

Summary
Recent studies have identified somatic alterations in the gene encoding for neurofibromin (NF1) in a subset of glioblastoma (GBM), usually associated with the mesenchymal molecular subtype. To understand the significance of NF1 genetic alterations in diffuse gliomas in general, we evaluated public databases and tested for NF1 copy number alterations in a cohort using fluorescence in situ hybridization. NF1 genetic loss (homozygous NF1 deletions or mutations with predicted functional consequences) was present in 30 (of 281) (11%) GBM and 21 (of 286) (7%) lower-grade gliomas in The Cancer Genome Atlas data. Furthermore, NF1 loss was associated with worse overall and disease-specific survival in the lower-grade glioma, but not GBM, Group in The Cancer Genome Atlas cohort. IDH1 or 2 mutations co-existed in lower-grade gliomas with NF1 loss (36%) but not in GBM. In our cohort studied by fluorescence in situ hybridization, NF1/17q (n = 2) or whole Ch17 (n = 3) losses were only identified in the GBM group (5/86 [6%]). Tumors with NF1/Ch17 loss were predominantly adult GBM (4/5); lacked EGFR amplification (0/4), strong p53 immunolabeling (1/5), or IDH1 (R132H) protein expression (0/5); but expressed the mesenchymal marker podoplanin in 4/5. NF1 genetic loss occurs in a subset of diffuse gliomas, and its significance deserves further exploration.
Keywords: Glioma, Neurofibromatosis, Glioblastoma, FISH, Neurofibromin, NF1
1. Introduction
Neurofibromatosis type 1 (NF1) is a genetic tumor-predisposing syndrome caused by germline mutations in the NF1 gene and is inherited in an autosomal dominant fashion [1,2]. The NF1 gene, located at chromosome region 17q11.2, encodes neurofibromin, a tumor suppressor that works primarily by regulating RAS but also modulates adenyl cyclase [3,4]. NF1 loss generally leads to increased activity in a variety of pro-tumorigenic pathways, particularly the mitogen-activated protein kinase pathway (Fig. 1). Patients with NF1 are predisposed to a variety of tumors affecting the central and peripheral nervous system. In the central nervous system, the predominant tumor type is pilocytic astrocytoma, which in this patient population has a propensity to involve the optic pathways. However, NF1 patients, especially adults, may develop gliomas of all types and grades [5]. In a large retrospective study of NF1-associated gliomas classified using current World Health Organization (WHO) criteria, 27% were diffusely infiltrating astrocytomas, and 22% were high grade (ie, anaplastic astrocytomas and glioblastomas) [6].
Fig. 1.
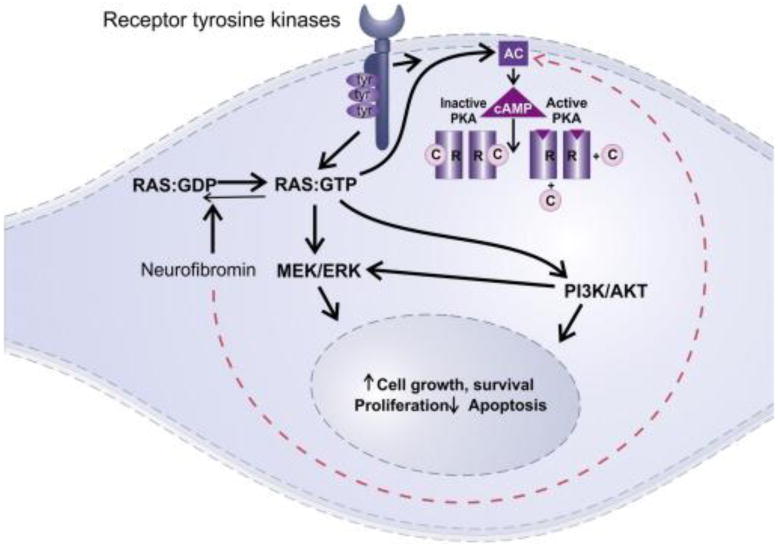
Cellular pathways affected by neurofibromin loss. Neurofibromin is an important tumor suppressor that negatively regulates RAS by driving it to the GDP bound state. Neurofibromin loss therefore leads to constitutive RAS activation and MEK/ERK signaling, resulting in pro-tumorigenic signals. Neurofibromin also has a lesser known function acting through adenyl cyclase (AC) (dotted arrow).
