

Abstract
Objective
The aim of this work was to determine whether atrophy of specific retinal layers and brain substructures are associated over time, in order to further validate the utility of optical coherence tomography (OCT) as an indicator of neuronal tissue damage in patients with multiple sclerosis (MS).
Methods
Cirrus high-definition OCT (including automated macular segmentation) was performed in 107 MS patients biannually (median follow-up: 46 months). Three-Tesla magnetic resonance imaging brain scans (including brain-substructure volumetrics) were performed annually. Individual-specific rates of change in retinal and brain measures (estimated with linear regression) were correlated, adjusting for age, sex, disease duration, and optic neuritis (ON) history.
Results
Rates of ganglion cell + inner plexiform layer (GCIP) and whole-brain (r = 0.45; p<0.001), gray matter (GM; r = 0.37; p<0.001), white matter (WM; r = 0.28; p = 0.007), and thalamic (r = 0.38; p < 0.001) atrophy were associated. GCIP and whole-brain (as well as GM and WM) atrophy rates were more strongly associated in progressive MS (r = 0.67; p<0.001) than relapsing-remitting MS (RRMS; r = 0.33; p = 0.007). However, correlation between rates of GCIP and whole-brain (and additionally GM and WM) atrophy in RRMS increased incrementally with step-wise refinement to exclude ON effects; excluding eyes and then patients (to account for a phenotype effect), the correlation increased to 0.45 and 0.60, respectively, consistent with effect modification. In RRMS, lesion accumulation rate was associated with GCIP (r = −0.30; p = 0.02) and inner nuclear layer (r = −0.25; p = 0.04) atrophy rates.
Interpretation
Over time GCIP atrophy appears to mirror whole-brain, and particularly GM, atrophy, especially in progressive MS, thereby reflecting underlying disease progression. Our findings support OCT for clinical monitoring and as an outcome in investigative trials.
Multiple sclerosis (MS) is regarded as an immune-mediated demyelinating disorder of the central nervous system. Although magnetic resonance imaging (MRI) is regarded as the gold-standard imaging modality for monitoring MS, the association between MRI parameters of inflammation and disability progression in MS is modest.1 Conversely, MRI estimates of neurodegeneration correlate better with disability progression.2,3 Indeed, it is now widely accepted that axonal and neuronal degeneration represents the principal pathological substrate underlying disability in MS.4–10 Nonconventional MRI techniques, including brain-substructure volumetrics, reveal that gray matter (GM) atrophy is a common, early feature of MS and may be better associated with disability than white matter (WM) atrophy.9–12 Such techniques, however, may lack sensitivity to assess progression at the individual level or in small numbers of patients. The development of novel techniques for objectively quantifying neurodegeneration in MS has therefore been an ongoing goal.
Optical coherence tomography (OCT) is an inexpensive, reproducible, well-tolerated, high-resolution imaging technique. Recently developed segmentation algorithms (now transitioning into clinical practice) allow reliable quantification of discrete retinal layers.13–16 Because the retina is unmyelinated, retinal axonal and neuronal measures are not confounded by myelin, making them ideal for assessing neuroaxonal degeneration. The retinal nerve fiber layer (RNFL) is the innermost retinal layer (Fig 1). RNFL axons (derived from ganglion cell neurons) coalesce at the optic discs to form the optic nerves and acquire myelin beyond the lamina cribrosa. Clinical and subclinical optic nerve involvement is common in MS. At postmortem examination, 94% to 99% of MS patients exhibit demyelinating lesions in the optic nerves.17,18 Over time, retrograde degeneration of optic nerve axons (owing to demyelination and transection)19–21 is captured by OCT and reflected by thinning of the peripapillary RNFL (pRNFL) and combined ganglion cell and inner plexiform layers (GCIP).22,23 Though this suggests a utility for OCT to monitor disease progression, a critical, yet unanswered, question is whether atrophy within specific retinal layers concomitantly mirrors global neurodegeneration in MS.
Figure 1.
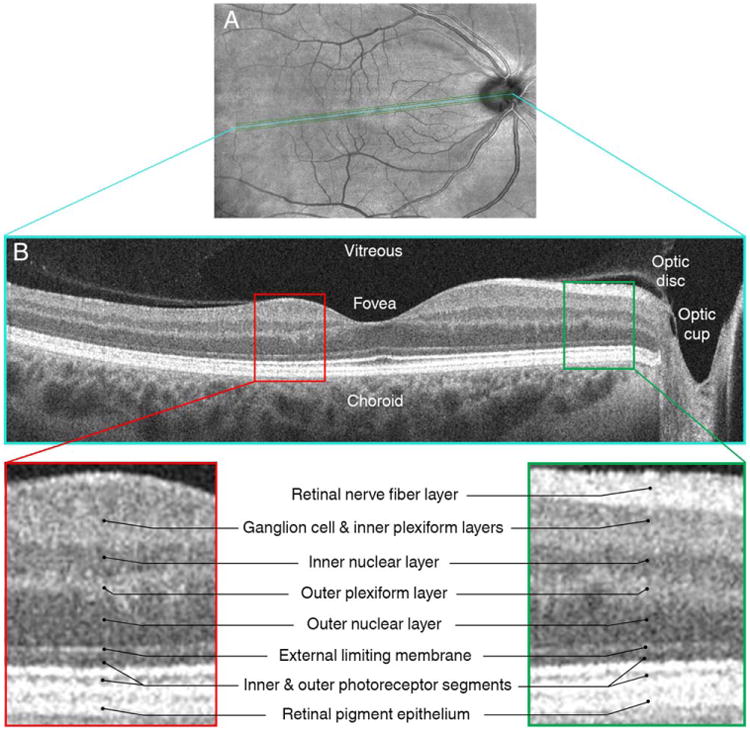
This figure illustrates an en face optical coherence tomography (OCT) fundus image (A) and a corresponding high-definition OCT section of the same retina in the area of the interposed line (panel B), acquired from the right eye of a multiple sclerosis patient. Insets represent magnified areas of the OCT scan illustrating differences in the reflectivity patterns of different retinal layers. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
OCT also reveals abnormalities of the inner nuclear layer (INL) in MS.13,24,25 Although postmortem analyses reveal neuronal loss in the INL of MS eyes,20 OCT data indicate that in vivo a sizeable proportion of MS eyes have thickening of the INL, suggesting that an inflammatory process might precede this neurodegeneration.25 Indeed, increased INL thickness in MS was associated with inflammatory disease activity.25 However, it remains to be determined whether INL thickness changes mirror ongoing global processes such as inflammation over time.
Studies assessing OCT-brain substructure relationships in MS to date have been primarily cross-sectional. Whereas pRNFL and GCIP thicknesses correlate with whole-brain volume,26–32 and INL thickness with lesion volume,33 the utility of tracking OCT measures, and the insight gleaned regarding ongoing global neurodegeneration and inflammation over time, requires a longitudinal study. The utility of OCT by MS subtype also remains unclear and in particular whether tracking OCT in progressive MS is more informative than in relapsing-remitting MS (RRMS). Finally, it remains to be determined whether a history of optic neuritis (ON) affects longitudinal retinal-brain relationships.28,32,33
The primary aims of this longitudinal observational study were to determine (1) whether atrophy within specific retinal layers over time is associated with concomitant atrophy of specific brain substructures and accumulation of lesions in MS, (2) how these relationships vary between RRMS and progressive MS, and (3) whether ON history affects these relationships.
