phosphorylates SGLT2 on Ser624 thereby increasing its activity [16]. Studies in cultured human proximal tubular cells reported that insulin, but not high glucose, enhanced SGLT2 protein expression. The insulin-induced increase in SGLT2 expression was associated with an increased reactive oxygen species content and attenuated by N-acetylcysteine [17]. Insulin binding in the kidney corresponds to sites of SGLT2 expression, and thus, the insulin release following ingestion of a glucose rich meal may activate SGLT2 to retain the ingested glucose in the body [18]. While hyperinsulinemia may contribute to the increased SGLT2 protein expression and activity in T2DM db/db mice, it cannot explain the increased renal SGLT2 expression in hypoinsulinemic T1DM Akita mice [13;14]. Other molecules implicated in the upregulation of SGLT2 activity include protein kinase C and protein kinase A, while angiotensin II AT1 receptors and hepatocyte nuclear factor HNF-1α have been proposed to increase SGLT2 expression in the diabetic state (for review see [12;19]). More studies are needed to better understand the nuances of the regulation of SGLT2 expression and activity.
Is there relevant extrarenal expression of SGLT2? This question is important for the clinical use of SGLT2 inhibitors, and consistent with previous reports in humans and mice [4], a recent study could not detect SGLT2mRNA in human small intestine, liver, heart, lung, fat tissue, and brain [9]. Obviously, negative results cannot exclude small amounts of SGLT2 mRNA expression, and as discussed below, new studies indicated that SGLT2 is expressed and functionally active in glucagon-secreting alpha cells of the pancreatic islets [20].
Why do SGLT2 inhibitors only excrete 50–60% of the filtered glucose?
If SGLT2 reabsorbs ~97% of filtered glucose, then it is surprising that humans treated with SGLT2 inhibitors only excrete 50–60% of the filtered glucose [4]. This is mimicked in SGLT2 knockout mice, in which mean fractional glucose excretion was 64% [7]. Studies in SGLT1−/− mice have indicated that SGLT2 inhibition strongly increases SGLT1-mediated glucose reabsorption [21], and demonstrated that the increase in SGLT1-mediated glucose reabsorption fully explained the fractional glucose reabsorption of 40–50% during SGLT2 inhibition in euglycemic mice [11] (Figure 2). Recent studies further indicated that the SGLT2 inhibitors reach their target mostly through glomerular filtration and from the luminal side [11;22].
SGLT2 inhibition and hypoglycemia, gluconeogenesis, beta cell preservation, and ketogenesis
According to a meta-analysis of patients with T2DM, SGLT2 inhibitors decreased Hb A1C levels by 0.5–0.7% at 12 weeks of treatment and this effect persisted for up to 52 weeks. Hypoglycemia was noted only when SGLT2 inhibitors were used in combination with other drugs, but not as monotherapy [23]. The blood glucose lowering effects of SGLT2 inhibition depended on both serum glucose concentration and GFR. These two parameters determine how much glucose is delivered to SGLT2 in the early proximal tubule and thus how much glucose can be excreted by SGLT2 inhibition. Therefore, lower values for each parameter are associated with lesser blood glucose lowering. Currently SGLT2 inhibitors are approved for use only in type 2 diabetes, with dapagliflozin approved for GFR > 60ml/min and canagliflozin and empagliflozin for GFR > 45 ml/min. Notably, SGLT2 inhibition in T2DM patients with stage 3 chronic kidney disease (CKD) (eGFR between 30–60 ml/min) was still able to reduce Hb A1C by 0.42% at 24 weeks [24]. Since SGLT2 inhibitors lower blood glucose and to some extent GFR (see below) and thereby their glucosuric potential, these effects are expected to help prevent and/or attenuate episodes of hypoglycemia.