A role for NF1 loss in sporadic astrocytomas has also been elucidated more recently. Sporadic pilocytic astrocytomas do not have alterations in NF1 [7], but rather BRAF fusions or mutations [8–11], as well as activating mutations in other oncogenes [12,13]. However, early clues that alterations in the NF1 gene may be important in the development of diffusely infiltrating gliomas were provided by mouse models in which Nf1 and Tp53 co-inactivation led to glioblastoma formation with high, if not complete, penetrance [14,15]. More recent large-scale comprehensive analyses have identified somatic alterations in the NF1 gene in a subset of glioblastoma [16,17], usually associated with the mesenchymal molecular subtype [18].
The mesenchymal molecular subtype was initially identified by gene expression profiling studies of highgrade gliomas that separated proneural, mesenchymal and proliferative classes. The mesenchymal signature defined a group with a relatively worse prognosis and relatively enriched in recurrent tumors, with frequent up-regulation of YKL40, CD44, and STAT3 [19]. Recent analysis of data from lower-grade gliomas (II-III) also highlighted the relevance of the glioblastoma molecular subtypes with respect to biology and prognosis [20]. However, the mesenchymal subclass was almost entirely restricted to glioblastomas in this study.
Germline NF1 mutations may be of many different types, including mutations altering splicing of mRNA, as well as nonsense, missense, and frameshift mutations [21,22]. Approximately 5% of NF1 patients have constitutional large gene deletions, which may be associated with a more severe phenotype [23,24]. Large constitutional deletions may also have an effect on tumor burden in a subset of patients [25]. At the current moment it is unclear what the role of NF1 genetic alterations, particularly deletions and mutations with predicted functional effects, is in the sporadic diffuse glioma subtypes. In the current study, we tested for NF1 copy number alterations in a cohort of diffuse gliomas, evaluated public databases, and searched for associations with pathologic and molecular features.
2. Materials and methods
2.1. The Cancer Genome Atlas data analysis
The cBIOPORTAL for cancer genomics website from Memorial Sloan Kettering Cancer Center (http://www.cbioportal.org/public-portal) was used to analyze The Cancer Genome Atlas (TCGA) data for NF1 alterations in diffuse gliomas (date of analysis 8 December 2014). Copy number/genotype platforms listed for the glioblastoma dataset are Affymetrix SNP6 and Agilent 224K/415K [26]. Data for homozygous deletions were obtained through the GISTIC algorithm. To exclude non-relevant “passenger” mutations or germline variants, we focused on mutations with predicted functional consequences as specified in cBIOPORTAL using previously published guidelines by Reva et al. [27]. In brief, mutations are assigned a functional impact score based on the extent of conservation of the specific amino acid residues across and within species. “Medium” and “High” scores (>∼1.9) are predicted to be functional (ie, relevant to disease). Details of cBIOPORTAL analysis have been previously published [28,29]. Frequencies were compared using the Fisher exact test. Survival analysis was performed using Kaplan-Meier curves and the log-rank test per the cBIOPORTAL algorithm. In addition, available scanned hematoxylin and eosin (H&E) slides were reviewed and IDH mutation status was abstracted in the subset of lower-grade glioma cases with NF1 alterations in cBIOPORTAL.
2.2. Primary tumor samples
A cohort of 130 diffuse gliomas obtained from two tissue microarrays constructed at Johns Hopkins Hospital were studied, including 86 glioblastomas (57 adults, 29 pediatric) and 44 lower-grade diffuse gliomas (grades II-III). Two to four 0.6 mm diameter cores per tumor were evaluated. Clinicopathologic features of these cases have been previously published [30]. All studies were performed under institutional review board approval and following all guidelines.
2.3. Immunohistochemistry
Immunohistochemical staining was performed in formalin-fixed paraffin-embedded tissue microarray sections using a podoplanin antibody (clone D2-40, DAKO, Carpinteria, CA) 1:200). A biotin-free polymer detection kit was used (Bond Polymer Refine Detection, Leica Microsystems, Bannock Burn, IL). At least 2 TMA scores per case were required for score assignment. Scoring was performed by 2 observers (M.A.V., F.J.R.) using a three-tiered scale based on staining intensity (0-3+) (Supplementary Fig. 1). Non-neoplastic gray and white matter were used as controls.
2.4. Fluorescence in situ hybridization
A custom-made fluorescence in situ hybridization (FISH) probe targeting the NF1 gene (17q11.2) with a CEP17 probe control was obtained from Empire Genomics (Buffalo, NY), and hybridized using previously described methods [31]. Sections were evaluated using an Olympus PROVIS (Center Valley, PA) fluorescence scope with appropriate filters. At least 50 non-overlapping nuclei were evaluated per case. Heterozygous deletion was defined as a target-to-control ratio of 0.80 or less. Monosomy was defined as loss of target and control probe in at least 60% of cells. Chromosome gain was defined as an extra copy of target and control probe in at least 30% of cells. Scoring and calls were made by an observer blinded as to the specific pathology of the specimens (M.A.V.), and confirmed by a second observer (F.J.R.).
