

Abstract
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that is involved in neuronal adaptions that underlie cocaine-induced sensitization and reward. mTOR exists in two functionally distinct multi-component complexes known as mTORC1 and mTORC2. In this study, we show that increased mTORC1 activity induced by cocaine is mediated by the dopamine D1 receptor (D1R). Specifically, cocaine treatment increased the phosphorylation on residues Thr2446 and Ser2481 but not on Ser2448 in the nucleus accumbens (NAc) and that this increase in phosphorylated mTOR levels was also apparent when complexed with its binding partner Raptor. Furthermore, the increase in phosphorylated mTOR levels, as well as phosphorylated 4E-BP1 and S6K, downstream targets of mTORC1 were blocked with SCH23390 treatment. Similar results were also observed in the dopamine-transporter knockout mice as the increase in phosphorylated mTOR Thr2446 and Ser2481 was blocked by SCH23390 but not with raclopride. To further validate D1R role in mTORC1 signaling, decrease in phosphorylated mTOR levels were observed in D1R knockout mice, whereas administration of SKF81297 elevated phosphorylated mTOR in the NAc. Lastly deletion of mTOR or Raptor in D1R expressing neurons reduced cocaine-induced locomotor activity. Together, our data supports a mechanism whereby mTORC1 signaling is activated by cocaine administration through the stimulation of D1R.
Keywords: Cocaine, mTOR, cell signaling, nucleus accumbens, dopamine, Raptor
1. Introduction
Cocaine addiction is a chronic, relapsing disease with a lack of effective pharmacologic treatment options (Vocci and Elkashef, 2005). Repeated exposure to cocaine causes enduring neuroadaptions within the mesolimbic dopaminergic system, which is thought to underlie the reward-related learning and memory processes required for addiction (Everitt and Wolf, 2002; Nestler, 2005). Cocaine acts to increase extracellular dopamine (DA) levels by blocking the dopamine transporter (DAT) leading to the activation of postsynaptic dopamine receptors (D1R and D2R). Although both D1R and D2R are critically involved in drug addiction and the reward pathway, it is the activation of D1R that is required for the facilitation of the cellular and behavioral effects of cocaine (Drago et al., 1994; Graham et al., 2007).
At the molecular level the neuroplasticity changes induced by cocaine are contributed by alterations in protein expression. While many studies have investigated differences in gene expression at the transcriptional level (Bannon et al., 2005; Rhodes and Crabbe, 2005), it is becoming apparent that translational control plays an essential role in drug addiction (Dayas et al., 2012). One of the major regulators of protein synthesis is the mammalian target of rapamycin (mTOR), an evolutionarily conserved Ser/Thr protein kinase that responds to multiple environment cues including growth factor, nutrient state and energy levels (Gingras et al., 2001; Sarbassov et al., 2005a). mTOR exists in two complexes, mTOR complex 1 or 2 (mTORC1 or mTORC2) but it is through mTORC1 which mTOR exerts translation control. mTORC1 is distinguished by its association with the regulatory-associated protein with mTOR (Raptor) (Kim et al., 2002) and its sensitivity to rapamycin. Within mTORC1, mTOR is able to phosphorylate p70 S6 kinase (S6K) and eIF4E-binding protein (4E-BP1), cascades that regulate the initiation step in mRNA translation (Holz et al., 2005; Sonenberg and Hinnebusch, 2009). Activation of S6K leads to the subsequent phosphorylation of S6, a component of the 40S subunit (Shahbazian et al., 2006), whereas 4E-BP1 results in the release of elF4E (Beretta et al., 1996; Diggle et al., 1996).
Several studies have shown that cocaine regulates mTOR signaling by utilizing the mTORC1 inhibitor, rapamycin. Systemic injections of rapamycin blocked the expression of cocaine-induced locomotion and conditional place preference (Bailey et al., 2012; Wu et al., 2011). Cocaine treatment has also been shown to increase phosphorylated S6K levels, a downstream target of mTORC1 in the nucleus accumbens (Wu et al., 2011). While these studies show that cocaine regulates mTOR signaling, it is unclear if the dopamine system is responsible for the activation of mTORC1 signaling. Thus, the aim of the present study was to examine the role of the D1R in cocaine-mediated activation of mTORC1 signaling. To address this, we assessed changes in mTORC1 activity in the NAc of cocaine treated mice and DAT knockout (DAT KO) mice. Additionally, cocaine-induced locomotor activity was evaluated in mice lacking mTOR or Raptor in D1R-expressing neurons.
2. Material and Methods
2.1 Animals
All mouse studies were conducted in accordance with the National Institutes of Health Guidelines for Animal Care and Use and with an approved animal protocol from the Duke University Animal Care and Use Committee. The generation of DAT KO mice and dopamine receptor 1 KO (D1R KO) mice has been described previously (Drago et al., 1994; Giros et al., 1996). The dopamine D1 receptor (DRD1) Cre mice were obtained from Nathaniel Heintz and Charles Gerfen [The Gene Expression Nervous System Atlas (GENSAT) Project, National Institute of Neurological Disorders and Stroke Contracts N01NS02331 and HHSN271200723701C to The Rockefeller University, NewYork]. The mTORflx/flx (stock 011009) and Raptorflx/flx (stock 013188) were purchased from The Jackson Laboratory (Bar Harbor, ME) (Risson et al., 2009; Sengupta et al., 2010). Experimental mice were generated by crossing homozygous floxed (mTORflx/flx or Raptorflx/flx) mice with heterozygous mice for cre (mTORflx/flxD1Cre or Raptorflx/flx D1Cre) to generate D1mTOR−/− and D1Raptor −/− knockout mice and there littermate controls D1mTOR+/+ and D1Raptor+/+, respectively. For all behavioral experiments adult male (8-16 weeks of age) mice were used. Male mice were used in the biochemical studies with the exception of the D1R KO experiment, where female D1R KO and their wildtype littermates were used. For biochemical and behavioral experiments with cocaine or mTOR inhibitors, C57Bl/6J mice (The Jackson Laboratory) were used. All mice were housed at a maximum of five per cage, provided with food and water ad libitum, and tested at 3-4 months of age.
2.2 Drugs
Cocaine (Sigma, St Louis, MO) was dissolved in a 0.9% NaCl solution. SCH23390, SKF81297, quinpirole and raclopride were purchased from Tocris Bioscience and dissolved in a 0.9% NaCl solution. Rapamycin was purchased from LC Laboratories and was dissolved in 4% ethanol and 5% Tween-20.
2.3 Drug Treatment and Experimental Paradigm
For the acute cocaine administration, C57Bl/6J mice were injected with saline (s.c.) on three consecutive days before the start of the study to acclimate the animals to the injection procedure. Mice received a single injection of cocaine (15mg/kg, i.p.) and protein was isolated from the NAc 60min following the injection. In the chronic study, C57Bl/6J mice received daily injections of either saline or cocaine (15mg/kg, i.p) for 5 consecutive days. Mice were sacrificed 60mins following the last cocaine/vehicle injection and the NAc was dissected.
