

Abstract
Wolbachia pipientis is possibly the most widespread endosymbiont of arthropods and nematodes. While all Wolbachia strains have historically been defined as a single species, 16 monophyletic clusters of diversity (called supergroups) have been described. Different supergroups have distinct host ranges and symbiotic relationships, ranging from mutualism to reproductive manipulation. In filarial nematodes, which include parasites responsible for major diseases of humans (such as Onchocerca volvulus, agent of river blindness) and companion animals (Dirofilaria immitis, the dog heartworm), Wolbachia has an obligate mutualist role and is the target of new treatment regimens. Here, we compare the genomes of eight Wolbachia strains, spanning the diversity of the major supergroups (A–F), analysing synteny, transposable element content, GC skew and gene loss or gain. We detected genomic features that differ between Wolbachia supergroups, most notably in the C and D clades from filarial nematodes. In particular, strains from supergroup C (symbionts of O. volvulus and D. immitis) present a pattern of GC skew, conserved synteny and lack of transposable elements, unique in the Wolbachia genus. These features could be the consequence of a distinct symbiotic relationship between C Wolbachia strains and their hosts, highlighting underappreciated differences between the mutualistic supergroups found within filarial nematodes.
Keywords: Wolbachia, GC skew, filarial nematodes, genome characteristics
1. Background
Wolbachia is one of the most widespread and studied genera of intracellular bacteria, encompassing endosymbionts of arthropods and nematodes [1,2]. All Wolbachia strains have historically been classified into a single species, Wolbachia pipientis [3,4]. This species, however, on the basis of single gene and multi-locus phylogenies [5,6], has been divided into 16 monophyletic supergroups, labelled A–Q (as supergroup G is possibly an artefacts we have not included it in the total of 16 considered here) [4,7,8]. The (A,B),(D,(C,F)) phylogenetic relationship among the most studied supergroups has recently been confirmed using whole-genome phylogenetic approaches, albeit only on a limited number of strains [9–11]. The taxonomic status of the major Wolbachia lineages is contentious [4,12]. While a ranking to species level has recently been proposed [13,14] based on genome analyses, this pivotal change in Wolbachia classification does not include all current supergroups and remains to be accepted by the Wolbachia community. Thus, in this work, we have used the historical Wolbachia nomenclature (one species, 16 supergroups).
The different Wolbachia supergroups are associated with distinct sets of hosts in arthropoda and nematoda. The nature of the association between Wolbachia strains and their hosts also varies greatly. The symbiosis between C and D supergroup strains and their filarial nematode hosts presents features associated with mutualism, including 100% prevalence [15], strict vertical inheritance [1,16] and metabolic integration [17–19]. Because filarial nematodes are responsible for major neglected tropical diseases of humans (including onchocerciasis or river blindness, caused by Onchocerca volvulus, and lymphatic filariasis, caused by Brugia malayi among other species), alongside an important infection of companion animals (heartworm, caused by Dirofilaria immitis), this obligate relationship has been exploited for novel anti-filarial treatments, such that the nematodes are sterilized or killed by antibiotics [20–22]. In contrast, A and B supergroup strains, infecting arthropod hosts, have less than 100% prevalence, display evidence of rampant lateral transfer and induce a variety of reproductive manipulation phenotypes, including cytoplasmic incompatibility, parthenogenesis, killing of male embryos and feminization of genetic males [2,23]. Wolbachia strains of the F supergroup have been observed in association with both arthropods and nematodes [4,24].
A recent genomic study, focused on two strains of Wolbachia belonging to either supergroup A or B and co-infecting Drosophila simulans, showed a lack of genetic exchange, suggesting their genetic isolation [14]. Are these results by Ellegaard et al. unique within the genus, or is genetic isolation common among Wolbachia lineages? If the different supergroups experienced independent evolution, then we can expect their genomes to present specific features as a consequence of their independent evolutionary histories.
Wolbachia strains have reduced genome size, a feature observed in most endosymbiont bacteria [25–27]. The process of genome reduction in endosymbionts can be classified in four stages [28], as follows. (i) Free-living bacteria: large genome size, few transposable elements, gene acquisition and loss, interstrain recombinations. (ii) Recently host-restricted bacteria: genome size smaller than free-living bacteria, many transposable elements, chromosome rearrangements and loss of genomic regions. (iii) Long-term obligate symbionts: further reduced genome size, stable chromosome and few or no transposable elements. (iv) Tiny-genome symbionts: very small genome size and high chromosome stability.
In this work, we compared the genomes of Wolbachia strains belonging to the A–D and F supergroups, in order to identify conserved and variable genomic features. We considered intragenomic recombinations, transposable elements, chromosome rearrangements, mutational bias and gene loss or gain. We found that Wolbachia strains belonging to supergroup C have conserved and distinct genomic features, probably the result of extensive periods of independent evolution.
2. Methods
2.1. Dataset
The genome assemblies of eight Wolbachia strains belonging to A–D and F supergroups (wMel, wRi, wPipPel, wDi, wOo, wBm, wLs and wCle) and of seven other Alphaproteobacteria (Caulobacter crescentus strain CB15, Cre; Anaplasma centrale strain Israel, Ace; Anaplasma phagocytophilum strain HZ, Aph; Ehrlichia chaffeensis strain Arkansas, Ech; Ehrlichia ruminantium strain Gardel, Eru; Neorickettsia risticii strain Illinois, Nri; Neorickettsia sennetsu strain Miyayama, Nse) were retrieved from public database (for more information about genome features, see table 1). Caulobacter crescentus was chosen because it is a complete genome of an alphaproteobacterium for which origin and terminus of replication were experimentally determined [29]. The genome assemblies included in the study are all complete or almost complete, with the exception of the genome of wLs, which is divided into 10 contigs. We included the genome of wLs in the study as a second representative of the nematode-associated Wolbachia supergroup D.