Gluconeogenesis is another important mechanism that serves to maintain normal blood glucose levels and prevent hypoglycemia. As expected for blood glucose lowering agents, SGLT2 inhibition increased plasma glucagon concentrations and endogenous glucose production in T2DM db/db mice [25], diabetic Tallyho/JngJ mice [25], insulin-resistant mice [20], and in patients with T2DM [26;27]. Importantly, evidence was provided that SGLT2 itself is expressed on glucagon-secreting alpha cells in the pancreas, that the expression is reduced in diabetic patients, and that inhibition of SGLT2 on pancreatic alpha cells can induce glucagon release [20]. Thus, SGLT2 inhibition can induce glucagon release and as a consequence hepatic gluconeogenesis by lowering blood glucose levels as well as direct effects on alpha cells (Figure 1).
While the central role of the liver in gluconeogensis is well established and recognized, the kidneys also produce between 15–55 gram glucose/day, and this process is upregulated in diabetes [28]. Phosphoenolpyruvate carboxykinase (PEPCK) is the main regulator of gluconeogensis and is upregulated in the kidneys of diabetic rats and humans. Studies in T1DM Akita mice confirmed that renal PEPCK mRNA was upregulated; however this effect was attenuated by SGLT2 inhibition [14]. Thus, SGLT2 inhibition may lower blood glucose levels in diabetes by inhibiting renal glucose reabsorption and renal gluconeogenesis (Figure 1). In contrast to the liver, still little is known about the regulation of gluconeogenesis in the kidney, and further studies are needed in this regard and to further define the involved mechanisms and the quantitative potential of SGLT2 inhibition.
Previous studies have shown that SGLT2 inhibition improved insulin sensitivity and increased β-cell mass in moderately hyperglycemic female Zucker diabetic fatty (ZDF) rats, i.e. in an early-stage T2DM model [29]. Using male ZDF rats it was shown that SGLT-2 inhibitors also evoked β-cell sparing effects in progressed T2DM with features of reduced β-cell mass [30]. Thus, SGLT2 inhibition, possibly by lowering glucose toxicity, may slow the loss of beta cells in T2DM and thereby the progression of the disease (Figure 1).
In May 2015 the FDA released a warning that SGLT2 inhibitors might increase the risk of ketoacidosis (http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm). Cases have been reported in both T1DM and T2DM diabetic patients. Potential mechanisms include lower insulin and higher glucagon levels in response to SGLT2 inhibition which can lead to increased lipolysis and ketogenesis, and potentially increased renal tubular reabsorption of ketones [31] (Figure 1). The recently reported EMPA-REG study (see more details below) found no difference in rates of ketoacidosis in patients treated with the SGLT2 inhibitor empagliflozin versus placebo over the course of 3 years [32]. Nevertheless, dedicated studies are needed to further define the clinical relevance and the involved mechanisms.
SGLT2 inhibition reduces hypertension, body weight, and cardiovascular mortality in T2DM
Hypertension and obesity are important cardiovascular risk factors. Consistent with preclinical data, a meta-analysis of T2DM patients treated with SGLT2 inhibitors showed a consistent small decrease in systolic blood pressure between 3–6 mm Hg [23]. This effect is likely due to a direct natriuretic and osmotic effect and body weight reduction by SGLT2 inhibition (Figure 1). Recent studies provided evidence that SGLT2 and NHE3 may be functionally linked such that SGLT2 inhibition may also inhibit the Na/H exchanger NHE3 in the proximal tubule [33;34] (Figure 2). Whereas such an interaction could be relevant for the effects of SGLT2 inhibition on GFR and blood pressure, more studies are required to further establish this interaction and its cardiovascular relevance. SGLT2 inhibition may also induce beneficial cardiovascular effects by lowering plasma uric acid levels. Whereas the involved transport mechanism and relevance remains to be determined, there is evidence that glycosuria rather than hyperglycemia increases uricosuria in T1DM patients [35].