To examine the role of the D1R in mTORC1 signaling upon cocaine administration, C57Bl/6J mice were injected with cocaine or saline daily for 5 days (15mg/kg). On the fifth day, mice were pretreated with an acute injection of SCH23390 (0.5mg/kg, i.p.) 15min prior to receiving the last cocaine or vehicle injection. Protein was isolated from the NAc 60min following the last cocaine or saline injection. In another set of experiments, C57Bl/6J mice were injected with SKF81297 (5mg/kg, i.p.), quinpirole (4mg/kg, i.p.) or appropriate vehicle daily for 5 days and sacrificed 60min later. D1R KO and their WT littermates were also sacrificed at the same time of day as the animals that received drugs via systemic injections.
To investigate if the dopamine system regulates mTORC1 signaling, we chose to utilize the hyperdopaminergic model, DAT KO mice. Naïve DAT KO and their wildtype (WT) littermates were sacrificed at the same time of day as the animals that received drugs via systemic injections. DAT KO and their WT littermates were also injected with SCH23390 (0.5mg/kg, i.p.), raclopride (1mg/kg, i.p.) or vehicle to address the role of D1R or D2R in a dopaminergic model system. Mice were sacrificed 30min following the injection and the NAc was immediately dissected.
In all experiments, mice were sacrificed by cervical dislocation and the NAc was immediately removed on an ice-cold platform using a 2mm micropunch. Bilateral punches were pooled together and considered one sample (n=1).
2.4 Protein Isolation and Western Blotting
Protein isolation, quantification and western blotting were performed as previously described for the NAc with some modifications (Ramsey et al., 2008). Primary antibodies were detected by infrared secondary antibodies using a LI-COR Odyssey imager. Briefly, tissue from the individual mice were homogenized using a dounce homogenizer in ice-cold lysis buffer (137mM NaCl, 20mM Tris (pH 8.0), 1% NP-40 and 10% glycerol and 0.1% sodium dodecyl sulfate) with the addition of a protease inhibitor tablet (Roche) and phosphatase inhibitor (Sigma-Aldrich). The homogenized tissue was sonicated, mixed with 5× loading buffer (125mM Tris (pH 6.8), 10% glycerol, 2% sodium dodecyl sulfate, 2% 2-mercaptoethanol and 0.01% bromophenol blue) and heated for 5min. Protein concentrations were determined using a bicinchoninic acid protein assay (Pierce). Protein extracts (20-50μg) were resolved on a polyacrylamide gel and transferred to a nitrocellulose membrane. Primary antibodies were detected by infrared secondary antibodies using a LI-COR Odyssey imager. The source and dilution of antibodies were as follows: mTOR (Cell Signaling #4517, 1:1000), phospho-mTOR Ser2448 (Millipore #09-213, 1:1000), phospho-mTOR Thr2446 (Millipore 09-345, 1:1000), phospho-mTOR Ser2481 (Millipore #09-343, 1:1000), elF-4E (Epitomics 1598-1, 1:500), phospho elF-4E (Epitomics 2227-1, 1:5000), 4E-BP1 (Cell Signaling #4923, 1000), phospho 4E-BP1 Thr37/46 (Cell Signaling #2855, 1000), Raptor (Cell Signaling #2280, 1:1000), p70 S6K (Cell Signaling #9202, 1:1000), phospho-p70 S6K Thr389 (Cell Signaling #9206, 1:1000), S6 (Cell Signaling #2317, 1:2000) and phospho-S6 Ser235/236 (Cell Signaling #4858, 1:1000). The band intensity for the individual blots were quantified using ImageJ analysis software (NIH). The phosphorylated levels for each sample were normalized to the corresponding total protein levels and the densitometry values were expressed as a percentage of control (vehicle).
2.5 Co-Immunoprecipitation
Protein was isolated from the NAc using a non-denaturing lysis buffer containing protease and phosphatase inhibitors. Following protein quantification, immunoprecipitation (IP) was performed using 750 μg of protein and sepharose beads conjugated to anti-Raptor IgG (Cell signaling Technology). IP samples and their lysate extracts were then analyzed by Western blotting as described above. The densitometry values were normalized as a ratio of mTOR (total or phosphorylated) to Raptor and expressed as a percentage of control.
2.6 Locomotor Activity
Locomotor activity was measured in an Accuscan activity monitor (Accuscan Instruments) as described previously (Ramsey et al., 2008). Briefly, locomotor activity was measured at 5min intervals and these data were analyzed for the total distance traveled in 5min increments for 120min or as indicated. All mice were allowed to acclimatize to the activity monitor for 30min before any drug treatments. Following acclimatization to the chambers, DAT KO mice and their WT littermates were injected with varying doses of rapamycin (2, 5 or 10mg/kg, i.p) or vehicle. D1mTOR−/− and D1Raptor−/− mice and their WT littermates were injected with cocaine (15g/kg, i.p).
2.7 Statistical Analysis
Data from the western blots were analyzed using one- or two-way ANOVA or Student's t-test where appropriate. For total locomotor activity, groups were compared using a two-way ANOVA. The time course for locomotor activity was compared by a three-way ANOVA with repeated measures with genotype and drug as between-subject factors and time as the within-subject factor. Analysis for Student's t-test, one- or two-way ANOVA was performed using GraphPad Prism and analysis for three-way ANOVA was performed with PAWS Statistics 18. Post hoc analysis were made with Bonferroni-Dunn test and values of p < 0.05 were considered statistically significant
3. Results
3.1 Cocaine activates mTORC1-mediated signaling in the NAc
Although cocaine has been shown to affect downstream targets of mTORC1, the effect on mTOR itself has not been fully addressed (Wu et al., 2011). Thus, initial experiments assessed the effects of acute and repeated cocaine on mTOR, by examining changes on three phosphorylation sites that function to regulate mTOR activity (Foster and Fingar, 2010). Here, we focused our studies on the NAc, a key component of the reward system that plays a critical role in the reward circuitry and development of drug addiction (Carelli, 2002; Koob and Volkow, 2010). Western blots showed that acute administration of cocaine significantly increased the phosphorylation of mTOR Ser2481 levels (t(6) = 4.838, p < 0.01) but no change in phosphorylated mTOR Ser2448 (t(6) = 0.8158, p > 0.05) or total mTOR was observed (t(6) = 0.2013, p > 0.05) (Figure 1a). There was a slight increase on phosphorylated mTOR Thr2446, however, this trend did not reach significance (t(6) = 1.5620, p > 0.05). Additionally, changes in mTOR signaling were also examined following repeated cocaine administration. It was found that daily cocaine administration for 5 days increased phosphorylated mTOR Thr2446 (t(10) = 6.550, p < 0.0001) and Ser2481 (t(10) = 5.833, p < 0.001) but no significant changes in Ser2448 (t(10) = 2.187, p > 0.05) or total mTOR levels (t(10) = 0.6974, p > 0.05) were detected (Figure 1b). Although both acute and repeated cocaine administration induced an increase in phosphorylated mTOR Ser2481 levels, we also observed an upregulation of phosphorylated mTOR Thr2446 following repeated cocaine administration.