Table 1.
List of the genomes included in this study. For each genome, information about the strain, the corresponding host and the genome are reported.
| Wolbachia strains (short name) | hosts | supergroups | no. contigs | contig length (nt) | sources |
|---|---|---|---|---|---|
| wMel | Drosophila melanogaster | A | 1 | 1 267 782 | NC_002978 |
| wRi | Drosophila simulans | A | 1 | 1 445 873 | NC_012416 |
| wPipPel | Culex quinquefasciatus | B | 1 | 1 482 455 | NC_010981 |
| wOo | Onchocerca ochengi | C | 1 | 957 990 | HE660029 |
| wDi | Dirofilaria immitis | C | 2 | 919 954, 1058 | http://dirofilaria.nematod.es |
| wBm | Brugia malayi | D | 1 | 1 080 084 | NC_006833 |
| wLs | Litomosoides sigmodontis | D | 10 | 605 213, 245 144, 135 750, 38 729, 16 626, 5094, 1163, 500, 375, 342 | http://litomosoides.nematod.es |
| wCle | Cimex lectularius | F | 1 | 125 0060 | AP013028 |
| Outgroup strains (short name) | strain names | supergroups | no. contigs | contig length (nt) | sources |
|---|---|---|---|---|---|
| Ace | Anaplasma centrale str. Israel | — | 1 | 1 206 806 | NC_013532 |
| Aph | Anaplasma phagocytophilum HZ | — | 1 | 1 471 282 | NC_007797 |
| Ech | Ehrlichia chaffeensis str. Arkansas | — | 1 | 1 176 248 | NC_007799 |
| Eru | Ehrlichia ruminantium str. Gardel | — | 1 | 1 499 920 | NC_006831 |
| Nri | Neorickettsia risticii str. Illinois | — | 1 | 879 977 | NC_013009 |
| Nse | Neorickettsia sennetsu str. Miyayama | — | 1 | 859 006 | NC_007798 |
| Ccr | Caulobacter crescentus CB15 | — | 1 | 4 016 947 | NC_002696 |
2.2. Origin of replication and genome orientation
The genomes of the Wolbachia strains included in the study were aligned with progressiveMauve [30]. For each genome, the position of the origin of replication (ORI) was inferred on the basis of the wMel and wBm ORI positions proposed by Ioannidis et al. [31]. Each genome assembly was oriented following the wMel and wBm ORI orientation, and organized to start with the ORI position. Below, we refer to these reorganized genomes as ‘ORI-starting’ genomes.
2.3. Analysis of genome rearrangements
Pairwise genome alignments of the wMel, wRi, wPipPel, wDi, wOo, wBm, wLs and wCle Wolbachia strains were produced and plotted with the software MUMmer v. 3.0 [32].
2.4. Transposable elements
Insertion sequences (ISs) and group II introns were identified and annotated in wDi (C supergroup), wLs (D supergroup) and wCle (F supergroup). Group II introns were identified following the methods of Leclercq et al. [33]. IS elements were identified using ISSaga [34], followed by manual curation of ISSaga output files. For wLs, most ISSaga hits were short and often formed groups of two to four hits located next to each other. This is typical of pseudo-genized and degraded IS elements. We attributed two consecutive hits to the same or to distinct IS copies using the following rules:
(1) IS family: if the two hits belong to different IS families, then they belong to distinct copies. Otherwise, go to criterion (2).
(2) Orientation: if the two hits are in opposite orientation, then they belong to distinct copies. Otherwise, go to criterion (3).
(3) Physical distance: if distance between the two hits is greater than 300 bp, then they belong to distinct copies. Otherwise, they belong to the same copy.
2.5. GC skew
The cumulative GC skew curve was calculated for each of the ORI-starting Wolbachia genome assemblies. It was calculated applying the formula ΣG − C/G + C, with a window size of 1000 nt and step size of 100 nt (analyses were performed with an in-house Perl script).
For each of the Wolbachia strains in the dataset, with the exception of wLs (fragmented in 10 contigs), the potential effect of genomic rearrangements on the current GC skew curve was evaluated. The following procedure was used: (i) the ORI-starting genome was aligned against the ORI-starting wDi genome with progressiveMauve; (ii) the detected syntenic blocks were sorted and oriented according to the ORI-starting wDi order; (iii) the cumulative GC skew curves were calculated for both the obtained reoriented genome and relative aligned wDi genome; and (iv) the mean absolute difference between the two curves was calculated. The mean distance values calculated for all Wolbachia strains were compared with the Wilcoxon–Mann–Whitney test with Bonferroni post hoc correction.
2.6. Mutational bias
The effect of mutational bias on the guanine and cytosine distribution along the genomes of Wolbachia strains C and F (wDi, wOo—C supergroup; wCle—F supergroup) was evaluated using Wolbachia strains A, B and D (wMel, wRi—A supergroup; wPipPel—B supergroup; wLs and wBm—D supergroup) as outgroups. A dataset of single-copy orthologous genes, shared among all the eight Wolbachia strains included in the study, was obtained with OrthoMCL [35] and in-house Perl scripts. Nucleotide gene sequences were aligned on the corresponding amino acid alignments, using Muscle [36] and in-house Perl scripts. For each gene, the number of mutations towards G and towards C for third position residues was evaluated for each pair of Wolbachia strains, using a custom Perl script. The mutational biases along wDi, wOo and wCle genomes were evaluated comparing each of them against all the other seven Wolbachia strains included in the study. The mutational biases on the Watson (forward) and Crick (reverse) strands (sensu lato) were evaluated by calculating the respective bias indexes. For genes located on the Watson strand, the bias index was computed as the ratio between the number of mutations towards G and the number of mutations towards C. Conversely, for genes located on the Crick strand, the bias index was computed as the ratio of the number of mutations towards C and the number of mutations towards G. The average of the middle positions of the genes with bias index more than one and less than one were compared with the Wilcoxon–Mann–Whitney test.