In both animal studies and human trials in T2DM, the glucosuric effect of SGLT2 inhibition was consistently associated with lower body weight. In obese rats and mice the reduction in body weight in response to SGLT2 inhibition was associated with lower body fat and increased lipolysis and fatty acid oxidation [36;37]. Lesser visceral and subcutaneous fat has been reported in T2DM patients treated with SGLT2 inhibitors [38;39] (Figure 1). In contrast to T2DM, lipolysis is increased and body fat and body weight are often reduced in poorly controlled T1DM. Notably, SGLT2 inhibition improved blood glucose in T1DM Akita mice and this was associated with increased body weight and epididymal fat adipocyte size [14], indicating that the effect of SGLT2 inhibition on body fat may be affected by the underlining pathophysiology.
The EMPA-REG trial compared the treatment of the SGLT2 inhibitor empagliflozin versus placebo for 3 years in addition to standard care in 7020 patients with T2DM and high cardiovascular risk [32]. Empagliflozin was associated with small improvements in blood glucose control and small reductions in weight, waist circumference, uric acid level, and systolic and diastolic blood pressure with no increase in heart rate and small increases in both LDL and HDL cholesterol. Importantly, empagliflozin treatment reduced the risk of death from cardiovascular disease, hospitalization for heart failure, and death from any cause (relative risk reductions of 38, 35, and 32%, respectively)[32].
Does SGLT2 inhibition provide kidney protection beyond lowering blood glucose levels?
Diabetes is a leading cause of CKD and end stage renal disease. SGLT2 inhibition may induce beneficial effects on the reno-cardiovascular system by lowering blood glucose, body weight, and blood pressure (see above). The pathophysiology of diabetic kidney disease is still poorly understood. T1DM and T2DM can lead to early kidney growth, glomerular hyperfiltration, and pro-inflammatory processes which have been associated with a greater risk for later nephropathy [12]. Beneficial effects on these parameters may have nephroprotective potential, and SGLT2 inhibition could affect these parameters through lowering blood glucose levels and/or because the kidneys sense hyperglycemia and glucotoxicity in part through SGLT2-mediated glucose uptake. To address this issue requires considering the metabolism, needs, and complexity of the in vivo renal system.
SGLT2 inhibition lowers hyperfiltration independent of lowering blood glucose
Hyperglycemia increases the glucose delivery to the proximal tubule and is associated with tubular growth. According to the tubular hypothesis of glomerular hyperfiltration in the diabetic kidney (for review see [12]) these changes increase proximal tubular reabsorption of glucose and Na via SGLT2 and SGLT1, of Na via Na/H exchanger NHE3, and secondarily of Cl, K and fluid; this in turn reduces the tubular Na, Cl, K and fluid load to the macula densa and distal tubule; in order to restore the NaCl and fluid load to the early distal tubule, the GFR is increased through the physiology of tubuloglomerular feedback as well as due to the reduction in Bowman space hydrostatic pressure which is a determinant of the filtration pressure and is sensitive to distal tubular fluid load (Figure 2). The quantitative role of glucose reabsorption in diabetic glomerular hyperfiltration and mechanistic insights have been demonstrated in rats by local application into the early proximal tubule of a non-selective SGLT inhibitor [40] and by systemic application of a selective SGLT2 inhibitor [41], as well as by studies in diabetic SGLT2 knockout mice [13]. All these maneuvers lowered GFR, and the GFR effect did not require a lowering of blood glucose levels, but was associated with an increased NaCl concentration at the macula densa [40;41] and an increase in the hydrostatic pressure in Bowman space [40] (Figures 1&2). In accordance, SGLT2 inhibition for 15 weeks prevented glomerular hyperfiltration in T1DM Akita mice [14]. Recent studies confirmed such an effect in humans: In an 8-week study of T1DM patients, SGLT2 inhibition decreased GFR by 19% in patients with baseline hyperfiltration; again, this effect was independent of blood glucose lowering [42]. SGLT2 inhibition for 3 weeks also induced a small reduction in eGFR and in albumin to creatinine ratios in T2DM patients with baseline GFR of 30–50 ml/min; these effects were mostly maintained over 26 weeks [43]. Similar effects were observed for up to 52 weeks of therapy in T2DM patients with CKD stage 2–3 [24]. Assuming that CKD is associated with deleterious glomerular hyperfiltration in remaining intact nephrons and therefore a high glucose delivery to the individual early proximal tubule, the reduction in GFR in response to SGLT2 inhibition is expected under these conditions in accordance with the tubular hypothesis [4] (Figure 2).