3. Results
3.1. NF1 mutations and homozygous deletions in glioblastomas and lower-grade gliomas in TCGA
We first looked at genomic data of diffuse gliomas from The Cancer Genome Atlas (TCGA) using the cBIOPORTAL for cancer genomics website from Memorial Sloan Kettering Cancer Center (http://www.cbioportal.org/public-portal) to determine the frequency of NF1 genetic alterations in diffuse gliomas of various grades (II-III). Genomic alterations (mutations with predicted functional consequences and homozygous deletions) were present in the NF1 gene in 30 (of 281) (11%) glioblastomas with sequencing and copy number data (homozygous deletion in 5, truncating mutations in 19, missense mutations with predicted functional effects in 4, in-frame heterozygous deletion in 1, and an additional single case with a homozygous deletion and a truncating mutation). A single case had a missense mutation with no predicted functional effects. None of these tumors had IDH1 or IDH2 mutations. Survival analysis did not demonstrate any differences when comparing glioblastomas with or without NF1 alterations (Fig. 2A and B). In the lower-grade glioma group, 21 (of 286) (7%) cases with sequencing and copy number data had inactivating alterations in the NF1 gene (homozygous deletions in 3, truncating mutations in 15, missense mutations with predicted functional effects in 3). A single case had a missense mutation with no predicted functional effects. Overall and disease-specific survival were worse in lower-grade gliomas with NF1 alterations compared to those without (P = .0001) (Fig. 2C and D).
Fig. 2.
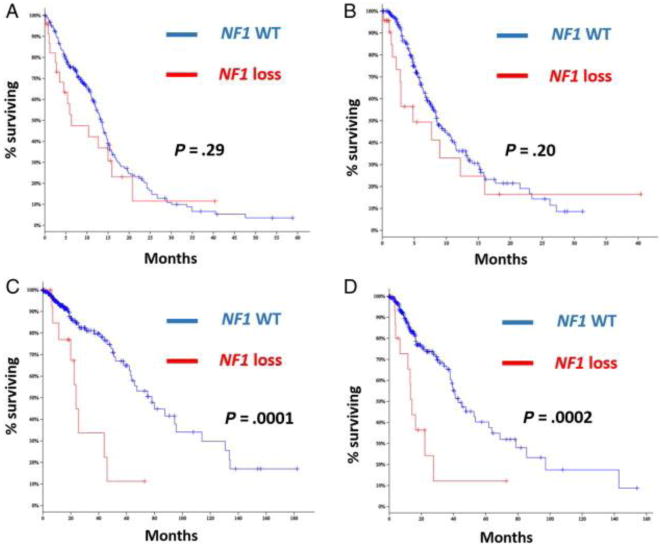
Overall and disease-specific survival by NF1 genetic alterations (loss) from the TCGA dataset. NF1 genetic loss shows no effect in overall (A) or disease-specific survival (B) in glioblastoma. However, in the lower-grade glioma group, NF1 loss was associated with significantly worse overall (C) and disease-specific survival (D) (log-rank test). (WT = wild type; NF1 loss includes homozygous deletions and predominantly mutations with functional consequences).
Histologically, this subgroup of lower-grade gliomas with NF1 genetic loss included low-grade astrocytoma (WHO grade II) (n = 10), low-grade oligodendroglioma (n = 2), oligoastrocytomas (WHO grade II-III) (n = 7), anaplastic astrocytoma (WHO grade III) (n = 1), and anaplastic oligodendroglioma (WHO grade III) (n = 1) per ICD-O-3-HISTOLOGY codes listed in cBIOPORTAL per case. Permanent H&E slides were available for review in 8 cases and confirmed a predominance of astrocytic histology, including the only 2 oligoastrocytomas with available slides. Of interest IDH1/2 mutations co-occurred in 8 (of 21) (38%) of these tumors (IDH1 [R132H] in 7, IDH2 [R172K] in 1). Three (of 5) of these patients with tumors with co-existing NF1 and IDH1/2 mutations and follow up >1 year were dead 22 to 46 months after diagnosis. FUBP1 and/or CIC mutations, typical of oligodendrogliomas, were present in only three (of 21) of these cases.