2.7. Gene loss and gain
Events of gene loss/gain that occurred in the genome of the ancestor of Wolbachia supergroup C were inferred on the basis of the pattern of gene presence/absence in the present strains. This presence/absence pattern was reconstructed, annotating the genomes of the eight Wolbachia strains included in the study and of six Anaplasmataceae outgroups, against the clusters of orthologous groups (COGs) database by PSI-BLAST with a p-value cut-off of 10–5. The loss and gain events occurred in the genome of the ancestor of Wolbachia supergroup C were inferred using the Gloome tool [37], mapping the pattern of presence/absence of functional COG annotations on a phylogenetic tree reconstructed from the literature [9–11,38]. The Gloome tool confers a probability value to each inferred event. Only events with a probability greater than 75% were considered reliable and thus manually checked.
3. Results
We are interested in the evolutionary dynamics of Wolbachia, an important genus of intracellular bacteria. Here, we explore the genomic signatures in eight Wolbachia strains from supergroups A to F, including intragenomic recombination, transposable elements, GC skew curve, mutational bias and gene loss or gain. We focus specifically on differences between two supergroup C genomes, wDi (from the dog heartworm, D. immitis) and wOo (from Onchocerca ochengi, a bovine parasite very closely related to O. volvulus); and two supergroup D genomes, wBm (from a human lymphatic filariasis parasite, B. malayi) and wLs (from a filarial model of rodents, Litomosoides sigmodontis).
3.1. Intragenomic recombinations
Wolbachia genomes have been reported to have undergone extensive rearrangement in comparison with other Rickettsiales [39]. We analysed eight genome assemblies belonging to Wolbachia strains from supergroups A to F [9,18,40–42]. An alignment of these high-quality genomic assemblies revealed conservation of synteny among the supergroup C genomes wDi and wOo, in marked contrast with very low levels of synteny within and between the other supergroups (figure 1). However, the wMel and wRi genomes also show conserved synteny, probably a consequence of their low evolutionary distance [9,42].
Figure 1.

Synteny conservation in supergroup C Wolbachia. A graphic representation of MUMmer v. 3.0 output is shown in the dot plots on the right. Red lines display collinear regions, whereas blue lines display inversions. Phylogenetic relationships among the Wolbachia strains are shown on the left.
3.2. Transposable elements
Synteny breakage and recombination is often associated with repeats and transposable elements. We therefore screened the Wolbachia genomes for classes of transposable element (electronic supplementary material, table S1; figure 2). We found no group II introns in the wDi (C supergroup) and wLs (D supergroup) genomes. However, ISs had a striking, disjointed pattern of presence. While wDi had only a single IS (similar to ISWpi16), wLs contained 210 IS copies. Supergroup A and B arthropod Wolbachia genomes also have many IS elements [43], albeit fewer than wLs. IS elements cover nearly 12% of the wLs genome, a higher percentage than in any other Wolbachia genome sequenced to date. Despite their high copy number, all wLs IS copies appear to be degraded and there is no apparent ‘live’ transpositional activity. Remarkably, 97% of the wLs IS copies (204/210) belong to a single IS type (ISWpi10). The six remaining copies belong to ISWpi5 (electronic supplementary material, table S2). Interestingly, the genome of wCle (F supergroup) is characterized by a high density (10%) and diversity (11 different types) of IS elements and the presence of group II introns (electronic supplementary material, table S1).
Figure 2.

Insertion sequences in Wolbachia genomes. (a) The known phylogenetic relationships among the Wolbachia strains are shown. (b) Results of insertion sequence (IS) analyses performed on the wLs, wBm, wDi, wOo, wCle, wMel, wRi and wPipPel Wolbachia strains are displayed as a histogram showing IS quantification. The known phylogenetic relationships among the Wolbachia strains are shown in (a). For each strain, the corresponding supergroup is colour-coded: orange, A; violet, B; green, C; blue, D; black, E and red, F.
Comparing the D supergroup genomes, no IS copy was found to be inserted at an orthologous site, despite the high number of IS copies. By contrast, in supergroup C, the single IS copy found in wDi is orthologous to the ISWpi16 copy found in wOo.
3.3. GC skew and mutational bias
Another feature described as characteristic of arthropod Wolbachia genomes is the absence of strong GC skew [39], in contrast with the pattern commonly observed in most free-living bacteria and in endosymbiotic bacteria such as Buchnera aphidicola [44,45]. The cumulative GC skew curve of the seven completely sequenced Wolbachia genomes included in the study (wMel, wRi, wPipPel, wDi, wOo, wBm and wCle) and of the Alphaproteobacterium outgroup, C. crescentus, were calculated (figure 2). In agreement with previous analyses on a smaller dataset [39], most Wolbachia genomes do not present any genome-wide pattern of GC skew (figure 3). However, the wDi genome has a strong pattern of GC skew (figure 3), which, among endosymbionts, is typically observed in bacteria with extremely reduced genomes.
Figure 3.