SGLT2 inhibition may lower early kidney growth and inflammation by lowering blood glucose
SGLT2 gene knockout decreased blood glucose levels from 470 to 300 mg/dl in streptozotocin-induced T1DM mice, but did not attenuate the increase in kidney weight, markers of kidney growth, inflammation or injury observed over a period of 4.5 months [13]. SGLT2 inhibition in T1DM Akita mice lowered blood glucose from 500 to 200 mg/dl; at such a strong blood glucose effect (see paper for explanations), SGLT2 inhibition attenuated kidney growth, glomerular growth, molecular markers of renal growth, albuminuria, and inflammation, apparently in proportion to blood glucose levels [14]. SGLT2 inhibition also reduced blood glucose from 400 to 200 mg/dl in T2DM ob/ob mice; again this was associated with a decrease in renal hypertrophy, albuminuria, and markers of inflammation and mesangial matrix expansion [15]. Furthermore, in T1DM eNOS knockout mice with matched high blood glucose levels across all groups, those with SGLT2 inhibition showed no benefit on glomerulosclerosis, tubular atrophy, or tubulointerstitial fibrosis, whereas those treated with an angiotensin II AT1 receptor antagonist did [44]. Together these studies are consistent with the notion that SGLT2 inhibition can lower early kidney growth and inflammation in the diabetic kidney primarily by lowering blood glucose (Figure 1).
What is the consequence of increasing glucose delivery to the segments downstream of the early proximal tubule by SGLT2 inhibition?
The main side effect of SGLT2 inhibition is an increased risk of genital infections and, to a relatively lesser extent, urinary tract infections (Figure 1). There is some remaining discussion on the precise role of glucosuria as a causative factor for these infections [45]. Importantly, upper urinary tract infection (pyelonephritis) is not increased by SGLT2 inhibitors. Humans with familial renal glycosuria due to mutations in SGLT2 do not show signs of general renal tubular dysfunction or other pathological changes; in accordance, genetic or pharmacologic inhibition of SGLT2 in non-diabetic rodents induced glucosuria with no changes in kidney injury markers or only minor increases in markers of tubular growth and protective mechanisms, respectively [4;5].
Using mathematical modeling of the rat proximal tubule, it was proposed that SGLT2 inhibition reduced proximal tubular Na reabsorption but increased oxygen consumption in the outer medullary S3 segment, by increasing Na and glucose delivery to this segment and inhibiting passive paracellular reabsorption and thereby increasing active transcellular reabsorption [46]. The model further predicted that diabetes increased proximal tubular oxygen consumption mostly as a consequence of glomerular hyperfiltration and increased tubular work load. Therefore, the net effect of SGLT2 inhibition on proximal tubular Na reabsorption and oxygen consumption was proposed to largely depend on the extent of the GFR-lowering effect [46]. Along these lines, acute non-selective SGLT inhibition improved cortical oxygen tension in STZ-induced T1DM rats, whereas it reduced medullary oxygen tension in control and diabetic rats [47]. These modeling and experimental data indicate that SGLT2 inhibition may enhance medullary hypoxia by inhibiting paracellular reabsorption in the S3 segment and redistribution of Na reabsorption to the S3 segment and/or medullary thick ascending limb, whereas such an effect is counteracted by the reduction in blood glucose and GFR (Figures 1&2). Longer-term experimental studies and studies in humans are needed as well as studies that address the question whether conditions that increase renal medullary hypoxia (e.g. ischemia-reperfusion or nephrotoxins) increase the sensitivity to SGLT2 inhibition. In this regard, the EMPA-REG trial found that the proportions of patients with adverse events, including acute renal failure, were similar in patients treated with SGLT2 inhibitor versus placebo. In a subgroup analysis of patients with GFR < 60 ml/min (CKD3) there was a favorable although not statistically significant trend toward the primary cardiovascular outcome in the empagliflozin treatment group [32].