Figure 1.
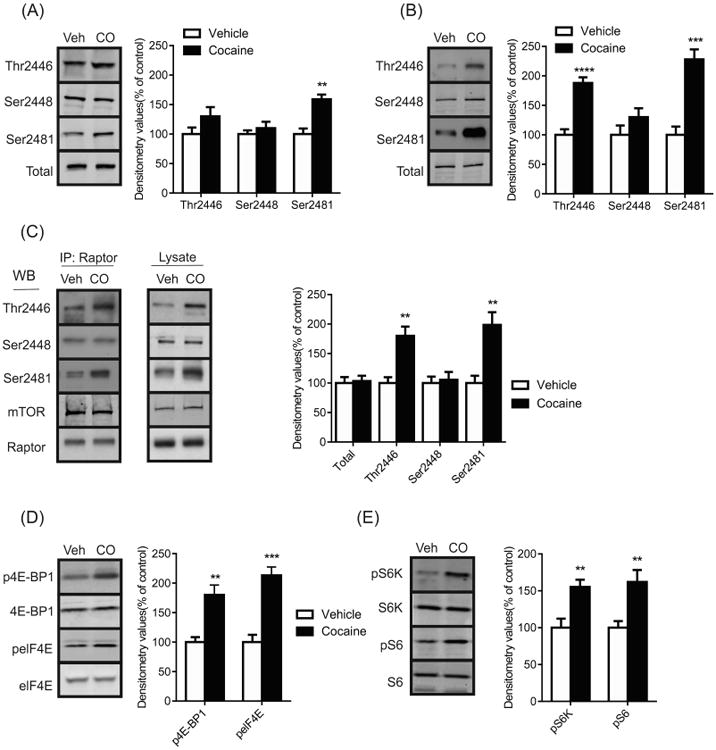
Cocaine activates mTORC1 signaling in the NAc of mice. Representative western blots and graphs of densitometry values for phosphorylated and total mTOR (Thr2446, Ser2448 and Ser2481) in the NAc of mice treated with (a) acute (15mg/kg) and (b) repeated cocaine (CO, 15mg/kg, 5 day) or vehicle (veh). (c) Representative images and graph of densitometry values for co-IPs showing an increased interaction between Raptor and phosphorylated mTOR in the NAc of mice administered with repeated cocaine. Samples were immunoprecipitated (IP) with Raptor conjugated sepharose beads and then subjected to western blotting (WB) using antibodies for phosphorylated mTOR Thr2446, Ser2448, Ser2481, mTOR and Raptor (Left panel, n = 4), whereas WB was performed using the same total lysate as the co-IPs (Right panel). Representative western blots and graphs of densitometry values for phosphorylated and total (d) 4E-BP1, elF4E (e) S6K and S6 in the NAc of mice treated with repeated cocaine (CO, 15mg/kg, 5 day) or vehicle (Veh). Data presented as mean ± SEM and expressed as a percentage of control (** p < 0.01, *** p < 0.001, **** p < 0.0001, two-tailed unpaired t test).
To address whether the increase in mTOR activity following cocaine treatment is specific to mTORC1 we investigated the association between mTOR and its binding partner, Raptor. Co-immunoprecipitations (Co-IPs) showed no change in the association between Raptor and mTOR following repeated cocaine treatment (Figure 1c; t(6) = 0.2586, p > 0.05). However, significant increases in the phosphorylation state of mTOR complexed with Raptor was observed with phosphorylated mTOR Thr2446 (t(6) = 4.353, p < 0.01) and Ser2481 (t(6) = 4.013, p < 0.01) but not with Ser2448 (t(6) = 0.3259, p > 0.05) (Figure 1c).
Lastly, the effect of repeated cocaine administration on downstream targets of mTORC1 were assessed; 4E-BP1 and S6K. Levels of phosphorylated 4E-BP1 (t(10) = 4.321, p < 0.01) and its target, phosphorylated elF4E (t(10) = 6.026, p < 0.001) were increased following cocaine administration (Figure 1d). In addition, upregulation of phosphorylated S6K (t(10) = 3.516, p < 0.01) and S6 levels (t(10) = 3.338, p < 0.01) were also observed in the cocaine samples (Figure 1e). Collectively, these results show that cocaine activates mTORC1 signaling in the NAc.
3.2 DAT KO mice show activation of mTORC1 signaling in the NAc
Since cocaine acts by increasing extracellular dopamine levels through the inhibition of the dopamine transporter we sought to determine if the dopamine system is responsible for the activation of mTORC1 signaling. To investigate whether the inhibition of the dopamine transporter can regulate mTORC1 signaling, the DAT KO mouse was utilized. DAT KO mice display a 5-fold increase in extracellular striatal DA levels and exhibit hyperactivity and stereotypic movement when placed in a novel environment (Gainetdinov et al., 1999; Giros et al., 1996). An increase in phosphorylated mTOR Thr2446 (t(14) = 8.770, p < 0.0001) and Ser2481 levels (t(14) = 3.040, p = 0.0088) were observed in the NAc of DAT KO mice compared with their wild type (WT) littermates (Figure 2a). Phosphorylated mTOR Ser2448 (t(14) = 1.704, p > 0.05) and total mTOR levels (t(14) = 2.051, p > 0.05) were unaffected in the NAc of DAT KO mice (Figure 2a). In addition, changes in mTORC1 signaling were evaluated in the DAT KO mice by investigating its downstream targets. Phosphorylated 4E-BP1 (t(14) = 3.717, p < 0.01) and elF4E levels (t(14) = 3.988, p < 0.01) were increased in DAT KO mice (Figure 2b), as well, phosphorylated S6K (t(14) = 3.727, p < 0.01) and S6 (t(14) = 4.682, p < 0.001) were also upregulated (Figure 2c). These results mimic the cocaine-induced response on mTORC1 signaling.
Figure 2.