Patients and Methods
Patients
Johns Hopkins University Institutional Review Board approval was acquired, and written informed consent was obtained from study participants. MS patients were recruited by unselected convenience sampling from the Johns Hopkins MS Center. MS diagnosis was confirmed by the treating neurologist (S.S., J.R., S.D.N., P.A.C.) based on the 2010 revised McDonald criteria.34 MS disease subtype was classified as RRMS, secondary-progressive (SPMS) or primary-progressive MS (PPMS).35 Patients with SPMS and PPMS were combined and classified as having progressive MS owing to the small sample size of each group. Disease duration, comorbidities, and history of ON, including the date and side of occurrence, were recorded. MS patients underwent clinical evaluation and OCT every 6 months and brain MRI annually (Fig 2). Expanded disability status scale (EDSS) scores were determined by Neurostatus-certified EDSS examiners at study visits (within 30 days of OCT and MRI examinations) and were available for 106 patients at the end of the study. Disability progression was defined as a 1-point or more, or a 0.5-point or more, increase in EDSS score from baseline to end-of-study EDSS examination if the baseline EDSS score was less than 6 or 6 or more, respectively.
Figure 2.
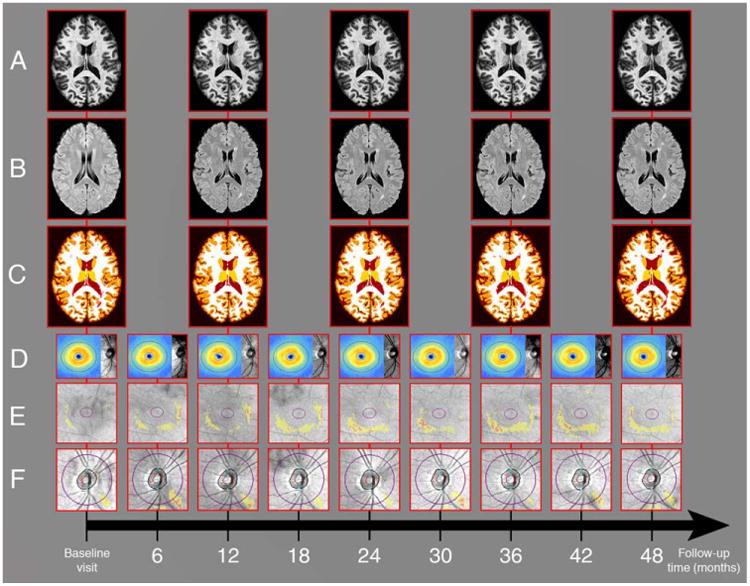
Our study protocol. Study participants underwent brain MRI annually for a median duration of 39 months. Illustrated are longitudinal three-dimensional MPRAGE (row A) and FLAIR (row B) brain MRI images of a study participant. Row C shows corresponding automated Lesion-TOADS brain segmentation masks used to compute brain substructure volumes. In addition, study participants also underwent OCT imaging every 6 months for a median duration of 46 months. Longitudinal OCT images of the right eye of the same study participant are shown, including ganglion cell layer plus inner plexiform layer (GCIP) thickness maps overlaid on OCT fundus photos (row D), and GCIP and retinal nerve fiber layer deviation maps (rows E and F, respectively; yellow areas represent thicknesses <5% percentile and red areas represent thicknesses <1% percentile relative to healthy controls). FLAIR = fluid-attenuated inversion recovery; MPRAGE = magnetization-prepared rapid acquisition of gradient echoes; MRI = magnetic resonance imaging; OCT = optical coherence tomography; TOADS = topology-preserving anatomy-driven segmentation.
Patients with acute ON within 6 months of baseline assessment were excluded. Patients who developed acute ON during study follow-up, and patients with diabetes, glaucoma, refractive errors of greater than or equal to ±6 diopters, or other ophthalmological or neurological disorders were also excluded from the study.
MRI
Brain MRI was performed with a 3-Tesla Philips Intera scanner (Philips Medical System, Best, Netherlands). Two-axial whole-brain sequences without gaps were used: multislice fluid-attenuated inversion recovery (FLAIR; acquired resolution: 0.8 × 0.8 × 2.2 or 0.8 × 0.8 × 4.4mm; echo time [TE]: 68ms; repetition time [TR]: 11 seconds; inversion time [TI]: 2.8 seconds; SENSE factor: 2; averages: 1); and three-dimensional [3D] magnetization-prepared rapid acquisition of gradient echoes (MPRAGE; acquired resolution: 0.8 × 0.8 × 1.2mm; TE: 6ms; TR: ∼10ms; TI: 835ms; flip angle: 8 degrees; SENSE factor: 2; averages: 1).
MRI images were analyzed with the TOADS (Topology-preserving Anatomy-Driven Segmentation) software package (http://www.nitrc.org/projects/toads-cruise).36,37 First, the skull and extracranial tissues were removed using an automated method applied to the baseline MPRAGE scan.38 The resulting brain mask was applied to the coregistered baseline FLAIR and follow-up images. Hence, the stripped baseline MPRAGE scans were rigidly registered to a single image atlas (“JHU_MNI_SS_T1”) that is available for download (http://www.mristudio.org), as previously described.39 Briefly this atlas has been registered to the standard Montreal Neurological Instute-152 atlas and resampled to 0.83-mm isotropic voxels. The baseline FLAIR and follow-up scans were also transformed to this space for the purpose of longitudinal assessment.
Subsequently, MPRAGE and FLAIR images were analyzed using Lesion-TOADS, an automated segmentation method, described elsewhere.38–40 This technique segments the brain into its component substructures while simultaneously delineating MS lesions, yielding volumes of the following: cortical GM, cerebral WM, caudate, putamen, thalamus, and brainstem. Cerebral volume fraction (CVF), analogous to brain parenchymal fraction, was calculated by dividing the summed volume of brain substructures by intracranial volume (ICV) and expressed as the percentage of ICV occupied by brain matter. FLAIR-WM lesion volume was measured using an automated image analysis method applied to Lesion-TOADS output, which uses a classification technique based on ensembles of decision trees to map MS lesions.41 All processing and segmentation results were manually reviewed by a trained observer (O.A.A.).
OCT
Retinal imaging was performed with spectral-domain Cirrus HD-OCT (model 4000, software version 6·0; Carl Zeiss Meditec, Dublin, California), as described elsewhere.42 Briefly, peripapillary and macular scans were obtained with the Optic Disc Cube 200 × 200 and Macular Cube 512 × 128 protocols, respectively. Scans with signal strength less than 7/10 or with artifact were excluded, in accordance with the OSCAR-1B criteria.43
Longitudinal measurements of pRNFL thickness were derived from the guided progression analysis feature that aligns follow-up optic disc cube scans to the baseline scan by translating and/or rotating the images as necessary.
Macular cube scans were analyzed using segmentation software, as described previously.13,44 Briefly, the segmentation performed in 3D yields the thicknesses of the macular RNFL, GCIP, INL (including the outer plexiform layer), and ONL (including the photoreceptor segments). Longitudinal segmentation measures were acquired from the same region on each scan, within an annulus of inner radius 0.54mm and outer radius 2.4mm, centered on the fovea. This segmentation protocol has been shown to be highly reproducible (inter-rater intraclass correlation coefficients: 0·91–0·99 for all measurements).13
Macular cube scans were also qualitatively assessed for macular microcysts and other retinal abnormalities, as described elsewhere.25
Statistical Analysis
Statistical analyses were performed using Stata software (version 13; StataCorp LP, College Station, TX). The Shapiro-Wilk test was used to assess the normality of distributions. Comparisons between MS subtypes were performed using the Wilcoxon rank-sum test for age, EDSS, disease duration, and follow-up duration, and chi-square (χ2) test for ON history and sex.
Ordinary least squares linear regression was used to obtain maximum likelihood (ML) estimates of subject-specific rates of change in brain compartment volumes and eye-specific rates of change in retinal layer thickness measurements over the study duration, with each subject acting as their own control. Partial correlation analyses were then performed to assess the relationships between rates of change in brain substructure volumes and retinal layer thicknesses (calculated as the average of the ML-estimated rates for both eyes) adjusting for age, sex, disease duration, and ON history. Bonferroni correction for multiple comparisons was performed. Fisher's r-to-z transformation was used to determine whether the correlation coefficient was significantly different between the relapsing and progressive groups.