Studies using a rat model of polycystic kidney disease (PCKD) found that SGLT2 inhibition for 6 weeks increased cyst volume and induced a 23% higher total kidney weight, without reducing creatinine clearance or changing renal cAMP content or increasing staining for Ki67 as a marker of cell proliferation [48]. This could be of relevance if prescribing SGLT2 inhibitors to PCKD patients. Further dedicated experimental and clinical studies and careful evaluation of ongoing longer term clinical studies will help to refine our understanding of the renal pharmacodynamics and safety of SGLT2 inhibitors (Figure 1).
Effects of SGLT2 inhibition beyond the reno-cardiovascular system
SGLT2 inhibition is a chronic therapy and effects on other clinical conditions are of great interest. In this regards, it was reported: 1) that glycemic control by SGLT2 inhibition for 10 weeks ameliorated cognitive dysfunction in obese T2DM mice [49], and 2) that SGLT2 expression was expressed in human pancreatic and prostate adenocarcinomas, and that SGLT2 inhibitors blocked glucose uptake and reduced tumor growth and survival in a xenograft model of pancreatic cancer. Thus, whereas imaging of functional SGLT2 by PET can help to diagnose and stage pancreatic and prostate cancers, SGLT2 inhibitors may serve for cancer therapy [50].
Conclusions
SGLT2 inhibitors induce a multitude of effects in the diabetic setting that have the potential to affect the progression of the primary disease as well as its cardiovascular consequences including beneficial effects on blood glucose, body weight, body fat, pancreatic beta cells, glomerular hyperfiltration, kidney growth and inflammation, and hypertension, associated with little risk for hypoglycemia. New studies provided further mechanistic clues, but also indicated the need to better understand the nuances of SGLT2 inhibition including effects on the outer medulla, in PCKD as well as their potential for ketoacidosis. In addition to EMPA-REG there are several studies under way looking at the effects of SGLT2 inhibition on cardiovascular and kidney outcomes including DECLARE, CANVAS, and CREDENCE (for review see [3]). Their results will come out in the next few years, complement the results from EMPA-REG, and help to further refine our understanding of the therapeutic potential and safety of SGLT2 inhibition.
Key Points.
-
-
SGLT2 inhibition on pancreatic alpha cells increases glucagon release which can help prevent/attenuate hypoglycemia
-
-
SGLT2 inhibitors induce a multitude of effects that have the potential to affect the progression of the primary diabetic disease as well as its cardiovascular consequences, with little risk for hypoglycemia
-
-
SGLT2 inhibition has the potential for nephroprotection through blood glucose-dependent and independent effects
-
-
need to better understand effects of SGLT2 inhibitors on renal outer medulla, PCKD, ketoacidosis, NHE3, uric acid excretion, and renal gluconeogenesis
Acknowledgments
Financial support and sponsorship
AN’s work was supported by a NIH T32 training grant. VV’s work was supported by the NIH (R01DK56248, R01HL094728, P30DK079337) and the Department of Veterans Affairs.
Footnotes
Conflicts of interest
VV has served as a consultant and received honoraria from Boehringer Ingelheim, Janssen Pharmaceutical, and Intarcia Therapeutics. VV’s work was supported by investigator-initiated research grants by Bristol-Myers Squibb, Astra-Zeneca, and Boehringer Ingelheim, Biberach. For the remaining authors none were declared.