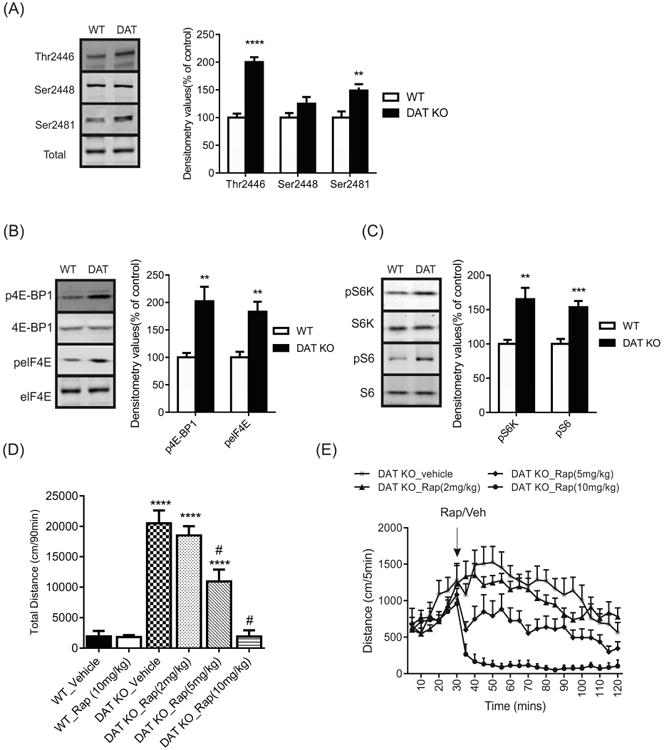
DAT KO mice show increased mTORC1-mediated signaling in the NAc. Representative western blots and graphs of densitometry values for phosphorylated and total (a) mTOR (Thr2446, Ser2448 and Ser2481) (b) 4E-BP1, elF4E (c) S6K and S6 in the NAc of DAT KO or wild type (WT) mice. Data presented as mean ± SEM and expressed as a percentage of control (n = 8; two-tailed unpaired t test). (d) Locomotor activity for DAT KO and WT mice treated with rapamycin (Rap; 2mg/kg, 5mg/kg, 10mg/kg) or vehicle (n = 9-12mice/group, two-way ANOVA). (e) Time course of locomotor activity for DAT KO mice treated with rapamycin or vehicle (n = 9-12mice/group, two-way ANOVA). Mice were administered vehicle or rapamycin following a 30min habituation to locomotor chambers. Data represented as mean ± SEM (** p < 0.01, *** p < 0.001, **** p < 0.0001 compared to WT_Vehicle; # p < 0.05 compared to DAT KO_Vehicle).
Next, to determine the involvement of mTORC1 in DA-dependent hyperactivity, DAT KO mice were treated with various doses of the mTORC1 inhibitor, rapamycin. Systemic administration of rapamycin dose dependently inhibited locomotor activity in DAT KO mice, whereas rapamycin in WT mice did not have a significant effect on locomotor activity (Figure 2d). A two-way ANOVA (drug by genotype) revealed a significant effect drug (F(3, 69) = 20.96, p < 0.0001), a significant effect of genotype (F(1, 69) = 152.4, p < 0.0001) and a significant interaction (F(3, 69) = 20.90, p < 0.0001). Figure 2e shows the time course of DAT KO mice treated with rapamycin, where doses of 5 and 10mg/kg inhibit locomotor activity (time F(23, 851) = 10.64, p < 0.0001; drug F(3, 37) = 17.69, p < 0.0001; interaction F(69, 851) = 4.595, p < 0.0001).
3.3 D1R regulates mTORC1 signaling
The striatal complex is comprised of medium spiny neurons that differentially express D1 or D2 dopamine receptors. Given that an activation of mTORC1 was observed in the NAc of DAT KO mice, we next assessed whether this effect was mediated by D1R or D2R. DAT KO mice were injected with the D1R antagonist, SCH23390, or the D2R antagonist, raclopride, and changes in phosphorylated mTOR levels were investigated. We observed the increase in phosphorylated mTOR 2446 levels in the NAc of DAT KO mice (compared to WT) was blocked by the administration of SCH23390 but not with raclopride (Figure 3a; genotype F(1, 18) = 56.37, p < 0.0001; treatment F(2, 18) = 7.406, p = 0.0045; interaction F(2, 18) = 3.320, p = 0.0592). Similarly, the upregulation of phosphorylated mTOR 2481 detected in DAT KO mice was also inhibited by SCH23390 but not with raclopride treatment (genotype F(1, 18) = 38.88, p < 0.0001; treatment F(2, 18) = 3.627, p = 0.0475; interaction F(2, 18) = 1.253, p > 0.05). Consistent with the previous experiment phosphorylated mTOR Ser2448 and total levels were unaltered in DAT KO mice and were unaffected by SCH23390 or raclopride treatment. These data indicate that regulation of mTOR in DAT KO mice is mediated by the D1R.
Figure 3.
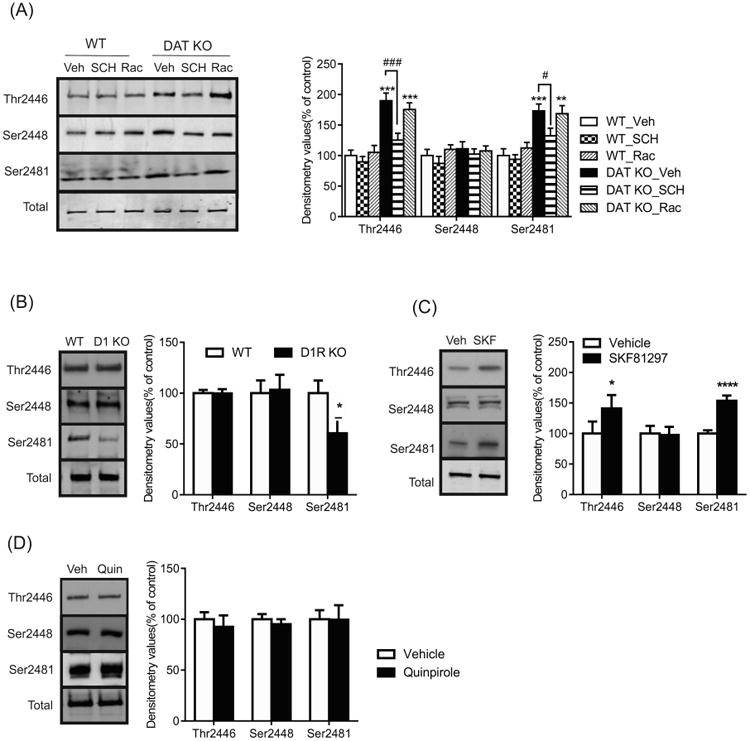
D1R regulates phosphorylated mTOR levels in the NAc. (a) Representative western blots and graphs of densitometry values for total and phosphorylated mTOR Thr2446, Ser2448 and Ser2481 levels in DAT KO mice treated with the D1R antagonist, SCH23390 (SCH, 0.5mg/kg), D2R antagonist, raclopride (Rac, 3mg/kg) or vehicle (n = 4mice/group, two-way ANOVA). (b) Representative western blots and graphs of densitometry values for total and phosphorylated mTOR levels in the NAc of D1R KO or WT mice (n = 4mice/group, two-tailed t-test). Representative western blots and graphs of densitometry values for total and phosphorylated mTOR levels in the NAc of C57BL/6J mice treated with (c) SKF81297 (n = 4-6mice/group) or (d) quinpirole (n = 4mice/group). Data presented as mean ± SEM and expressed as percentage of control (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 compared to WT/vehicle; # p < 0.05, ### p < 0.001 compared to DAT KO_Veh).