For validation purposes, separate subject- and eye-specific rates of change were computed using empirical Bayes prediction from mixed-effects linear regression models. ML estimates are unbiased in that they rely solely on the observed data (for each subject and eye) to generate subject- and eye-specific rates of change. One of the limitations with respect to this approach, however, is that some of the clusters may not contain a sufficient number of observations to yield stable estimates, resulting in the so-called “bouncing beta” phenomenon. Therefore, we undertook further analytical steps in order to validate our findings and account for this possibility. First, we analyzed the results using multilevel mixed-effects linear regression and used empirical Bayes prediction to assign values to the random subject and eye-specific slopes for both the MRI and OCT measures. In contrast to ML estimation, empirical Bayes predictions combine information from the prior distribution with the likelihood in order to obtain the posterior distribution of the random slopes given the observed responses. The end result of this approach will be shrinkage of the random subject and eye-specific slopes toward the mean of the prior distribution utilizing the James-Stein method of “borrowing-strength,” mainly for uninformative clusters. The details of this method are beyond the scope of this article; however, it is worth noting that empirical Bayes predictions are conditionally biased toward the mean of the prior distribution (i.e., zero).
Three statistical models were used to study the effects of ON history on longitudinal OCT-MRI relationships. Model 1 calculated subject-specific rates of change in retinal layer thickness (obtained from ML estimation) as the average of the rates for both eyes, with the subsequent correlation analyses adjusted for ON history. Model 2 excluded eyes with a previous history of ON, and in model 3 subjects with a past history of ON in either eye were excluded. Analysis of covariance (ANCOVA) was performed to assess whether ON history resulted in effect modification of the longitudinal relationship between GCIP and CVF rates of change, accounting for within-subject intereye correlation, and adjusting for age, sex, and disease duration. Interaction terms of time by ON history, disease duration, baseline retinal layer thickness, presence of new contrast-enhancing or T2 lesions at any time point during the study, and EDSS progression were used to assess whether rates of retinal layer change varied based on different clinical parameters. To study the effects of clinical relapses during the period of observation, linear spline breakpoints were inserted at the time of symptom onset in the group who experienced clinical relapses in order to directly compare rates of change in brain MRI and OCT measures before versus after a relapse. Statistical significance was defined as p < 0.05.
Results
Study Population
One hundred seven MS patients (71 RRMS and 36 progressive MS: 24 SPMS and 12 PPMS) participated in the study. Median durations of OCT and MRI follow-up for the entire cohort were 46.2 and 38.7 months, respectively, and were similar between MS subtypes. At study enrollment, 39 RRMS (55%) and 9 progressive MS patients (25%) had a previous history of ON. All progressive MS patients with a history of ON had SPMS. The proportion of subjects with a previous history of ON was not significantly different between RRMS and SPMS (p = 0.14). None of the study participants had ON during the 6-month period preceding baseline evaluation or developed ON during study follow-up. INL microcysts were identified in 4 RRMS patients at baseline (bilateral in one case). In one patient, INL microcysts became visible during follow-up. Of the 6 eyes exhibiting INL microcysts, 5 (80%) had a previous history of ON. Baseline demographic and clinical characteristics are summarized in Table 1. Table 2 shows the estimated rates of change of brain compartment volumes and retinal layer thicknesses for study participants over the follow-up duration.
Table 1. Demographics and Clinical Characteristics of Study Participants.
| Overall n = 107 | RRMS n = 71 | Progressive MS n = 36 | p | |
|---|---|---|---|---|
| Baseline demographic and clinical characteristics | ||||
| Age, yr; mean (SD) | 44.2 (12.1) | 38.9 (10.3) | 54.6 (8.2) | <0.01a |
| Female; n (%) | 75 (70.1) | 51 (71.8) | 24 (66.7) | 0.58b |
| Disease duration, yr; median (Q1–3) | 10 (4–16) | 7 (3–11) | 16 (11–24) | <0.01a |
| EDSS; median (Q1–3)d | 3 (2–6) | 2 (1.5–3.0) | 6 (5.5–6.5) | <0.01a |
| Disease-modifying therapy; n (%) | <0.01c | |||
| • Interferon beta 1a | 25 (23) | 20 (28) | 5 (14) | |
| • Interferon beta 1b | 7 (7) | 4 (6) | 3 (8) | |
| • Glatiramer acetate | 29 (27) | 21 (30) | 8 (22) | |
| • Natalizumab | 24 (22) | 23 (32) | 1 (3) | |
| • None | 22 (21) | 3 (4) | 19 (53) | |
| Clinical and radiological characteristics during follow-up | ||||
| Follow-up duration, mo; median (mean; SD; range) | ||||
| • OCT | 46.2 (41.4; 14.0; 7–60) | 46.2 (41.2; 14.5; 7–60) | 45.5 (41.7; 12.9; 12–58) | 0.99a |
| • MRI | 38.7 (36.6; 16.3; 11–63) | 37.8 (35.4; 16.9; 11–62) | 43.5 (39.0; 15.0; 11–63) | 0.40a |
| Nonocular relapses; n (%) | 24 (22.4) | 23 (32.4) | 1 (2.8) | <0.01c |
| New gadolinium-enhancing lesions; n (%) | 29 (27.1) | 24 (33.8) | 5 (13.9) | 0.03b |
| New T2 lesions; n (%) | 42 (39.3) | 36 (50.7) | 6 (16.7) | <0.01b |
Wilcoxon rank-sum test.
Chi-square test.
Fisher's exact test.
Available for 66 RRMS and 34 PMS patients.
AON = acute optic neuritis; EDSS = expanded disability status scale; OCT = optical coherence tomography; MRI = magnetic resonance imaging; RRMS = relapsing-remitting multiple sclerosis; SD = standard deviation; Q1 = first quartile; Q3 = third quartile.
Table 2. Rates of Change in MRI and OCT Measures During The Studya.
| Measures | Overall n = 107 | RRMS n = 71 | Progressive MS n = 36 | P RRMS vs. Progressive MSc | |
|---|---|---|---|---|---|
| Mean annualized change in MRI volume; %/year (SD)b | Cortical GM | −0.61 (1.2) | −0.64 (1.3) | −0.53 (1.0) | 0.49 |
| Cerebral WM | −0.39 (1.0) | −0.44 (1.1) | −0.30 (0.8) | 0.43 | |
| Thalamus | −1.16 (2.4) | −1.19 (2.7) | −1.10 (1.6) | 0.86 | |
| Caudate | 0.23 (3.5) | 0.25 (4.1) | 0.19 (2.2) | 0.73 | |
| Putamen | −0.95 (2.4) | −0.93 (2.7) | −1.0 (1.5) | 0.99 | |
| Brainstem | −0.73 (1.5) | −0.84 (1.7) | −0.52 (1.3) | 0.40 | |
| FLAIR lesion | 24.0 (48.5) | 22.9 (41.8) | 26.2 (60.5) | 0.09 | |
| CVF | −0.63 (0.9) | −0.68 (0.9) | −0.54 (0.8) | 0.36 | |
| Mean change in OCT thickness; μm/year (SD)c | pRNFL | −0.36 (1.0) | −0.41 (1.0) | −0.28 (1.2) | 0.32 |
| GCIP | −0.34 (0.7) | −0.31 (0.6) | −0.39 (0.9) | 0.22 | |
| INL | −0.04 (0.2) | 0.01 (0.69) | −0.14 (0.71) | 0.78 | |
| ONL | 0.03 (0.7) | −0.06 (0.5) | 0.20 (1.0) | 0.09 |
Generated using ordinary least squares linear regression.