Reference List
- 1.Guariguata L, Whiting DR, Hambleton I, et al. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Scheen AJ. Pharmacodynamics, efficacy and safety of sodium-glucose co-transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs. 2015;75:33–59. doi: 10.1007/s40265-014-0337-y. [DOI] [PubMed] [Google Scholar]
- 3.Inzucchi SE, Zinman B, Wanner C, et al. SGLT-2 inhibitors and cardiovascular risk: proposed pathways and review of ongoing outcome trials. Diab Vasc Dis Res. 2015;12:90–100. doi: 10.1177/1479164114559852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vallon V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med. 2015;66:255–270. doi: 10.1146/annurev-med-051013-110046. [DOI] [PubMed] [Google Scholar]
- 5.Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–89. doi: 10.1177/1479164114561992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 7.Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22:104–112. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balen D, Ljubojevic M, Breljak D, et al. Revised immunolocalization of the Na+-D-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol. 2008;295:C475–C489. doi: 10.1152/ajpcell.00180.2008. [DOI] [PubMed] [Google Scholar]
- 9*.Vrhovac I, Balen ED, Klessen D, et al. Localizations of Na-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch. 2015;467:1881–1898. doi: 10.1007/s00424-014-1619-7. The study used well-validated antibodies to localize SGLT2 and SGLT1 in human kidney. [DOI] [PubMed] [Google Scholar]
- 10.Gorboulev V, Schurmann A, Vallon V, et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes. 2012;61:187–196. doi: 10.2337/db11-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Rieg T, Masuda T, Gerasimova M, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol. 2014;306:F188–F193. doi: 10.1152/ajprenal.00518.2013. The study demonstrated that SGLT2 and SGLT1 can account for all renal glucose reabsorption, and that the increase in SGLT1-mediated glucose reabsorption explains why SGLT2 inhibition only excretes 50–60% of the filtered glucose in euglycemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351–375. doi: 10.1146/annurev-physiol-020911-153333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Vallon V, Rose M, Gerasimova M, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304:F156–F167. doi: 10.1152/ajprenal.00409.2012. The study showed that genetic models of T1DM and T2DM have increased renal SGLT2 expression. SGLT2 knockout lowers the diabetes-induced glomerular hyperfiltration independent of lowering blood glucose levels. SGLT2 inhibition in the absence of a strong effect on hyperglycemia is not sufficient to attenuate early kidney growth and inflammation in diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Vallon V, Gerasimova M, Rose MA, et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol. 2014;306:F194–F204. doi: 10.1152/ajprenal.00520.2013. The study showed an increased renal SGLT2 expression but reduced SGLT1 expression in a genetic model of T1DM. A strong effect of SGLT2 inhibition on hyperglycemia is associated with beneficial effects on early kidney growth and inflammation. The studies provided first evidence that SGLT2 inhibition may lower renal gluconeogenesis in diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15*.Gembardt F, Bartaun C, Jarzebska N, et al. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am J Physiol Renal Physiol. 2014;307:F317–F325. doi: 10.1152/ajprenal.00145.2014. The study showed that a strong effect of SGLT2 inhibition on hyperglycemia is associated with beneficial effects on early kidney growth and inflammation in diabetic mice. [DOI] [PubMed] [Google Scholar]
- 16.Ghezzi C, Wright EM. Regulation of the human Na+ dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol. 2012;303:C348–C354. doi: 10.1152/ajpcell.00115.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura N, Matsui T, Ishibashi Y, et al. Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol Metab Syndr. 2015;7:48. doi: 10.1186/s13098-015-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiwari S, Riazi S, Ecelbarger CA. Insulin’s impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am J Physiol Renal Physiol. 2007;293:F974–F984. doi: 10.1152/ajprenal.00149.2007. [DOI] [PubMed] [Google Scholar]