To further examine the role of D1R in mTOR signaling, levels of phosphorylated mTOR were investigated in mice lacking the D1R or by targeting the D1R pharmacologically. Levels of phosphorylated mTOR Ser2481 (t(6) = 2.046, p < 0.05) were found to be decreased in the NAc of D1R KO mice, whereas levels of phosphorylated mTOR Thr2446 (t(6) = 0.048, p > 0.05) and Ser2448 (t(6) = 0.1698, p > 0.05) showed no variation compared with their WT littermates (Figure 3b). C57Bl/6J mice treated chronically with SKF81297 showed elevated phosphorylated mTOR Thr2446 (t(8) = 2.705, p < 0.05) and Ser2481 levels (t(8) = 10.71, p < 0.0001) but Ser2448 (t(8) = 0.2521, p > 0.05) was unaffected (Figure 3c). In addition, mice were injected with quinpirole (D2/D3R agonist) but no significant alternations in phosphorylated mTOR levels were observed (Figure 3d).
Given that D1R regulates the phosphorylation state of mTOR, we sought to investigate whether the changes in mTORC1 signaling induced by repeated cocaine were mediated by the D1R. Following repeated cocaine administration (5 days), mice were pretreated with SCH23390 before the final cocaine injection and changes in phosphorylated mTOR levels were analyzed. Western blots revealed that increased phosphorylated mTOR Thr2446 observed in the NAc of cocaine treated mice was blocked by the administration of SCH23390 (Figure 4a; drug F(1,12) = 8.536, p < 0.05; SCH23390 pretreatment F(1,12) = 36.70, p < 0.0001). Similarly, phosphorylated mTOR Ser2481 levels were attenuated in cocaine treated mice that were administered SCH23390 compared with cocaine treatment alone (Figure 4a; drug F(1,12) = 8.729, p < 0.05; SCH23390 pretreatment F(1,12) = 33.66 p < 0.0001). Levels of phosphorylated mTOR Ser2448 (drug F(1,12) = 0.1528, p > 0.05; SCH23390 pretreatment F(1,12) = 0.5313, p > 0.05) were unchanged by the pretreatment of SCH23390. Examining downstream targets of mTORC1 revealed a reduction in p4E-BP1 (drug F(1,12) = 18.08, p < 0.01; SCH23390 pretreatment F(1,12) = 27.90, p < 0.001; interaction F(1,12) = 10.80, p < 0.01), pelf4E (drug F(1,12) = 48.92, p < 0.0001; SCH23390 pretreatment F(1,12) = 61.65, p < 0.0001; interaction F(1,12) = 29.36, p < 0.0001), pS6K (drug F(1,12) = 29.22, p < 0.001; SCH23390 pretreatment F(1,12) = 29.91, p < 0.001; interaction F(1,12) = 8.329, p < 0.05) and pS6 (drug F(1,12) = 16.51, p < 0.001; SCH23390 pretreatment F(1,12) = 10.58, p < 0.01; interaction F(1,12) = 19.81, p < 0.01) in the NAc of mice pretreated with SCH23390 compared with cocaine treatment alone (Figure 4b-c). Overall, these data show that cocaine mediated changes in the mTORC1 signaling via the D1R.
Figure 4.
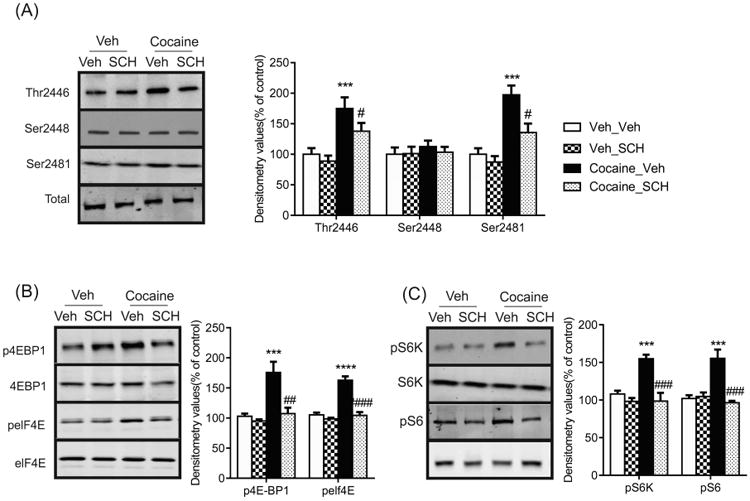
Inhibition of D1R blocks activation of the mTORC1 signaling pathway by repeated cocaine treatment. Following 5 days of cocaine treatment, C57BL/6J mice were pretreated with SCH23390 (0.5mg/kg) or vehicle prior to the last cocaine injection. Representative western blots and graphs of densitometry values for phosphorylated and total (a) mTOR (Thr2446, Ser2448 and Ser2481) (b) 4E-BP1, elF4E (c) S6K and S6. Data presented as mean ± SEM and expressed as percentage of control (n = 4 mice/group; *** p < 0.001, **** p < 0.0001 compared to Veh_Veh; # p < 0.05, ## p < 0.001, ### p < 0.001 compared to Cocaine_Veh; two-way ANOVA).
3.4 D1R-mediated activation of mTOR regulates cocaine-induced locomotion
The previous experiments showed that cocaine activates mTORC1 signaling through the activation of the D1R. To test this effect behaviorally, we generated the genetic deletion of mTOR in D1R-expressing neurons by crossing mTORflx/flx mice to mice harboring a Cre recombinase under the D1R promoter, generating mTORflx/flxD1Cre (D1mTOR-/-) and their littermate controls D1mTOR+/+. Decreased levels of mTOR in the NAc of D1mTOR-/- mice are shown by western blots (Figure 5a). The D1mTOR-/- and D1mTOR+/+ mice did not differ in basal locomotor activity (Figure 5b). In response to cocaine administration, D1mTOR-/- mice had reduced locomotor activity compared to D1mTOR+/+ mice (Figure 5b). Two-way ANOVA shows a significant effect of drug (F(1,40) = 8.055, p < 0.0001), drug-genotype interaction (F(1,40) = 7.345, p < 0.01) but no effect of genotype (F(1,40) = 3.066, p > 0.05). The time course for cocaine treatment showed that locomotor activity of D1mTOR-/- mice was attenuated compared to their WT littermates (Figure 5c; time-drug-genotype interaction F(1,23) = 3.387, p < 0.05).
Figure 5.