Percentage changes utilize the baseline study visit measures as reference.
Generated using multivariate linear regression for MRI measures and mixed-effects linear regression (accounting for within-subject intereye correlation) for OCT measures. All models were adjusted for age at baseline visit, sex, and disease duration.
MRI=magnetic resonance imaging; OCT = optical coherence tomography; RRMS = relapsing remitting multiple sclerosis; SD = standard deviation; GM = gray matter; WM = white matter; CVF = cerebral volume fraction; GCIP = ganglion cell and inner plexiform layers; INL = inner nuclear and outer plexiform layers; ONL = outer nuclear layer including the photoreceptor segments.
Longitudinal OCT-MRI Relationships Across the MS Cohort
Partial correlation analyses revealed distinctive relationships between the rates of change of retinal layer thicknesses and brain substructure volumes over time (Table 3). Longitudinally, faster rates of pRNFL and GCIP thinning were associated with faster rates of whole brain (CVF), cortical GM, thalamic, and brainstem atrophy. The relationships between the rates of GCIP thinning (but not pRNFL thinning) and CVF, cortical GM, and thalamic atrophy retained significance after adjustment for multiple comparisons (r = 0.45; adjusted p<0.003, r = 0.37; adjusted p = 0.003, and r = 0.38; adjusted p = 0.003, respectively). In contrast, pRNFL thinning remained significantly associated with caudate and brainstem atrophy, after adjustment for multiple comparisons (r = 0.44; adjusted p<0.003, and r = 0.33; adjusted p=0.03, respectively). Supplementary Table 1 presents results from partial correlation analyses obtained when empirical Bayes predictions of subject-specific OCT and MRI rates of change were used in place of ML estimates from OLS regression.
Table 3. Partial Correlation Coefficients Between Rates of Change in MRI- and OCT-Derived Measures Across Whole MS Cohort Over the Study Durationa.
| MRI-derived brain compartment volume rate of change | OCT-Derived Retinal Layer Thickness Rate of Changeb | |||
|---|---|---|---|---|
| pRNFL | GCIP | INL | ONL | |
| Cortical GM | 0.207c | 0.371c,d | 0.092 | −0.028 |
| Cerebral WM | 0.144 | 0.285c | 0.036 | −0.066 |
| Thalamus | 0.273c | 0.379c,d | 0.228c | −0.242c |
| Caudate | 0.438c,d | 0.155 | −0.015 | −0.144 |
| Putamen | 0.146 | 0.147 | 0.186 | −0.228c |
| Brainstem | 0.329c,d | 0.210c | −0.019 | −0.094 |
| Lesions | 0.014 | −0.128 | −0.120 | 0.169 |
| CVF | 0.291c | 0.449c,d | 0.085 | −0.068 |
Partial correlation coefficients were adjusted for age, sex, MS subtype, disease duration, and history of ON.
The average rate of change for both eyes was used to compute the subject-specific rate of change in retinal layer thickness.
Indicates an unadjusted p value <0.05.
Indicates a p value <0.05 after Bonferroni adjustment for multiple comparisons.
CVF = cerebral volume fraction; GCIP = ganglion cell and inner plexiform layers; GM = gray matter; INL = inner nuclear and outer plexiform layers; ONL = outer nuclear layer including the photoreceptor segments; MRI = magnetic resonance imaging; OCT = optical coherence tomography; pRNFL = peripapillary retinal nerve fiber layer; WM = white matter.
Longitudinal GCIP-MRI Relationships Stratified by MS Subtype
The association between the rate of GCIP thinning and whole brain (CVF) atrophy was consistent across MS subtypes (Fig 3). GCIP thinning remained independently correlated with the rate of CVF atrophy, with the correlation being more pronounced in the progressive MS cohort (r = 0.67; p < 0.001), as compared to the RRMS cohort (r = 0.33; p = 0.007). The difference in the correlation between rates of GCIP and CVF atrophy between RRMS and progressive MS was significant (p = 0.04), confirming a stronger relationship in progressive MS, as compared to RRMS. Analysis of specific brain substructures by MS subtype revealed that the longitudinal relationships between GCIP and CVF atrophy were primarily related to cortical GM and cerebral WM volume loss in both RRMS and progressive MS, as well as thalamic volume loss in the RRMS cohort (Table 4).
Figure 3.
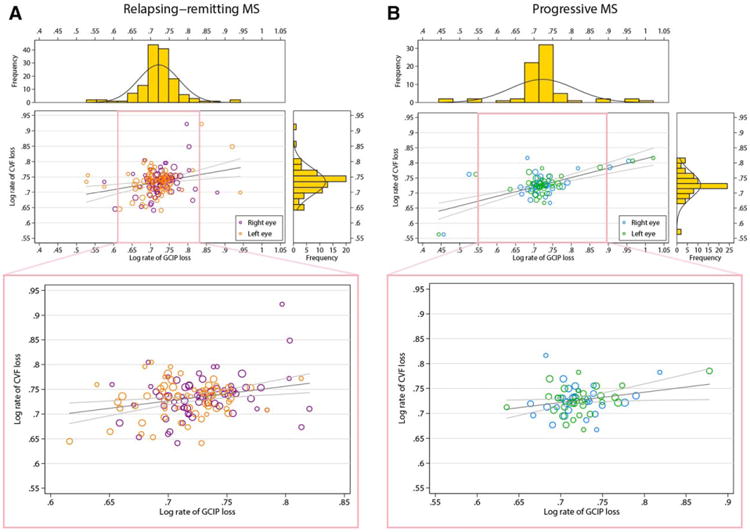
Relationship between rates of GCIP (x-axis) and cerebral volume fraction loss (CVF; y-axis) for the RRMS patients followed in the study (A). (B) Same relationship between rates of GCIP and CVF loss for the PMS patients followed in the study. The observations reported are log transformations of the original data (=log10[−x +5], where x is the respective annualized rate of change in % for CVF and mm for GCIP, i.e., increasing log values being associated with faster rates of GCIP and brain parenchymal fraction loss). The dimension of each circle is proportional to the duration of MRI follow-up. Gray lines represent regression lines and 95% confidence limits indicating the utility of GCIP thinning for predicting simultaneous brain atrophy. Pink insets illustrate the association after limiting the regression analysis to ±2 standard deviations from the mean of the log rate of GCIP change in order to visually demonstrate that the relationships observed are not simply the derivative of outlier effects. Side histograms illustrate the distribution of observations for the corresponding axis. CVF = cerebral volume fraction; GCIP = ganglion cell+inner plexiform layer; MRI = magnetic resonance imaging; RRMS = relapsing-remitting multiple sclerosis; PMS = progressive multiple sclerosis.
Table 4. Partial Correlation Coefficients Between Rates of Change in GCIP Thickness and MRI-Derived Measures Stratified by MS Subtype and ON Modela.
| MS Subtype | ON Modela | Cortical GM | Cerebral WM | Thalamus | Caudate | Putamen | Brainstem | CVF |
|---|---|---|---|---|---|---|---|---|
| Total | ||||||||
| Model 1: n = 107 | Model 1 | 0.361 | 0.275 | 0.380 | 0.163 | 0.147 | 0.202 | 0.437 |
| Model 2: n = 95 | Model 2 | 0.416 | 0.267 | 0.245 | 0.070 | 0.100 | 0.300 | 0.492 |
| Model 3: n = 59 | Model 3 | 0.500 | 0.433 | 0.351 | 0.070 | 0.225 | 0.355 | 0.614 |
| RRMS | ||||||||
| Model 1: n = 71 | Model 1 | 0.312 | 0.201 | 0.466 | 0.265 | 0.131 | 0.111 | 0.328 |
| Model 2: n = 61 | Model 2 | 0.431 | 0.211 | 0.296 | 0.182 | 0.085 | 0.291 | 0.450 |
| Model 3: n = 32 | Model 3 | 0.575 | 0.334 | 0.575 | 0.299 | 0.375 | 0.353 | 0.602 |
| Progressive MS | ||||||||
| Model 1: n = 36 | Model 1 | 0.528 | 0.619 | 0.270 | −0.211 | 0.090 | 0.305 | 0.673 |
| Model 2: n = 34 | Model 2 | 0.546 | 0.583 | 0.286 | −0.154 | 0.127 | 0.293 | 0.670 |
| Model 3: n = 27 | Model 3 | 0.535 | 0.637 | 0.355 | −0.285 | 0.110 | 0.261 | 0.677 |
All models were adjusted for baseline age, sex, and disease duration. Model 1 uses the average of the rates of retinal layer thickness change from both eyes, regardless of ON history, but is additionally adjusted for a past history of ON. Model 2 excludes eyes with a past history of ON from the calculation of the rate of change of retinal layer thickness. Model 3 excludes subjects with a previous history of ON in either eye.