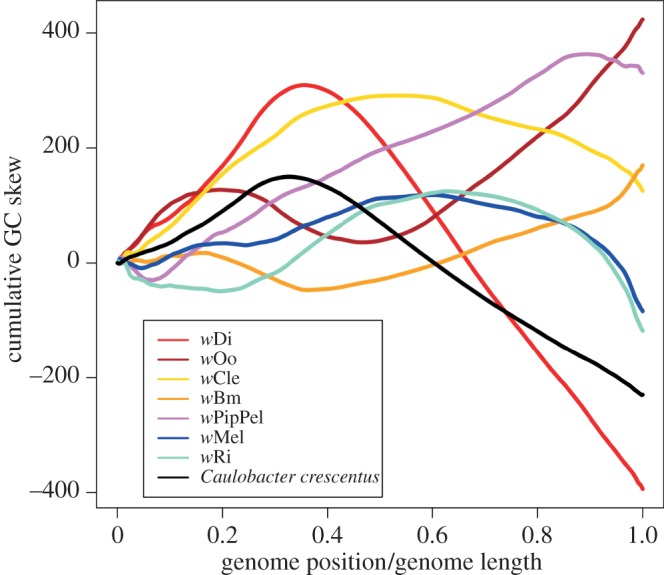
Cumulative GC skew curves. GC skew was calculated with window size of 1000 nucleotides and step size of 100 nucleotides. The curve for Caulobacter crescentus is coloured in black, whereas the curves for Wolbachia strains are coloured as follows: wMel, blue; wRi, azure; wPipPel, pink; wDi, red, wOo, dark red; wBm, orange; wCle, yellow.
This pattern of cumulative GC skew in wDi could have originated uniquely in wDi or could be an ancestral feature of Wolbachia, lost by most lineages. To test the hypothesis that the wDi GC skew pattern is ancestral, we evaluated whether its absence in the other six complete Wolbachia genomes included in the study could have been caused by genome rearrangements. We reordered each genome to conform the wDi gene order and recalculated the GC skew on the ‘pseudo-ancestral’ genome (figure 4). While rearrangement of supergroup A–C and F genomes did not reveal any hidden GC skew pattern, in the rearranged wOo genome (belonging to the C supergroup), we observed a trend similar to that of wDi (figure 4). No better fit was observed between native wDi and the other five rearranged Wolbachia genomes included in the analysis (wMel, wRi, wPipPel, wBm and wCle; electronic supplementary material, figure S1).
Figure 4.

Cumulative GC skew curves of six reoriented Wolbachia genomes (red) compared with the wDi genome (blue). Genomes were reordered on the basis of the wDi gene order using a progressiveMauve genome alignment.
Based on the GC skew analysis presented above, the occurrence of genome rearrangements could explain the difference in GC distribution between wDi and the other C supergroup Wolbachia genome included in the study (i.e. wOo), but cannot explain the differences between wDi and the genomes of strains belonging to other supergroups. We thus hypothesized that, during the evolution of the C supergroup, a mutational bias led to the asymmetric distribution of GC observed in the wDi genome. Indeed, in the wDi genome, the Watson strand of the genes localized on the first part of the genome tends to be mutated towards G more than towards C, opposite to what was detected in the genes localized on the second part of the genome, as shown in figure 5.
Figure 5.

GC mutational bias. The figure displays information about the (a) wDi, (b) wOo and (c) wCle genomes, including GC skew, cumulative GC skew and mutational bias calculated using the wBm genome as reference (see Material and methods). In each graph, the position along the genome is reported on the x-axis; the GC skew curve is reported with an orange line and the cumulative GC skew with a red line. Genes shared among all the Wolbachia strains included in the study are represented with blue/azure coloured points: blue genes have a positive GC mutational bias index, whereas azure genes have a negative GC mutational bias index (see Materials and methods). The horizontal boxplots indicate the average position of genes with positive GC mutational bias index (blue) and with negative GC mutational bias index (azure).
GC skew is thought to arise from biased substitution processes driven by the replicational structure of the circular chromosome. This model explains the opposite mutational biases observed in the genes in the first and in the second part of the wDi genome (figure 5a). Following this model, we infer that the position of the mutational bias switch, near the middle of the wDi genome (figure 5a), corresponds to the position of the terminus of replication, but this should be verified experimentally. No mutational bias was observed for the other analysed strains, wOo (supergroup C) and wCle (supergroup F; figure 5b,c).
3.4. Gene loss and gain in the C Wolbachia ancestor
Wolbachia genomes vary in size from approximately 0.9 to approximately 1.4 Mb. These size differences could have arisen from either gain of genetic material (including transposable elements and phages) or loss, or both. Gene loss and gain have a strong impact on Wolbachia strains’ metabolic capability. Indeed, the genome stability observed in C Wolbachia strains, in particular in the wDi strain, could be the consequence of specific events of gene loss occurring during the evolution of Wolbachia supergroup C.
We identified the putative events of gene loss and gain in the ancestor of the Wolbachia supergroup C, on the basis of the COG annotation of the genomes of the 14 Anaplasmataceae strains included in the study (of which eight belong to Wolbachia, two to Anaplasma, two to Ehrlichia and two to Neorickettsia). Mapping this COG presence/absence pattern on the Anaplasmataceae tree, 22 loss events and no gain events were inferred at node of the C Wolbachia strain ancestor (figure 6; electronic supplementary material, table S2). The replication, recombination and repair pathway was affected by a particularly intense erosion process, from which the C Wolbachia ancestor lost eight members (figure 6; electronic supplementary maerial, table S3).
Figure 6.