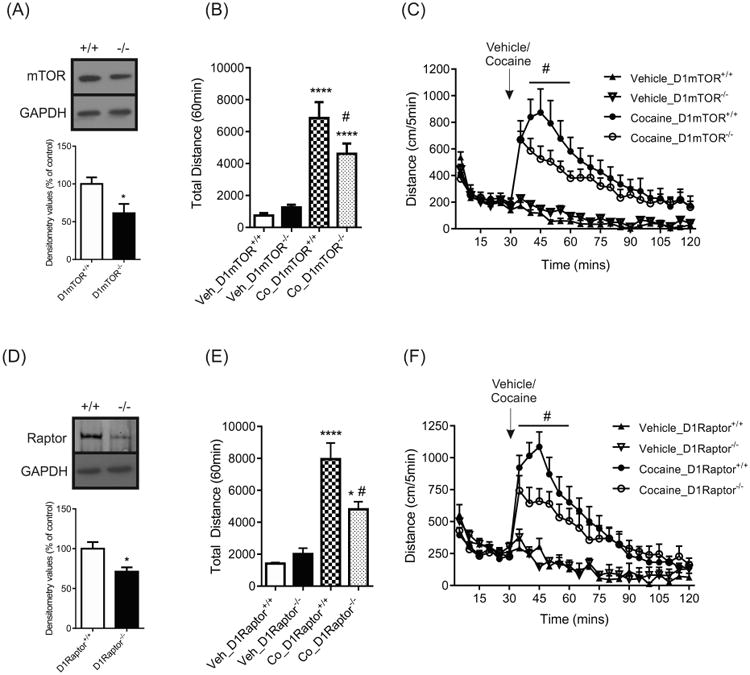
mTOR or Raptor deletion in D1 neurons display lower locomotor response upon cocaine administration. (a) Representative western blots and graphs of densitometry values of total mTOR levels in the NAc of D1mTOR+/+ and D1mTOR-/- mice (n = 4mice/group; * p < 0.05, two-tailed unpaired t test). (b) Total locomotor activity for D1mTOR+/+ and D1mTOR-/- mice treated with cocaine (Co, 15mg/kg) or vehicle (Veh; n = 11/group, **** p < 0.0001 compared to Veh_D1mTOR+/+, # p < 0.05 Co_D1mTOR+/+ vs Co_D1mTOR-/-; Two-way ANOVA) (c) Time course of locomotor activity for D1mTOR+/+ and D1mTOR-/- mice treated with cocaine (# p < 0.05 cocaine_ D1mTOR+/+ vs cocaine_ D1mTOR-/-, Three-way ANOVA with repeated measures). (d) Representative western blots and graphs of densitometry values of total Raptor levels in the NAc of D1Raptor+/+ and D1Raptor-/- mice (n = 4mice/group; * p < 0.05, two-tailed unpaired t test). (e) Total locomotor activity for D1Raptor+/+ and D1Raptor-/- mice treated with cocaine (15mg/kg) or vehicle. (Veh; n = 5-9mice/group, * p < 0.05, **** p < 0.0001 compared to Veh_D1Raptor+/+, # p < 0.05 Co_D1Raptor+/+ vs Co_D1Raptor-/-; Two-way ANOVA). (f) Time course of locomotor activity for D1Raptor+/+ and D1Raptor-/- mice treated with cocaine. (# p < 0.05 cocaine_ D1Raptor+/+ vs cocaine_ D1Raptor-/-, Three-way ANOVA with repeated measures).
Given that mTOR participates in two signaling complexes (mTORC1 and mTORC2), we wanted to test whether this mTOR function might also be associated with mTORC1 signaling. To target mTORC1 we utilized mice that lacked Raptor in D1R-expressing neurons, D1Raptor-/- mice and their WT littermates, D1Raptor+/+. A decrease in Raptor level was detected in D1Raptor-/- compared with their littermates (Figure 5d). D1Raptor-/- and D1Raptor+/+ mice exhibited similar basal locomotor activity (Figure 5e; genotype F(1, 22) = 2.557, p > 0.05). In response to cocaine, D1Raptor-/- mice displayed a blunted effect in their locomotor activity compared to D1Raptor+/+ mice (Figure 5e; drug F(1, 22) = 34.38, p < 0.0001; drug-genotype interaction F(1, 22) = 5.502, p < 0.05). The time course showed that D1Raptor-/- mice exhibit an attenuated response to cocaine compared to D1Raptor+/+ mice (Figure 5F; time-drug-genotype interaction F(1,23) = 2.897, p < 0.05).
4. Discussion
The current study demonstrates that cocaine leads to a D1R-mediated activation of mTORC1 signaling. Activation of mTORC1 signaling was observed in the NAc of cocaine treated mice and in DAT KO mice, whereas administration of a D1R antagonist blocked the activation of mTORC1 signaling in these dopaminergic animal models. Furthermore, we showed that conditional deletion of mTOR or Raptor in D1R-expessing neurons attenuates cocaine-induced locomotion compared to its WT littermates. Overall the results demonstrate the involvement of mTORC1 signaling in mediating partial dopamine D1R dependent behaviors.
Our findings showed that the regulation of mTOR in the NAc of mice treated with repeated cocaine and in the DAT KO mice was mediated through phosphorylation on specific residues, Thr2446 and Ser2481, further validating a role for the dopamine system in regulating the mTOR signaling pathway. Moreover, treatment with SCH23390 in the repeated cocaine model and in DAT KO mice blocked the increase in phosphorylated mTOR levels on Thr2446 and Ser2481. Even though a direct comparison of post-injection times is sometimes difficult to interpret when using different model systems (transgenic and pharmacological) the results demonstrate a role for D1R in regulating specific phosphorylation sites in these hyperdopaminergic models. The C-terminus of mTOR contains the phosphorylation sites Thr2446, Ser2448 and Ser2481 that lie within or near a repressor domain and consequently correlate with an increase in activity (Hoeffer and Klann, 2010). These sites are regulated by several different kinase including downstream effectors of the mTOR pathway itself or by autophosphorylated. For example, phosphorylation of Ser2481 is an autophosphorylation site that directly monitors its catalytic activity for both mTORC1 and mTORC2 (Manning, 2012; Soliman et al., 2010). The Thr2446 site of mTOR has been shown to be a target of AMP activated protein kinase (AMPK) and S6K (Cheng et al., 2004; Holz and Blenis, 2005). Considering S6K is an effector of mTOR, it has been hypothesized that the phosphorylation of Thr2446 by S6K is part of a positive feedback loop (Holz and Blenis, 2005). Another interesting observation was the lack of change on the Ser2448 site following cocaine treatment or in the DAT KO mice, a site that is regulated by Akt and S6K (Chiang and Abraham, 2005; Reynolds et al., 2002). The specific phosphorylation pattern of mTOR induced by cocaine and in the DAT KO mice is indicative of upstream signaling, as each phosphorylation site is regulated by different mechanisms.
The increase in phosphorylated mTOR levels induced by cocaine or in the DAT KO mice correlates with an increase in kinase activity but does not distinguish between the two distinct mTOR complexes, mTORC1 and mTORC2. However, cocaine administration increased the levels of phosphorylated mTOR that is associated with Raptor, revealing an enhanced activity of mTORC1. Additionally, increased phosphorylated S6K and 4E-BP1 levels, direct targets of mTORC1 were observed in the NAc of cocaine treated mice and DAT KO mice. The mTOR complexes are also distinguished by their differing sensitivities to rapamycin, as mTORC1 but not mTORC2 is inhibited by acute rapamycin treatment (Dowling et al., 2010). Thus, the inhibitory action of rapamycin on locomotor activity in DAT KO mice and on cocaine sensitization (Wu et al., 2011) is attributed to the blockade of mTORC1. Furthermore, D1mTOR and D1Raptor knockout mice displayed similar locomotor activity from cocaine treatment, suggesting the behavioral effect was due to mTORC1. Although it is shown that cocaine activates mTORC1, it does not rule out the possible involvement of mTORC2 as there is crosstalk between these signaling cascades. For example mTORC2 activation can be mediated through inhibitory effects by mTORC1-driven mechanisms (Dibble et al., 2009; Julien et al., 2010). mTORC2 is known to phosphorylate Akt Ser473, SGK1 and PKC, which control cell survival and cytoskeletal organization (Garcia-Martinez and Alessi, 2008; Sarbassov et al., 2004; Sarbassov et al., 2005b). Nevertheless, it is shown that cocaine increased mTORC1 activity through the activation of the D1R.