CVF = cerebral volume fraction; GCIP = ganglion cell and inner plexiform layers; GM = gray matter; MRI = magnetic resonance imaging; ON = optic neuritis; RRMS = relapsing-remitting multiple sclerosis; WM = white matter.
Effect of Clinical Parameters on GCIP-MRI Relationships
The effect of ON history on longitudinal GCIP-MRI relationships was studied using three statistical models that incrementally excluded eyes and then subjects with a previous history of ON. Across the cohort, the linear association between GCIP thinning and CVF atrophy increased incrementally for each step-wise refinement to exclude ON effects. A stratified analysis revealed that this effect was primarily derived from the RRMS cohort (see Table 4; Fig 4).
Figure 4.
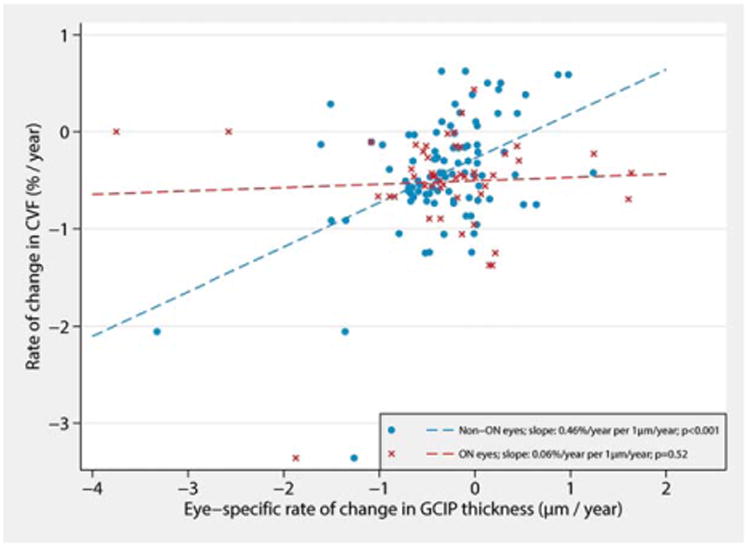
Effect modification of the relationship between the rates of change in GCIP thickness and whole brain volume in the RRMS group by history of ON. Faster rates of GCIP thinning in eyes without a history of ON were associated with faster rates of whole brain atrophy over the study duration (blue). In contrast, changes in GCIP thickness of eyes with a past history of ON (red) were not significantly related to rates of whole brain atrophy. *Results account for within-subject, intereye correlation and are adjusted for age, sex, and disease duration. CVF=cerebral volume fraction; GCIP=ganglion cell+inner plexiform layer; ON=optic neuritis; RRMS=relapsing-remitting multiple sclerosis.
This was confirmed by the results of an ANCOVA model examining whether longitudinal GCIP thinning in eyes with versus without a history of ON had differential predictive utilities in terms of whole-brain atrophy (Table 5 and Supplementary Table 2). An annual loss of 1μm of GCIP thickness in RRMS eyes without ON history was predictive of 0.46% annual CVF loss (95% confidence interval [CI]: 0.27, 0.64; p < 0.001). In contrast, a 1-μm annual loss of GCIP thickness in RRMS eyes with a history of ON was only associated with 0.06% annual CVF loss (95% CI: −0.12, 0.24; p = 0.52). Similar effect modification by ON history was not observed in progressive MS (p = 0.45), and more specifically in the SPMS cohort (p = 0.43). Analysis of the impact of disease duration on GCIP-CVF relationships did not reveal similar effect modification associations in the RRMS cohort (p = 0.43).
Table 5. Comparison of the Relationship Between GCIP Thinning and CVF Loss in Eyes With and Without a Past History of ON.
| MS Subtype | Past ON History | Number of Eyes | Estimated % Yearly CVF Loss Per 1μ;m/year GCIP Thinninga | 95% CI | p | Non-ON vs ON Eyes, p |
| RRMS | Non-ON eyes | 93 | 0.455 | 0.27, 0.64 | <0.001 | 0.002 |
| ON eyes | 49 | 0.059 | −0.12, 0.24 | 0.52 | ||
| PMS | Non-ON eyes | 61 | 0.268 | 0.18, 0.36 | <0.001 | 0.45 |
| ON eyes | 11 | 0.608 | −0.26, 1.48 | 0.17 |
Estimated using multilevel linear regression accounting for within-subject intereye correlation and adjusting for baseline age, sex, and disease duration.
CI = confidence interval; CVF = cerebral volume fraction; GCIP = ganglion cell and inner plexiform layers; ON eyes = eyes with a past history of optic neuritis; Non-ON eyes = eyes without a past history of optic neuritis; RRMS = relapsing-remitting multiple sclerosis; PMS = progressive multiple sclerosis.
Analysis of the rate of change in GCIP thickness over time by ON history revealed a trend for eyes with a past history of ON to have less-pronounced GCIP atrophy over time. On average, eyes with a past history of ON had an annualized rate of GCIP loss 0.13 μm/year less than eyes without a history of ON (p = 0.02). Rates of GCIP atrophy over time also appeared to be strongly related to baseline GCIP thickness (Fig 5). For every 1-μm increase in baseline GCIP thickness, MS eyes tended to lose an additional 0.016μm/year of their GCIP thickness over time (p < 0.001). Including baseline GCIP thickness in a two-level mixed-effects linear regression model explained a greater percentage of the variance in GCIP slopes of change over follow-up time (R2 = 0.22), as opposed to a categorical indicator of previous optic neuritis history (R2 = 0.11).
Figure 5.