- 19.Poulsen SB, Fenton RA, Rieg T. Sodium-glucose cotransport. Curr Opin Nephrol Hypertens. 2015;24:463–469. doi: 10.1097/MNH.0000000000000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Bonner C, Kerr-Conte J, Gmyr V, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med. 2015;21:512–517. doi: 10.1038/nm.3828. The study provides first evidence that SGLT2 is expressed on pancreatic alpha cells, such that SGLT2 inhibition directly stimulates glucagon release. [DOI] [PubMed] [Google Scholar]
- 21.Powell DR, DaCosta CM, Gay J, et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab. 2013;304:E117–E130. doi: 10.1152/ajpendo.00439.2012. [DOI] [PubMed] [Google Scholar]
- 22.Ghezzi C, Hirayama BA, Gorraitz E, et al. SGLT2 inhibitors act from the extracellular surface of the cell membrane. Physiol Rep. 2014;2 doi: 10.14814/phy2.12058. pii: e12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monami M, Nardini C, Mannucci E. Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2014;16:457–466. doi: 10.1111/dom.12244. [DOI] [PubMed] [Google Scholar]
- 24.Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:369–384. doi: 10.1016/S2213-8587(13)70208-0. [DOI] [PubMed] [Google Scholar]
- 25.Neschen S, Scheerer M, Seelig A, et al. Metformin supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by suppressing endogenous glucose production in diabetic mice. Diabetes. 2015;64:284–290. doi: 10.2337/db14-0393. [DOI] [PubMed] [Google Scholar]
- 26.Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124:509–514. doi: 10.1172/JCI70704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrannini E, Muscelli E, Frascerra S, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124:499–508. doi: 10.1172/JCI72227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. doi: 10.1111/j.1464-5491.2009.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macdonald FR, Peel JE, Jones HB, et al. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab. 2010;12:1004–1012. doi: 10.1111/j.1463-1326.2010.01291.x. [DOI] [PubMed] [Google Scholar]
- 30**.Hansen HH, Jelsing J, Hansen CF, et al. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J Pharmacol Exp Ther. 2014;350:657–664. doi: 10.1124/jpet.114.213454. This study shows that SGLT2 inhibitors can evoke β-cell sparing effects in a rodent model of progressed T2DM with features of reduced β-cell mass. [DOI] [PubMed] [Google Scholar]
- 31.Taylor SI, Blau JE, Rother KI. SGLT2 Inhibitors May Predispose to Ketoacidosis. J Clin Endocrinol Metab. 2015;100:2849–2852. doi: 10.1210/jc.2015-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015 doi: 10.1056/NEJMc1600827. This study shows that the SGLT2 inhibitor empagliflozin reduced the risk of death from cardiovascular disease, hospitalization for heart failure, and death from any cause in T2DM patients with high cardiovascular risk. [DOI] [PubMed] [Google Scholar]
- 33**.Pessoa TD, Campos LC, Carraro-Lacroix L, et al. Functional Role of Glucose Metabolism, Osmotic Stress, and Sodium-Glucose Cotransporter Isoform-Mediated Transport on Na+/H+ Exchanger Isoform 3 Activity in the Renal Proximal Tubule. J Am Soc Nephrol. 2014;25:2028–2039. doi: 10.1681/ASN.2013060588. This study proposes a functional link between SGLT and NHE3 in the proximal tubule such that SGLT inhibition also inhibits NHE3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Fu Y, Gerasimova M, Mayoux E, et al. SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: possible implications for the prevention of glomerular hyperfiltration. Diabetes. 2014;63(supplement 1):A132. This preliminary study found that SGLT2 inhibition acutely increased the urinary anion gap and its application for 10 days induced a modest metabolic acidosis, which may reflect NHE3 inhibition; chronic SGLT2 inhibition enhanced phosphorylation of renal NHE3 at S552 and S605 in T1DM mice, an effect previously associated with reduced NHE3 activity. [Google Scholar]