Several studies using drugs of abuse have investigated mTORC1 signaling, as mTOR has been implicated in reward-related behaviors. In addition to cocaine, alcohol administration has also been shown to activate mTORC1-mediated signaling in the NAc of mice (Neasta et al., 2010) and increases in phosphorylated S6K and S6 following morphine treatment has been reported in the VTA (Mazei-Robison et al., 2011). In contrast, amphetamine administration does not affect mTORC1 signaling as amphetamine does not induce changes in the phosphorylation state of mTOR Ser2448, S6K or 4E-BP1 levels (Biever et al., 2015; Rapanelli et al., 2014). Thus, the activation of mTORC1 signaling may not be a common mechanism shared by all psychostimulants or in general drugs of abuse.
mTOR has been shown to be important in long-term potentiation, long-term depression and synaptic plasticity (Costa-Mattioli et al., 2009; Jaworski and Sheng, 2006). Changes in synaptic plasticity induced by drugs of abuse are thought to underlie the neuroadaptive processes that drive addiction and relapse behavior (Kauer and Malenka, 2007). Nicotine mediated changes in structural plasticity have been shown to be mediated by mTORC1 signaling (Collo et al., 2013) and morphological changes induced by morphine have been associated with mTORC2 (Mazei-Robison et al., 2011). Since mTOR plays a major role in synaptic plasticity, mTOR may be involved in the neuroadaptions associated with the addiction process by influencing translation (Dayas et al., 2012; Neasta et al., 2014). The overall increase in mTORC1 activity due to changes in mTOR phosphorylation induced by cocaine treatment is sufficient to allow mTORC1 to phosphorylate its targets, S6K and 4E-BP1 and consequently is expected to increase cap-dependent initiation of mRNA translation. Furthermore, mTORC1 pathway plays an important role in regulating mRNA translation during synaptic plasticity and memory formation (Hoeffer and Klann, 2010). There is strong evidence that activation of mTORC1 signaling is necessary for the translation of mRNA, which appears to be critical in reward and relapse vulnerability, including Ca2+/Calmodulin-dependent kinase II alpha (CamKIIα), AMPA receptor subunits (GluRs), microtubule-associated protein (MAP2), post-synaptic density protein 95 (PSD-95) and glutamate receptor interaction protein (Hou and Klann, 2004; Mameli et al., 2007).
mTOR has been shown in rodents to be important in drug-motivated behaviors. In addition to cocaine, infusions of rapamycin in the NAc have been shown to block the development of methamphetamine-induced conditional place preference (Narita et al., 2005). Rapamycin treatment has also been shown to attenuate the expression of locomotor sensitization and CPP to alcohol (Neasta et al., 2010). Additionally, rapamycin has been shown to affect cocaine and morphine reconsolidation of reward memory in rats, a process thought to be required for drug relapse (Lin et al., 2014; Shi et al., 2014). Clinically, acute rapamycin has been shown to significantly reduce craving in heroin addicts (Shi et al., 2009). All of these studies have utilized rapamycin, suggesting a major role for mTORC1 in reward and relapse behaviors. Here we went a step further suggesting a major role of mTORC1 in D1R-expressing neurons in cocaine-mediated behaviors. Our work is consistent with the concept that the rewarding effects of cocaine are generally associated with D1R signaling. The systemic administration of a D1R antagonist attenuates both the development and expression of cocaine-sensitization (Cabib et al., 1991; Fontana et al., 1993; Le et al., 1997). More recently, a study utilizing D1R KO mice with a viral D1R re-expression approach, demonstrated that D1R-expression in the core of the NAc mediates cocaine-sensitization (Gore and Zweifel, 2013). Together, our data supports a mechanism whereby cocaine leads to the activation of mTORC1 signaling through the stimulation of D1R. This cascade may be a mechanism by which cocaine exposure induces its rewarding effects and may provide a new approach for the development of therapies for cocaine addiction.
Highlights.
mTORC1 signaling is activated by cocaine through the dopamine D1 receptor.
Cocaine increased mTOR phosphorylation on residues Thr2446 and Ser2481.
DAT KO mice show increased mTORC1 signaling in the nucleus accumbens.
Blockade of D1R inhibits mTORC1 signaling in cocaine treated and DAT KO mice.
Mice lacking Raptor in D1R neurons display attenuated cocaine-induced locomotion.