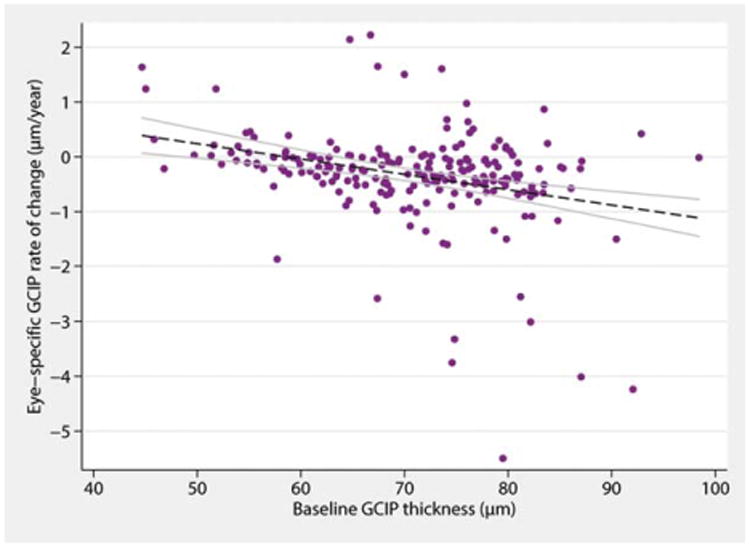
Effect of GCIP thickness at baseline on the rate of GCIP atrophy over time in MS. In general, the lower the GCIP thickness is at baseline, the lower the rate of GCIP atrophy is over time. GCIP=ganglion cell + inner plexiform layer; MS = multiple sclerosis. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Another factor influencing the rate of GCIP change during the observation period was the occurrence of clinical relapses. Comparing the slopes of change in GCIP thickness before and after a relapse in a subgroup of 24 patients experiencing nonocular relapses, a more pronounced rate of GCIP thinning was observed in the post-relapse period as compared to before (difference in slopes [after – before]: −0.30μm/year; p = 0.042). No statistically significant differences were observed for other brain substructures or retinal layers assessed (results not shown). The results were consistently observed after performing a sensitivity analysis examining patients with relapses who were observed for at least 1 year before and after the occurrence of a relapse (n = 14; difference in slopes [after – before]: −0.39 mm/year; p = 0.028). Similarly, patients who developed contrast-enhancing lesions (CEL) at any time point during the study exhibited an additional loss of 0.41um of GCIP thickness and 94.8mm3 of thalamic volume per year, as opposed to patients without CEL development (p< 0.01 for both comparisons). No statistically significant differences in retinal layer atrophy were observed between groups with versus without EDSS progression.
Longitudinal OCT-FLAIR Lesion Volume Relationships
In addition to correlating with rates of brain atrophy, a faster rate of GCIP thinning was also associated with a more rapid rate of increase in FLAIR-lesion volume only in RRMS (r = −0.30; p = 0.02). Although rates of change in INL and ONL thicknesses were not associated with rates of change in whole-brain or brain-substructure volumes (see Table 3), the rate of INL atrophy only in RRMS was significantly associated with the rate of FLAIR lesion accumulation (r = −0.25; p = 0.04; data not shown).
Discussion
Results of this study provide support that thinning of the pRNFL and GCIP mirror concomitant global neurodegeneration and brain atrophy in MS, thereby further validating the potential utility of OCT, an inexpensive, reproducible, noninvasive, and well-tolerated imaging technique for the purpose of tracking MS patients over time.15,16 Our results suggest that tracking GCIP atrophy may be of greater utility than tracking pRNFL atrophy and are in accordance with previous observations that GCIP thickness may have improved structure-function relationships as compared to pRNFL thickness in MS.45 This may relate to (1) better reproducibility, (2) astrogliosis within the RNFL confounding p-RNFL measurements, and (3) reduced susceptibility of the GCIP to edema during optic nerve inflammation.13,44,46 Importantly, faster rates of GCIP atrophy were associated with faster rates of whole-brain, thalamic, and GM atrophy over the study duration, with these relationships remaining significant even after correction for multiple comparisons. Although robust in RRMS, these relationships were particularly striking in progressive MS. Our findings complement and expand upon previous work demonstrating that rates of GCIP thinning may be faster in MS patients exhibiting disease activity (non-ocular relapses, new T2 lesions, and new contrast-enhancing lesions) and disability progression.22 Indeed, rates of GCIP atrophy and FLAIR lesion accumulation were also related in the RRMS group. Tracking GCIP thickness in MS appears to predominantly reflect global neurodegeneration, highlighting the importance of our study findings, given that neurodegeneration is regarded as the primary basis for disability in MS. Whether retinal atrophy similarly mirrors global brain atrophy in other putative neurodegenerative disorders, such as dementias, remains to be investigated. Should outcomes from ongoing investigations corroborate this, it would have far-reaching implications for the field of neurodegeneration in general.
Previous cross-sectional studies suggest that pRNFL and GCIP thicknesses derived from eyes with a history of ON might not correlate with brain and/or brain-substructure volumes, unlike those from eyes without a previous history of ON.28,32,33 Interestingly, our study findings suggest that this pattern of effect modification of ON also extends to the longitudinal association between GCIP thinning and brain atrophy in RRMS. Our findings underscore the disparate predictive utility of GCIP thinning in eyes with versus those without a history of ON for simultaneous brain atrophy. History of ON therefore needs to be integrated into the interpretation of OCT measures for the purpose of tracking disease progression in RRMS. From a pathobiological perspective, the effect modification of ON may be owing to a number of potential reasons. Subsequent to ON, there is acute (within 3 months) disproportionate retinal tissue injury,44,47 thereby potentially masking the relationship between ongoing local retinal and more global neurodegeneration. Moreover, given that there is ultimately less retinal tissue available owing to GCIP loss resulting from the ON event, the rate of retinal atrophy thereafter may be reduced, thus skewing its association with brain atrophy. Indeed, we found that previous ON history and baseline GCIP thickness were both significant predictors of the overall rate of GCIP thickness change over time, with baseline GCIP thickness explaining a greater percentage of the slope variance. This finding has broad implications for clinical trials utilizing OCT as an outcome measure, especially in the RRMS setting. In such a scenario, it might be necessary to match the different arms of a trial by previous optic neuritis history or, more preferably, by baseline GCIP thickness in order to minimize the effect that the baseline amount of available tissue in the GCIP layer has on subsequent rates of atrophy during follow-up. Another practical consideration for clinical trial purposes would be to enrich trial arms with patients who have evidence of disease activity, such as relapses or CELs in the year preceding enrollment. Our results have revealed that subjects with those characteristics tended to have greater rates of GCIP atrophy during follow-up, and their enrichment in clinical trials could shed light on whether neuroprotective interventions have the capacity to slow retinal layer degeneration in MS. Eventually, after ON, the rates of brain and GCIP atrophy may become realigned again in the future, especially as patients transition to a more progressive disease course, thus potentially explaining the lack of effect modification of ON history in the SPMS cohort. It must also be acknowledged, however, that there is underpowering of eyes with ON history in the SPMS cohort included in this study, and therefore this may have accounted for the lack of effect modification for ON history observed in the SPMS cohort.
Unlike the effect of optic neuropathy on the retina, brain inflammation in MS typically results in tissue injury in discrete areas, which are unlikely to affect complete substructure regions, or therefore dramatically alter their total volumes. Thus, brain inflammation could result in dilution (rather than masking) of the relationships between brain and retinal atrophy, possibly contributing to the observation that correlations between GCIP thinning and whole-brain atrophy are less robust in RRMS than in more neurodegenerative progressive MS. Furthermore, GCIP atrophy was also associated with FLAIR lesion accumulation in RRMS. It is possible that axonal injury in the posterior visual pathways, or even outside the visual pathways, might result in trans-synaptic degeneration of retinal ganglion cells.30,48,49 Alternatively, during brain inflammation in MS, there may be more diffuse opening of the blood–brain barrier than appreciated clinically or radiologically, with subclinical inflammation within the optic nerves perhaps accounting for this observation.22
MS subtype analyses also revealed that rates of INL atrophy and FLAIR lesion accumulation, but not brain atrophy, were associated over time in RRMS. This suggests that tracking INL atrophy in RRMS, but not in the less-inflammatory progressive MS subtypes, may provide information regarding ongoing global inflammation. Whereas dynamic macular “microcysts” (although they may not actually be cysts), predominantly of the INL, have been observed to occur in a proportion of MS eyes, similar findings, however, have also been observed in other inflammatory and noninflammatory disease processes, suggesting they are nonspecific.50–53 Nonetheless, in RRMS, a relationship between increased INL thickness and FLAIR lesion volume cross-sectionally has been previously proposed,33 and increased INL thickness at baseline (even in the absence of visible macular microcysts) has been shown to be associated with clinical and radiological disease activity.25 It is possible that increases in INL thickness herald inflammatory activity in MS, and as this activity ensues, the INL volume subsequently declines.