- 35.Lytvyn Y, Skrtic M, Yang GK, et al. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am J Physiol Renal Physiol. 2015;308:F77–F83. doi: 10.1152/ajprenal.00555.2014. [DOI] [PubMed] [Google Scholar]
- 36.Yokono M, Takasu T, Hayashizaki Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi: 10.1016/j.ejphar.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki M, Takeda M, Kito A, et al. Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr Diabetes. 2014;4:e125. doi: 10.1038/nutd.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolinder J, Ljunggren O, Johansson L, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16:159–169. doi: 10.1111/dom.12189. [DOI] [PubMed] [Google Scholar]
- 39.Bolinder J, Ljunggren O, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–1031. doi: 10.1210/jc.2011-2260. [DOI] [PubMed] [Google Scholar]
- 40.Vallon V, Richter K, Blantz RC, et al. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–2576. doi: 10.1681/ASN.V10122569. [DOI] [PubMed] [Google Scholar]
- 41.Thomson SC, Rieg T, Miracle C, et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–R83. doi: 10.1152/ajpregu.00357.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129:587–597. doi: 10.1161/CIRCULATIONAHA.113.005081. This study confirms in hyperfiltering diabetic patients that SGLT2 inhibition can lower GFR without lowering blood glucose levels. [DOI] [PubMed] [Google Scholar]
- 43.Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15:463–473. doi: 10.1111/dom.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Gangadharan KM, Gross S, Mudaliar H, et al. Inhibition of kidney proximal tubular glucose reabsorption does not prevent against diabetic nephropathy in type 1 diabetic eNOS knockout mice. PLoS One. 2014;9:e108994. doi: 10.1371/journal.pone.0108994. This study reports that in the absence of lowering blood glucose SGLT2 inhibition does not induce beneficial effects on early kidney growth and inflammation in experimental diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geerlings S, Fonseca V, Castro-Diaz D, et al. Genital and urinary tract infections in diabetes: Impact of pharmacologically-induced glucosuria. Diabetes Res Clin Pract. 2014;103:373–381. doi: 10.1016/j.diabres.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 46*.Layton AT, Vallon V, Edwards A. Modeling oxygen consumption in the proximal tubule: effects of NHE and SGLT2 inhibition. Am J Physiol Renal Physiol. 2015;308:F1343–F1357. doi: 10.1152/ajprenal.00007.2015. This study uses mathematical modeling to propose that SGLT2 inhibition increases oxygen consumption in the S3 segment in the outer medulla by shifting Na and glucose reabsorption to this segment and increasing active transcellular reabsorption. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47*.Neill O, Fasching A, Pihl L, et al. Acute SGLT inhibition normalizes oxygen tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol. 2015 doi: 10.1152/ajprenal.00689.2014. ajprenal. This study indicates that non-selective SGLT inhibition can increase renal medullary hypoxia in non-diabetic and diabetic rats. [DOI] [PubMed] [Google Scholar]
- 48*.Kapoor S, Rodriguez D, Riwanto M, et al. Effect of Sodium-Glucose Cotransport Inhibition on Polycystic Kidney Disease Progression in PCK Rats. PLoS One. 2015;10:e0125603. doi: 10.1371/journal.pone.0125603. This study used a rat model of polycystic kidney disease (PCKD) to report that SGLT2 inhibition increased cyst volume and total kidney weight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin B, Koibuchi N, Hasegawa Y, et al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. 2014;13:148. doi: 10.1186/s12933-014-0148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50**.Scafoglio C, Hirayama BA, Kepe V, et al. Functional expression of sodium-glucose transporters in cancer. Proc Natl Acad Sci USA. 2015;112:E4111–E4119. doi: 10.1073/pnas.1511698112. This study indicates a functional role of SGLT2 in cancer diagnostics and therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]