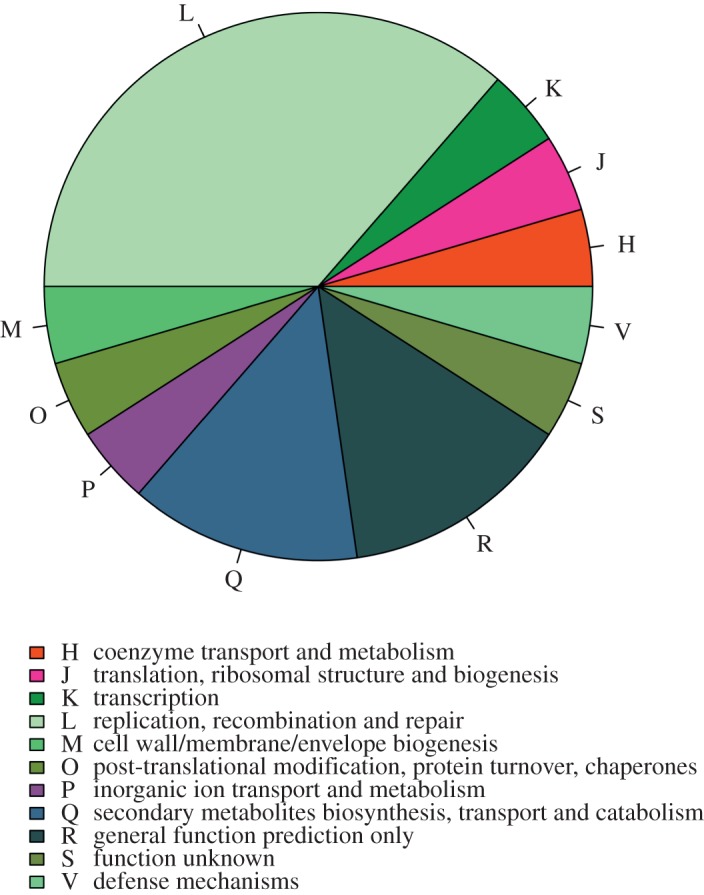
COG classification of the genes identified as putatively lost during the evolution of the wDi and wOo ancestor.
4. Discussion
Bacteria belonging to the alphaproteobacterial genus Wolbachia have been classified into 16 supergroups, mainly on the basis of 16S rDNA phylogenetic analyses. This classification groups Wolbachia strains coherently with the host taxonomy and ecology. Phylogenomic analyses have further organized most of the Wolbachia diversity into two monophyletic clusters of supergroups: (A + B) and (C + D + F) [9–11]. While recombination has been observed between strains belonging to the same supergroup, each supergroup may be relatively genetically isolated. Indeed, no recombination was detected between wHa (supergroup A) and wNo (supergroup B), despite their coinfection of the same arthropod species [14]. We can expect that Wolbachia strains belonging to a genetically isolated supergroup should present conserved genomic signatures, as a consequence of their independent evolutionary patterns. We sought to detect structural genomic differences between supergroups, with a particular focus on the (C + D + F) cluster.
Early comparisons of Wolbachia genomes revealed an extreme lack of synteny between strains from supergroups A and B, and wBm (supergroup D) [39]. Several additional Wolbachia genomes belonging to supergroups C, D and F are now available: specifically wDi and wOo (supergroup C), wLs (supergroup D) and wCle (supergroup F). This has allowed us to further investigate synteny patterns in the (C + D + F) cluster. Here, we find that the genomes of supergroup C show an elevated level of synteny, compared with the supergroup D genomes included in the study (figure 1). This disjointed pattern suggests that supergroup D genomes may be evolving differently from those of the strains of supergroup C. Similar results, on a slightly different genome dataset, where recently obtained by Ramírez-Puebla et al. [13].
IS elements are present in extremely variable numbers in different bacterial lineages, and are known to promote intragenomic recombination, causing the interruption of synteny conservation [46]. Wolbachia genomes vary dramatically in terms of their IS content. Supergroup C genomes show a paucity of IS elements, whereas genomes of supergroups A, B, D and F have many IS elements, a pattern consistent with a possible role for IS in synteny breakage in some Wolbachia genomes. The low number of IS elements observed in the C Wolbachia genomes (ranging from one to six—see electronic supplementary material, table S1) is consistent with the amounts observed in genomes of other long-term, vertically inherited obligate symbionts [28]. Conversely, the genomes of arthropod Wolbachia strains included in the study (strains from supergroups A, B and F) contain a higher number of IS elements (ranging from 105 to 181—see electronic supplementary material, table S1), many of which are potentially capable of transposition. This is typical of endosymbionts that undergo at least some horizontal transmission [47]. Interestingly, supergroup D genomes (wBm and wLs) contain a high number of IS elements (respectively 52 and 210—see electronic supplementary material, table S1), but they are all disrupted and on their way to being lost, as part of the reductive genome evolution of these vertically inherited endosymbionts [28]. This is consistent with a scenario in which IS transpositional activity ceased a long time ago in these Wolbachia strains, as previously noted for other endosymbionts with a similar lifestyle [28].
In general, lifestyle is thought to be a major factor influencing mobile DNA evolution in intracellular bacteria [47,48]. In Wolbachia, the mutualistic supergroup C and D strains are only vertically inherited in their nematode hosts, whereas supergroup A and B strains experience a combination of vertical and horizontal transmission. Horizontal transmission should enable more frequent contact and genetic exchanges with other microorganisms, thereby maintaining a flux of intact IS copies and generating higher IS diversity. The supergroup F genome (from wCle) is also from a strain exhibiting mutualistic interactions with its host, but wCle displays high IS diversity, like the non-mutualistic supergroup A and B strains. This suggests that wCle might have recently shifted to mutualism and still shows transposable element patterns of its non-mutualistic ancestor.
Intragenomic recombinations can affect the distribution of guanine and cytosine along bacterial genomes. Studies on free-living bacterial genomes showed that in many cases, during genome replication, the Watson and Crick strands are subjected to asymmetric cumulative mutation pressures [49,50]. Indeed, intragenomic recombinations randomize the cumulative effect of this mutation pressure. For this reason, the strong asymmetry distribution of cytosine and guanine observed in the wDi genome (figure 3) suggests that it experienced a long period of chromosome stability, in contrast with other Wolbachia genomes. We reordered the other Wolbachia genomes and compared them with wDi to identify any residual ancestral GC skew signatures that had not yet been erased during subsequent evolution. The reoriented wOo genome showed stronger GC skew than the natively ordered genome, albeit less pronounced than that of wDi, and was more similar to the wDi curve than that of other reoriented Wolbachia genomes (figure 4).