Acknowledgments
This work was supported in part by the Canadian Institutes of Health Research (CIHR) Fellowship awarded to LPS and by National Institutes of Health Grant 5R37-MH-073853 to MGC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey J, Ma D, Szumlinski KK. Rapamycin attenuates the expression of cocaine-induced place preference and behavioral sensitization. Addict Biol. 2012;17:248–258. doi: 10.1111/j.1369-1600.2010.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon M, Kapatos G, Albertson D. Gene expression profiling in the brains of human cocaine abusers. Addict Biol. 2005;10:119–126. doi: 10.1080/13556210412331308921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proceedings of the National Academy Sciences of the United States of America. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- Biever A, Puighermanal E, Nishi A, David A, Panciatici C, Longueville S, Xirodimas D, Gangarossa G, Meyuhas O, Herve D, Girault JA, Valjent E. PKA-dependent phosphorylation of ribosomal protein S6 does not correlate with translation efficiency in striatonigral and striatopallidal medium-sized spiny neurons. J Neurosci. 2015;35:4113–4130. doi: 10.1523/JNEUROSCI.3288-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib S, Castellano C, Cestari V, Filibeck U, Puglisi-Allegra S. D1 and D2 receptor antagonists differently affect cocaine-induced locomotor hyperactivity in the mouse. Psychopharmacology (Berl) 1991;105:335–339. doi: 10.1007/BF02244427. [DOI] [PubMed] [Google Scholar]
- Carelli RM. The nucleus accumbens and reward: neurophysiological investigations in behaving animals. Behav Cogn Neurosci Rev. 2002;1:281–296. doi: 10.1177/1534582302238338. [DOI] [PubMed] [Google Scholar]
- Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem. 2004;279:15719–15722. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- Collo G, Bono F, Cavalleri L, Plebani L, Mitola S, Merlo Pich E, Millan MJ, Zoli M, Maskos U, Spano P, Missale C. Nicotine-induced structural plasticity in mesencephalic dopaminergic neurons is mediated by dopamine D3 receptors and Akt-mTORC1 signaling. Mol Pharmacol. 2013;83:1176–1189. doi: 10.1124/mol.113.084863. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayas CV, Smith DW, Dunkley PR. An emerging role for the Mammalian target of rapamycin in “pathological” protein translation: relevance to cocaine addiction. Front Pharmacol. 2012;3:13. doi: 10.3389/fphar.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle TA, Moule SK, Avison MB, Flynn A, Foulstone EJ, Proud CG, Denton RM. Both rapamycin-sensitive and -insensitive pathways are involved in the phosphorylation of the initiation factor-4E-binding protein (4E-BP1) in response to insulin in rat epididymal fat-cells. Biochem J. 1996;316(Pt 2):447–453. doi: 10.1042/bj3160447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433–439. doi: 10.1016/j.bbapap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Drago J, Gerfen CR, Lachowicz JE, Steiner H, Hollon TR, Love PE, Ooi GT, Grinberg A, Lee EJ, Huang SP, et al. Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc Natl Acad Sci U S A. 1994;91:12564–12568. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana D, Post RM, Weiss SR, Pert A. The role of D1 and D2 dopamine receptors in the acquisition and expression of cocaine-induced conditioned increases in locomotor behavior. Behav Pharmacol. 1993;4:375–387. [PubMed] [Google Scholar]
- Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Caron MG. Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol Psychiatry. 1999;46:303–311. doi: 10.1016/s0006-3223(99)00122-5. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. Control of translation by the target of rapamycin proteins. Prog Mol Subcell Biol. 2001;27:143–174. doi: 10.1007/978-3-662-09889-9_6. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gore BB, Zweifel LS. Genetic reconstruction of dopamine D1 receptor signaling in the nucleus accumbens facilitates natural and drug reward responses. J Neurosci. 2013;33:8640–8649. doi: 10.1523/JNEUROSCI.5532-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DL, Hoppenot R, Hendryx A, Self DW. Differential ability of D1 and D2 dopamine receptor agonists to induce and modulate expression and reinstatement of cocaine place preference in rats. Psychopharmacology (Berl) 2007;191:719–730. doi: 10.1007/s00213-006-0473-5. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280:26089–26093. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34:205–219. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Tomkins D, Higgins G, Quan B, Sellers EM. Effects of 5-HT3, D1 and D2 receptor antagonists on ethanol- and cocaine-induced locomotion. Pharmacol Biochem Behav. 1997;57:325–332. doi: 10.1016/s0091-3057(96)00333-4. [DOI] [PubMed] [Google Scholar]
- Lin J, Liu L, Wen Q, Zheng C, Gao Y, Peng S, Tan Y, Li Y. Rapamycin prevents drug seeking via disrupting reconsolidation of reward memory in rats. Int J Neuropsychopharmacol. 2014;17:127–136. doi: 10.1017/S1461145713001156. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Lujan R, Luscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Manning BD. Comment on “A dynamic network model of mTOR signaling reveals TSC-independent mTORC2 regulation”: building a model of the mTOR signaling network with a potentially faulty tool. Sci Signal. 2012;5:lc3. doi: 10.1126/scisignal.2003250. author reply lc4. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Koo JW, Friedman AK, Lansink CS, Robison AJ, Vinish M, Krishnan V, Kim S, Siuta MA, Galli A, Niswender KD, Appasani R, Horvath MC, Neve RL, Worley PF, Snyder SH, Hurd YL, Cheer JF, Han MH, Russo SJ, Nestler EJ. Role for mTOR signaling and neuronal activity in morphine-induced adaptations in ventral tegmental area dopamine neurons. Neuron. 2011;72:977–990. doi: 10.1016/j.neuron.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Akai H, Kita T, Nagumo Y, Narita M, Sunagawa N, Hara C, Hasebe K, Nagase H, Suzuki T. Involvement of mitogen-stimulated p70-S6 kinase in the development of sensitization to the methamphetamine-induced rewarding effect in rats. Neuroscience. 2005;132:553–560. doi: 10.1016/j.neuroscience.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Neasta J, Barak S, Hamida SB, Ron D. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J Neurochem. 2014;130:172–184. doi: 10.1111/jnc.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neasta J, Ben Hamida S, Yowell Q, Carnicella S, Ron D. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc Natl Acad Sci U S A. 2010;107:20093–20098. doi: 10.1073/pnas.1005554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Laakso A, Cyr M, Sotnikova TD, Salahpour A, Medvedev IO, Dykstra LA, Gainetdinov RR, Caron MG. Genetic NMDA receptor deficiency disrupts acute and chronic effects of cocaine but not amphetamine. Neuropsychopharmacology. 2008;33:2701–2714. doi: 10.1038/sj.npp.1301663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapanelli M, Frick LR, Pogorelov V, Ota KT, Abbasi E, Ohtsu H, Pittenger C. Dysregulated intracellular signaling in the striatum in a pathophysiologically grounded model of Tourette syndrome. Eur Neuropsychopharmacol. 2014;24:1896–1906. doi: 10.1016/j.euroneuro.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds THt, Bodine SC, Lawrence JC., Jr Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem. 2002;277:17657–17662. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Crabbe JC. Gene expression induced by drugs of abuse. Curr Opin Pharmacol. 2005;5:26–33. doi: 10.1016/j.coph.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, Richard-Bulteau H, Vignaud A, Baas D, Defour A, Freyssenet D, Tanti JF, Le-Marchand-Brustel Y, Ferrier B, Conjard-Duplany A, Romanino K, Bauche S, Hantai D, Mueller M, Kozma SC, Thomas G, Ruegg MA, Ferry A, Pende M, Bigard X, Koulmann N, Schaeffer L, Gangloff YG. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005a;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005b;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–1104. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Jun W, Zhao LY, Xue YX, Zhang XY, Kosten TR, Lu L. Effect of rapamycin on cue-induced drug craving in abstinent heroin addicts. Eur J Pharmacol. 2009;615:108–112. doi: 10.1016/j.ejphar.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Shi X, Miller JS, Harper LJ, Poole RL, Gould TJ, Unterwald EM. Reactivation of cocaine reward memory engages the Akt/GSK3/mTOR signaling pathway and can be disrupted by GSK3 inhibition. Psychopharmacology (Berl) 2014;231:3109–3118. doi: 10.1007/s00213-014-3491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem. 2010;285:7866–7879. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vocci FJ, Elkashef A. Pharmacotherapy and other treatments for cocaine abuse and dependence. Curr Opin Psychiatry. 2005;18:265–270. doi: 10.1097/01.yco.0000165596.98552.02. [DOI] [PubMed] [Google Scholar]
- Wu J, McCallum SE, Glick SD, Huang Y. Inhibition of the mammalian target of rapamycin pathway by rapamycin blocks cocaine-induced locomotor sensitization. Neuroscience. 2011;172:104–109. doi: 10.1016/j.neuroscience.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]