This study has a number of limitations. Because the majority of included patients had RRMS, more accurate characterization of the associations between retinal and brain atrophy by MS subtype is warranted, requiring the enrollment of greater numbers of progressive MS patients, of both the SPMS and PPMS subtypes. Larger and longer longitudinal studies would help address these limitations and establish the validity of our findings. Furthermore, the cohort included in this study is a heterogeneous cohort, both in terms of clinical characteristics and disease-modifying therapies. Therefore, it is necessary to exercise caution when extrapolating results from the current study for the purpose of designing future clinical trials, which would more likely be structured toward recruitment of homogenous MS cohorts. Moreover, virtually all RRMS patients in the current study cohort were on disease-modifying therapies, and, as a result, it is likely that our results underestimate true rates of retinal atrophy; retinal rates of atrophy might be hypothetically higher in untreated MS populations. In addition, there was variability in terms of the classes of disease-modifying therapies patients were receiving not only at baseline, but also for the duration of study follow-up. This mix in disease-modifying therapies throughout the study duration precluded assessment of the effects of MS treatments on our results. Future studies including more homogenously treated MS subgroups would allow for more accurate assessment of the effects of disease modifying therapies on the relationships between rates of retinal and brain atrophy. Such information would be of great utility and assist in guiding future clinical trial designs that incorporate OCT as an outcome measure.
In summary, results of this study indicate that GCIP and brain atrophy in MS closely parallel each other over time, suggesting a role for OCT as a valuable biomarker not only for the purpose of tracking patients clinically, but also in clinical trials for objective investigation of putative neuroprotective and/or neurorestorative therapies. Although GCIP and brain atrophy are associated in RRMS (especially after refinement for ON history; a factor that should be borne in mind in the interpretation of GCIP measures longitudinally), the associations between GCIP and brain atrophy in progressive MS appear to be exceptional. Although our findings require independent verification, and should be replicated across larger MS cohorts, they nonetheless help to answer many previously unanswered questions, particularly with respect to how OCT measures relate to global MS processes longitudinally.
Supplementary Material
Acknowledgments
This study was funded by the Race to Erase MS (to S.S.), National Institutes of Health (5R01NS082347-02 [to P.A.C.] and R01NS070906 [to D.P.]), National Multiple Sclerosis Society (TR 3760-A-3 [to P.A.C.] and RG 4212-A-4 [to L.J.B. subcontracted to P.A.C.]), National Eye Institute (R01 EY 014993 and R01 EY 019473 [to L.J.B. subcontracted to P.A.C.]), Braxton Debbie Angela Dillon and Skip (DADS) Donor Advisor Fund (to P.A.C., E.M.F., and L.J.B.), and the Intramural Research Program of NINDS (to D.S.R.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
Potential Conflicts of Interest: S.S. has served on a scientific advisory board for Biogen Idec and Genzyme. J.N.R. was a consultant for Genzyme and received research support from Biogen Idec, Novartis, and Sun Pharmaceuticals. Since the completion of this work, he has now become an employee of MedImmune. J.O. has received personal compensation for consulting or speaking from EMD-Serono, Genzyme, Biogen Idec, and Novartis and has received research funding from Genzyme. S.D.N. has received personal compensation for consulting from Biogen Idec, Novartis, and Genzyme and has received research funding from Biogen Idec and Novartis. J.L.P. has received consulting fees from and holds stock in Diagnosoft, Inc. P.V.Z. has received grant funding from, has technology licensed to, and is a paid lecturer for Philips Healthcare. L.J.B. has received speaking and consulting honoraria from Biogen Idec, Bayer, and Novartis. E.M.F. has received speaker and consulting fees from Novartis, Genzyme, Acorda, and TEVA. P.A.C. has received personal compensation for consulting and serving on scientific advisory boards from Vertex, Vaccinex, Merck, and AbbVie and has received research funding from Biogen IDEC, MedImmune, and Novartis.
References
- 1.Kappos L, Moeri D, Radue EW, et al. Predictive value of gadolinium-enhanced magnetic resonance imaging for relapse rate and changes in disability or impairment in multiple sclerosis: A meta-analysis. Gadolinium MRI Meta-analysis Group. Lancet. 1999;353:964–969. doi: 10.1016/s0140-6736(98)03053-0. [DOI] [PubMed] [Google Scholar]
- 2.Fisher E, Rudick RA, Simon JH, et al. Eight-year follow-up study of brain atrophy in patients with MS. Neurology. 2002;59:1412–1420. doi: 10.1212/01.wnl.0000036271.49066.06. [DOI] [PubMed] [Google Scholar]
- 3.Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014;75:43–49. doi: 10.1002/ana.24018. [DOI] [PubMed] [Google Scholar]
- 4.van Waesberghe JH, Kamphorst W, De Groot CJ, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol. 1999;46:747–754. doi: 10.1002/1531-8249(199911)46:5<747::aid-ana10>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- 5.De Stefano N, Narayanan S, Francis GS, et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58:65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- 6.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 7.Miller DH. Biomarkers and surrogate outcomes in neurodegenerative disease: lessons from multiple sclerosis. NeuroRx. 2004;1:284–294. doi: 10.1602/neurorx.1.2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minneboo A, Uitdehaag BM, Jongen P, et al. Association between MRI parameters and the MS severity scale: a 12 year follow-up study. Mult Scler. 2009;15:632–637. doi: 10.1177/1352458509102617. [DOI] [PubMed] [Google Scholar]
- 9.Calabrese M, Atzori M, Bernardi V, et al. Cortical atrophy is relevant in multiple sclerosis at clinical onset. J Neurol. 2007;254:1212–1220. doi: 10.1007/s00415-006-0503-6. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese M, Agosta F, Rinaldi F, et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch Neurol. 2009;66:1144–1150. doi: 10.1001/archneurol.2009.174. [DOI] [PubMed] [Google Scholar]
- 11.Simon JH. Brain atrophy in multiple sclerosis: what we know and would like to know. Mult Scler. 2006;12:679–687. doi: 10.1177/1352458506070823. [DOI] [PubMed] [Google Scholar]
- 12.Inglese M, Oesingmann N, Casaccia P, Fleysher L. Progressive multiple sclerosis and gray matter pathology: an MRI perspective. Mt Sinai J Med. 2011;78:258–267. doi: 10.1002/msj.20247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saidha S, Syc SB, Ibrahim MA, et al. Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain. 2011;134(pt 2):518–533. doi: 10.1093/brain/awq346. [DOI] [PubMed] [Google Scholar]
- 14.Petzold A, de Boer JF, Schippling S, et al. Optical coherence tomography in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol. 2010;9:921–932. doi: 10.1016/S1474-4422(10)70168-X. [DOI] [PubMed] [Google Scholar]
- 15.Frohman EM, Fujimoto JG, Frohman TC, et al. Optical coherence tomography: a window into the mechanisms of multiple sclerosis. Nat Clin Pract Neurol. 2008;4:664–675. doi: 10.1038/ncpneuro0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saidha S, Eckstein C, Ratchford JN. Optical coherence tomography as a marker of axonal damage in multiple sclerosis. CML - Multiple Sclerosis. 2010;2:33–43. [Google Scholar]
- 17.Toussaint D, Perier O, Verstappen A, Bervoets S. Clinicopathological study of the visual pathways, eyes, and cerebral hemispheres in 32 cases of disseminated sclerosis. J Clin Neuroophthalmol. 1983;3:211–220. [PubMed] [Google Scholar]
- 18.Ikuta F, Zimmerman HM. Distribution of plaques in seventy autopsy cases of multiple sclerosis in the united states. Neurology. 1976;26(6 pt 2):26–28. doi: 10.1212/wnl.26.6_part_2.26. [DOI] [PubMed] [Google Scholar]