The analysis of mutational bias on the Watson strand of the wDi genome shows that on the genes localized in the first part of the wDi genome, mutations towards G are positively selected in comparison with mutations towards C, whereas an inverse pattern is seen in the genes localized on the second part of the wDi genome (figure 5). The combination between high genome stability and GC mutational bias probably led to the current asymmetrical distribution of GC along the wDi genome. Interestingly, just a weak GC mutational bias can be observed in the wOo genome (figure 5), which currently maintains the GC distribution originated during the evolution of the wOo-wDi ancestor. This result suggests that the wOo genome replicates with a very low mutation rate: not enough to generate significant mutational bias, but also not enough to erase the ancestral GC distribution signal conserved in the wDi genome.
Klasson & Andersson [45] described an asymmetric distribution of G and C in the genome of the aphid endosymbiont B. aphidicola, and hypothesized that the lack of recA and mutational bias could be the causes of this GC distribution pattern. Indeed, intragenomic recombination can lead to bacterial death, in the absence of an adequate homologous recombination pathway. recA, one of the most important genes involved in the homologous recombination pathway, is lacking in all supergroup C genomes [18,51]. By contrast, in supergroup D, the homologous recombination pathway is complete in the only closed genome available, wBm [40,51], supporting the hypothesis of higher genome plasticity. However, wBm may be exceptional, as other supergroup D genomes appear to have a deficient homologous recombination pathway [51]. It must be noted that these genomes are not closed, thus additional complete genome sequences from supergroup D strains are needed to determine whether wBm is unusual in its recA status and rearrangement history.
Is the wDi genome representative of the ancestor of all Wolbachia? We suggest not. It is likely that the loss of the recA pathway in the last common ancestor of supergroup C and the general loss of IS elements resulted in a halt to genome rearrangement, and this stability then permitted a build-up of GC skew and mutational bias in the stabilized genome. Limited subsequent rearrangements observed in wOo have obscured, but not erased, the signatures of evolutionary stability.
The process of gene loss is one of the most important phenomena in the evolution of intracellular bacteria [52]. Within the Wolbachia genus, this process is exacerbated in filarial strains, where gene acquisition from other bacterial species has not been described. In our analysis, recA was identified as being lost from supergroup C, as expected, but we also identified a number of other losses in the supergroup C lineage associated with a variety of other processes. The physiological linkage between these gene losses, if any, is unclear.
5. Conclusion
In conclusion, our analyses present evidence supporting the hypothesis that Wolbachia supergroups are not just phylogenetic lineages. Evidence of genetic isolation and convergent evolution had been reported for two strains belonging to Wolbachia supergroups A and B [14]. Here, we report evidence that supergroup C strains share a suite of genomic features (very low number of genomic rearrangements, paucity of IS elements, strong GC asymmetric distribution) that is commonly observed in endosymbiotic bacteria with extremely reduced genomes, which have long-lasting relationships with their host. These features are absent in the other lineages of Wolbachia included in the study. Genomic analyses enabled us to infer the evolutionary pathway that originated this suite of features. Our results are not sufficient to conclude if the different genomic features observed in C and D supergroup genomes are the result of different selective pressures, or if the two supergroups are in two different stages of the genome reduction process typical of bacterial endosymbionts. Additional genomes will help to shed light on this matter.
Nematode Wolbachia strains live in mutualistic association with the host, and are considered important targets for anti-filarial pharmaceutical treatments [41]. In this work, we report genomic evidence that C and D Wolbachia supergroup strains experienced a long period of independent evolution. We can hypothesize that the observed differences between the C and D Wolbachia strain genomes are a consequence of different specific symbiotic relationships with the filarial hosts, probably resulting in specific host–Wolbachia metabolic complementarities. If our results are supported by analyses of additional Wolbachia genomes, the mutualism of C and D Wolbachia strains with filarial nematodes should be considered separately, with potential implications for anti-Wolbachia strategies, as drugs effective against one supergroup may not always be equally potent against the other.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
F.C. and C.B. thank Massimo Pajoro for interesting and inspiring discussions.
Authors' contributions
F.C., R.C., C.B., M.B., A.D., D.S. participated in the design of the study; F.C. performed the analyses on synteny, ORI position, genomes reorientation, GC skew, mutational bias, and gene loss and gain; R.C. carried out the transposable elements identification and analysis; F.C., M.M. and D.S. interpreted and refined the bioinformatic analyses; F.C., R.C. and D.S. drafted the manuscript; M.B., B.L.M. and C.B. critically revised the manuscript.
Competing interests
The authors declare no competing interests.
Funding
R.C. was supported by a European Research Council Starting Grant (FP7/2007–2013, grant no. 260729 EndoSexDet).