- 19.Shindler KS, Ventura E, Dutt M, Rostami A. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp Eye Res. 2008;87:208–213. doi: 10.1016/j.exer.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green AJ, McQuaid S, Hauser SL, et al. Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration. Brain. 2010;133:1591–1601. doi: 10.1093/brain/awq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerrison JB, Flynn T, Green WR. Retinal pathologic changes in multiple sclerosis. Retina. 1994;14:445–451. doi: 10.1097/00006982-199414050-00010. [DOI] [PubMed] [Google Scholar]
- 22.Ratchford JN, Saidha S, Sotirchos ES, et al. Active MS is associated with accelerated retinal ganglion cell/inner plexiform layer thinning. Neurology. 2013;80:47–54. doi: 10.1212/WNL.0b013e31827b1a1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talman LS, Bisker ER, Sackel DJ, et al. Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann Neurol. 2010;67:749–760. doi: 10.1002/ana.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelfand JM, Nolan R, Schwartz DM, et al. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain. 2012;135(pt 6):1786–1793. doi: 10.1093/brain/aws098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saidha S, Sotirchos ES, Ibrahim MA, et al. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol. 2012;11:963–972. doi: 10.1016/S1474-4422(12)70213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon-Lipkin E, Chodkowski B, Reich DS, et al. Retinal nerve fiber layer is associated with brain atrophy in multiple sclerosis. Neurology. 2007;69:1603–1609. doi: 10.1212/01.wnl.0000295995.46586.ae. [DOI] [PubMed] [Google Scholar]
- 27.Grazioli E, Zivadinov R, Weinstock-Guttman B, et al. Retinal nerve fiber layer thickness is associated with brain MRI outcomes in multiple sclerosis. J Neurol Sci. 2008;268:12–17. doi: 10.1016/j.jns.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Siger M, Dziegielewski K, Jasek L, et al. Optical coherence tomography in multiple sclerosis: thickness of the retinal nerve fiber layer as a potential measure of axonal loss and brain atrophy. J Neurol. 2008;255:1555–1560. doi: 10.1007/s00415-008-0985-5. [DOI] [PubMed] [Google Scholar]
- 29.Dorr J, Wernecke KD, Bock M, et al. Association of retinal and macular damage with brain atrophy in multiple sclerosis. PLoS One. 2011;6:e18132. doi: 10.1371/journal.pone.0018132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfueller CF, Brandt AU, Schubert F, et al. Metabolic changes in the visual cortex are linked to retinal nerve fiber layer thinning in multiple sclerosis. PLoS One. 2011;6:e18019. doi: 10.1371/journal.pone.0018019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young KL, Brandt AU, Petzold A, et al. Loss of retinal nerve fibre layer axons indicates white but not grey matter damage in early multiple sclerosis. Eur J Neurol. 2013;20:803–811. doi: 10.1111/ene.12070. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann H, Freing A, Kaufhold F, et al. Optic neuritis interferes with optical coherence tomography and magnetic resonance imaging correlations. Mult Scler. 2013;19:443–450. doi: 10.1177/1352458512457844. [DOI] [PubMed] [Google Scholar]
- 33.Saidha S, Sotirchos ES, Oh J, et al. Relationships between retinal axonal and neuronal measures and global central nervous system pathology in multiple sclerosis. JAMA Neurol. 2013;70:34–43. doi: 10.1001/jamaneurol.2013.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. national multiple sclerosis society (USA) advisory committee on clinical trials of new agents in multiple sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 36.Bazin PL, Pham DL. Topology-preserving tissue classification of magnetic resonance brain images. IEEE Trans Med Imaging. 2007;26:487–496. doi: 10.1109/TMI.2007.893283. [DOI] [PubMed] [Google Scholar]
- 37.Han X, Pham DL, Tosun D, et al. CRUISE: Cortical reconstruction using implicit surface evolution. Neuroimage. 2004;23:997–1012. doi: 10.1016/j.neuroimage.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 38.Carass A, Cuzzocreo J, Wheeler MB, et al. Simple paradigm for extra-cerebral tissue removal: algorithm and analysis. Neuroimage. 2011;56:1982–1992. doi: 10.1016/j.neuroimage.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiee N, Bazin PL, Zackowski KM, et al. Revisiting brain atrophy and its relationship to disability in multiple sclerosis. PLoS One. 2012;7:e37049. doi: 10.1371/journal.pone.0037049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiee N, Bazin PL, Ozturk A, et al. A topology-preserving approach to the segmentation of brain images with multiple sclerosis lesions. Neuroimage. 2010;49:1524–1535. doi: 10.1016/j.neuroimage.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geremia E, Clatz O, Menze BH, et al. Spatial decision forests for MS lesion segmentation in multi-channel magnetic resonance images. Neuroimage. 2011;57:378–390. doi: 10.1016/j.neuroimage.2011.03.080. [DOI] [PubMed] [Google Scholar]
- 42.Warner CV, Syc SB, Stankiewicz AM, et al. The impact of utilizing different optical coherence tomography devices for clinical purposes and in multiple sclerosis trials. PLoS One. 2011;6:e22947. doi: 10.1371/journal.pone.0022947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tewarie P, Balk L, Costello F, et al. The OSCAR-IB consensus criteria for retinal OCT quality assessment. PLoS One. 2012;7:e34823. doi: 10.1371/journal.pone.0034823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Syc SB, Saidha S, Newsome SD, et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012;135(pt 2):521–533. doi: 10.1093/brain/awr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saidha S, Syc SB, Durbin MK, et al. Visual dysfunction in multiple sclerosis correlates better with optical coherence tomography derived estimates of macular ganglion cell layer thickness than peripapillary retinal nerve fiber layer thickness. Mult Scler. 2011;17:1449–1463. doi: 10.1177/1352458511418630. [DOI] [PubMed] [Google Scholar]
- 46.Syc SB, Warner CV, Hiremath GS, et al. Reproducibility of high-resolution optical coherence tomography in multiple sclerosis. Mult Scler. 2010;16:829–839. doi: 10.1177/1352458510371640. [DOI] [PubMed] [Google Scholar]
- 47.Costello F, Coupland S, Hodge W, et al. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol. 2006;59:963–969. doi: 10.1002/ana.20851. [DOI] [PubMed] [Google Scholar]
- 48.Reich DS, Smith SA, Gordon-Lipkin EM, et al. Damage to the optic radiation in multiple sclerosis is associated with retinal injury and visual disability. Arch Neurol. 2009;66:998–1006. doi: 10.1001/archneurol.2009.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabilondo I, Martinez-Lapiscina EH, Martinez-Heras E, et al. Trans-synaptic axonal degeneration in the visual pathway in multiple sclerosis. Ann Neurol. 2014;75:98–107. doi: 10.1002/ana.24030. [DOI] [PubMed] [Google Scholar]
- 50.Sotirchos ES, Saidha S, Byraiah G, et al. In vivo identification of morphologic retinal abnormalities in neuromyelitis optica. Neurology. 2013;80(15):1406–1414. doi: 10.1212/WNL.0b013e31828c2f7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaufhold F, Zimmermann H, Schneider E, et al. Optic neuritis is associated with inner nuclear layer thickening and microcystic macular edema independently of multiple sclerosis. PLoS One. 2013;8:e71145. doi: 10.1371/journal.pone.0071145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barboni P, Carelli V, Savini G, et al. Microcystic macular degeneration from optic neuropathy: not inflammatory, not trans-synaptic degeneration. Brain. 2013;136(pt 7):e239. doi: 10.1093/brain/awt014. [DOI] [PubMed] [Google Scholar]
- 53.Wolff B, Basdekidou C, Vasseur V, et al. Retinal inner nuclear layer microcystic changes in optic nerve atrophy: a novel spectral-domain OCT finding. Retina. 2013;33:2133–2138. doi: 10.1097/IAE.0b013e31828e68d0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.