References
- 1.Bandi C, Anderson TJ, Genchi C, Blaxter ML. 1998. Phylogeny of Wolbachia in filarial nematodes. Proc. R. Soc. Lond. B 265, 2407–2413. (doi:10.1098/rspb.1998.0591) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Werren JH, Baldo L, Clark ME. 2008. Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. (doi:10.1038/nrmicro1969) [DOI] [PubMed] [Google Scholar]
- 3.Hertig M. 2009. The rickettsia, Wolbachia pipientis (gen. et sp.n.) and associated inclusions of the mosquito, Culex pipiens. Parasitology 28, 453 (doi:10.1017/S0031182000022666) [Google Scholar]
- 4.Lo N, Paraskevopoulos C, Bourtzis K, O'Neill SL, Werren JH, Bordenstein SR, Bandi C. 2007. Taxonomic status of the intracellular bacterium Wolbachia pipientis. Int. J. Syst. Evol. Microbiol. 57, 654–657. (doi:10.1099/ijs.0.64515-0) [DOI] [PubMed] [Google Scholar]
- 5.Werren JH, Zhang W, Guo LR. 1995. Evolution and phylogeny of Wolbachia: reproductive parasites of arthropods. Proc. R. Soc. Lond. B 261, 55–63. (doi:10.1098/rspb.1995.0117) [DOI] [PubMed] [Google Scholar]
- 6.Bordenstein SR, Paraskevopoulos C, Dunning Hotopp JC, Sapountzis P, Lo N, Bandi C, Tettelin H, Werren JH, Bourtzis K. 2009. Parasitism and mutualism in Wolbachia: what the phylogenomic trees can and cannot say. Mol. Biol. Evol. 26, 231–241. (doi:10.1093/molbev/msn243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Augustinos AA, et al. 2011. Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS ONE 6, e28695 (doi:10.1371/journal.pone.0028695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glowska E, Dragun-Damian A, Dabert M, Gerth M. 2015. New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 30, 140–146. (doi:10.1016/j.meegid.2014.12.019) [DOI] [PubMed] [Google Scholar]
- 9.Comandatore F, et al. 2013. Phylogenomics and analysis of shared genes suggest a single transition to mutualism in Wolbachia of nematodes. Genome Biol. Evol. 5, 1668–1674. (doi:10.1093/gbe/evt125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T. 2014. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc. Natl Acad. Sci. USA 111, 10 257–10 262. (doi:10.1073/pnas.1409284111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerth M, Gansauge M-T, Weigert A, Bleidorn C. 2014. Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nat. Commun. 5, 5117 (doi:10.1038/ncomms6117) [DOI] [PubMed] [Google Scholar]
- 12.Pfarr K, Foster J, Slatko B, Hoerauf A, Eisen JA. 2007. On the taxonomic status of the intracellular bacterium Wolbachia pipientis: should this species name include the intracellular bacteria of filarial nematodes? Int. J. Syst. Evol. Microbiol. 57, 1677–1678. (doi:10.1099/ijs.0.65248-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramírez-Puebla ST, Servín-Garcidueñas LE, Ormeño-Orrillo E, Vera-Ponce de León A, Rosenblueth M, Delaye L, Martínez J, Martínez-Romero E. 2015. Species in Wolbachia? Proposal for the designation of ‘Candidatus Wolbachia bourtzisii’, ‘Candidatus Wolbachia onchocercicola’, ‘Candidatus Wolbachia blaxteri’, ‘Candidatus Wolbachia brugii’, ‘Candidatus Wolbachia taylori’, ‘Candidatus Wolbachia collembolicola’ and ‘Candidatus Wolbachia multihospitum’ for the different species within Wolbachia supergroups. Syst. Appl. Microbiol. 38, 390–399. (doi:10.1016/j.syapm.2015.05.005) [DOI] [PubMed] [Google Scholar]
- 14.Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SGE. 2013. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 9, e1003381 (doi:10.1371/journal.pgen.1003381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor MJ, Bandi C, Hoerauf A. 2005. Wolbachia bacterial endosymbionts of filarial nematodes. Adv. Parasitol. 60, 245–284. (doi:10.1016/S0065-308X(05)60004-8) [DOI] [PubMed] [Google Scholar]
- 16.Casiraghi M, Anderson TJ, Bandi C, Bazzocchi C, Genchi C. 2001. A phylogenetic analysis of filarial nematodes: comparison with the phylogeny of Wolbachia endosymbionts. Parasitology 122, 93–103. (doi:10.1017/S0031182000007149) [DOI] [PubMed] [Google Scholar]
- 17.Hosokawa T, Koga R, Kikuchi Y, Meng X-Y, Fukatsu T. 2010. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl Acad. Sci. USA 107, 769–774. (doi:10.1073/pnas.0911476107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darby AC, et al. 2012. Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res. 22, 2467–2477. (doi:10.1101/gr.138420.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill AC, Darby AC, Makepeace BL. 2014. Iron necessity: the secret of Wolbachia’s success? PLoS Negl. Trop. Dis. 8, e3224 (doi:10.1371/journal.pntd.0003224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langworthy NG, Renz A, Mackenstedt U, Henkle-Dührsen K, de Bronsvoort MB, Tanya VN, Donnelly MJ, Trees AJ. 2000. Macrofilaricidal activity of tetracycline against the filarial nematode Onchocerca ochengi: elimination of Wolbachia precedes worm death and suggests a dependent relationship. Proc. R. Soc. Lond. B 267, 1063–1069. (doi:10.1098/rspb.2000.1110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazzocchi C, Mortarino M, Grandi G, Kramer LH, Genchi C, Bandi C, Genchi M, Sacchi L, McCall JW. 2008. Combined ivermectin and doxycycline treatment has microfilaricidal and adulticidal activity against Dirofilaria immitis in experimentally infected dogs. Int. J. Parasitol. 38, 1401–1410. (doi:10.1016/j.ijpara.2008.03.002) [DOI] [PubMed] [Google Scholar]
- 22.Johnston KL, Ford L, Umareddy I, Townson S, Specht S, Pfarr K, Hoerauf A, Altmeyer R, Taylor MJ. 2014. Repurposing of approved drugs from the human pharmacopoeia to target Wolbachia endosymbionts of onchocerciasis and lymphatic filariasis. Int. J. Parasitol. Drugs Drug Resist. 4, 278–286. (doi:10.1016/j.ijpddr.2014.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordaux R, Bouchon D, Grève P. 2011. The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends Genet. 27, 332–341. (doi:10.1016/j.tig.2011.05.002) [DOI] [PubMed] [Google Scholar]
- 24.Keiser PB, Coulibaly Y, Kubofcik J, Diallo AA, Klion AD, Traoré SF, Nutman TB. 2008. Molecular identification of Wolbachia from the filarial nematode Mansonella perstans. Mol. Biochem. Parasitol. 160, 123–128. (doi:10.1016/j.molbiopara.2008.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M, Aksoy S. 2002. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32, 402–407. (doi:10.1038/ng986) [DOI] [PubMed] [Google Scholar]
- 26.Sassera D, et al. 2011. Phylogenomic evidence for the presence of a flagellum and cbb3 oxidase in the free-living mitochondrial ancestor. Mol. Biol. Evol. 28, 3285–3296. (doi:10.1093/molbev/msr159) [DOI] [PubMed] [Google Scholar]
- 27.Rao Q, et al. 2015. Genome reduction and potential metabolic complementation of the dual endosymbionts in the whitefly Bemisia tabaci. BMC Genomics 16, 226 (doi:10.1186/s12864-015-1379-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCutcheon JP, Moran NA. 2011. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. (doi:10.1038/nrmicro2670) [DOI] [PubMed] [Google Scholar]
- 29.Shaheen SM, Ouimet M-C, Marczynski GT. 2009. Comparative analysis of Caulobacter chromosome replication origins. Microbiology 155, 1215–1225. (doi:10.1099/mic.0.025528-0) [DOI] [PubMed] [Google Scholar]
- 30.Darling ACE, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. (doi:10.1101/gr.2289704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ioannidis P, Dunning Hotopp JC, Sapountzis P, Siozios S, Tsiamis G, Bordenstein SR, Baldo L, Werren JH, Bourtzis K. 2007. New criteria for selecting the origin of DNA replication in Wolbachia and closely related bacteria. BMC Genomics 8, 182 (doi:10.1186/1471-2164-8-182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (doi:10.1186/gb-2004-5-2-r12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leclercq S, Giraud I, Cordaux R. 2011. Remarkable abundance and evolution of mobile group II introns in Wolbachia bacterial endosymbionts. Mol. Biol. Evol. 28, 685–697. (doi:10.1093/molbev/msq238) [DOI] [PubMed] [Google Scholar]
- 34.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. 2011. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12, R30 (doi:10.1186/gb-2011-12-3-r30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. (doi:10.1101/gr.1224503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen O, Pupko T. 2011. Inference of gain and loss events from phyletic patterns using stochastic mapping and maximum parsimony: a simulation study. Genome Biol. Evol. 3, 1265–1275. (doi:10.1093/gbe/evr101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawahara M, Rikihisa Y, Isogai E, Takahashi M, Misumi H, Suto C, Shibata S, Zhang C, Tsuji M. 2004. Ultrastructure and phylogenetic analysis of ‘Candidatus Neoehrlichia mikurensis’ in the family Anaplasmataceae, isolated from wild rats and found in Ixodes ovatus ticks. Int. J. Syst. Evol. Microbiol. 54, 1837–1843. (doi:10.1099/ijs.0.63260-0) [DOI] [PubMed] [Google Scholar]
- 39.Klasson L, et al. 2008. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 25, 1877–1887. (doi:10.1093/molbev/msn133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foster J, et al. 2005. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 3, e121 (doi:10.1371/journal.pbio.0030121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godel C, et al. 2012. The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J. 26, 4650–4661. (doi:10.1096/fj.12-205096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klasson L, et al. 2009. The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc. Natl Acad. Sci. USA 106, 5725–5730. (doi:10.1073/pnas.0810753106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerveau N, Leclercq S, Leroy E, Bouchon D, Cordaux R. 2011. Short- and long-term evolutionary dynamics of bacterial insertion sequences: insights from Wolbachia endosymbionts. Genome Biol. Evol. 3, 1175–1186. (doi:10.1093/gbe/evr096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lobry JR. 1996. Origin of replication of Mycoplasma genitalium. Science 272, 745–746. (doi:10.1126/science.272.5262.745) [DOI] [PubMed] [Google Scholar]
- 45.Klasson L, Andersson SGE. 2006. Strong asymmetric mutation bias in endosymbiont genomes coincide with loss of genes for replication restart pathways. Mol. Biol. Evol. 23, 1031–1039. (doi:10.1093/molbev/msj107) [DOI] [PubMed] [Google Scholar]
- 46.Wagner A, Lewis C, Bichsel M. 2007. A survey of bacterial insertion sequences using IScan. Nucleic Acids Res. 35, 5284–5293. (doi:10.1093/nar/gkm597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bordenstein SR, Reznikoff WS. 2005. Mobile DNA in obligate intracellular bacteria. Nat. Rev. Microbiol. 3, 688–699. (doi:10.1038/nrmicro1233) [DOI] [PubMed] [Google Scholar]
- 48.Moran NA, Plague GR. 2004. Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14, 627–633. (doi:10.1016/j.gde.2004.09.003) [DOI] [PubMed] [Google Scholar]
- 49.Grigoriev A. 1998. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 26, 2286–2290. (doi:10.1093/nar/26.10.2286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocha EPC. 2004. The replication-related organization of bacterial genomes. Microbiology 150, 1609–1627. (doi:10.1099/mic.0.26974-0) [DOI] [PubMed] [Google Scholar]
- 51.Badawi M, Giraud I, Vavre F, Grève P, Cordaux R. 2014. Signs of neutralization in a redundant gene involved in homologous recombination in Wolbachia endosymbionts. Genome Biol. Evol. 6, 2654–2664. (doi:10.1093/gbe/evu207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koonin EV, Wolf YI. 2012. Evolution of microbes and viruses: a paradigm shift in evolutionary biology? Front. Cell. Infect. Microbiol. 2, 119 (doi:10.3389/fcimb.2012.00119) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.