When looking at TCGA tumors with copy number alteration data only, NF1 deletions were present in (7/563 [1.2%]) glioblastomas compared to lower-grade gliomas (4/512 [0.7%]), but this did not achieve statistical significance (P = .55, Fisher exact test).
3.2. Large NF1 copy number alterations occur in a subset of glioblastomas
Next we searched for copy number alterations in a cohort of diffuse gliomas grades II-IV from JHH, in order to confirm our observations above regarding NF1 genetic loss in public databases. We chose FISH, a technique readily applicable to formalin-fixed, paraffin-embedded tissues and suitable for identification of tumor areas with genetic alterations, even focally. In addition, this technique allows for the identification of large, possibly more deleterious, deletions. NF1/17q or Ch 17 gains/polysomies were relatively common and identified in 40 (31%) of 130 cases across tumors tested, including 2 of 11 diffuse astrocytomas, 6/16 anaplastic astrocytomas, 7 of 17 oligodendroglial tumors, and 25 (29%) of 86 glioblastomas (GBM) (8/29 pediatric GBM, 17/57 adult GBM). Of greater functional interest, NF1/17q (n = 2) or whole Ch17 (n = 3) losses were only identified in the GBM group (6% [5/86]) (Fig. 3 and Table 1).
Fig. 3.
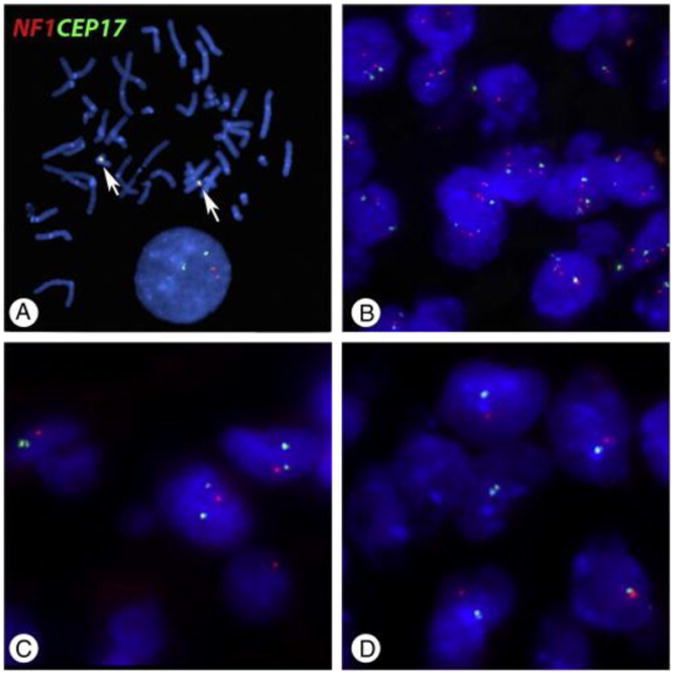
Copy number alterations in the NF1 gene in diffuse gliomas. A, Normal human metaphases demonstrate proper targeting of the probes for the NF1 locus (Orange 5-TAMRA dUTP) and centromere 17 (Green 5-Fluorescein dUTP) (arrows) (original magnification ×1000). B, Nuclei are stained blue (DAPI). The most frequent alteration was polysomy involving chromosome 17 (original magnification ×600). Conversely, the more functionally relevant alteration of NF1/17q loss (original magnification ×1000) (C) or monosomy 17 (original magnification ×1000)(D) were present in a small subset of glioblastomas.
Table 1. Pathologic and molecular features of gliomas with NF1/Ch17 loss.
| NF1/Ch17loss n= 5 | Ch17 intact n = 125 | |
|---|---|---|
| Glioma subtype | GBM 5/5 (100%) | 81/125 (65%) |
| Adult GBM 4/5 | 53/125 (42%) | |
| Pediatric GBM 1/5 | 28/125 (22%) | |
| Diffuse (II-III) (0) | 44/125 (35%) | |
| Podoplanin IHC | 4/5 | 55/122 (45%) |
| EGFR amp | 0/4 | 17/90 (19%) |
| p53 IHC | 1/5 | 32/96 (33%) |
| IDH1 (R132H) IHC | 0/5 | 37/125 (30%) |
3.3. Clinicopathologic and molecular features of glioblastomas with NF1 losses
When looking at the GBM subset with NF1/Ch17 loss in the JHH cohort, the tumors were predominantly adult GBM (4/5). Pathologic examination revealed that 2 of the 5 tumors were gliosarcomas (Table 2). The diagnosis of gliosarcoma was performed using current WHO criteria, specifically demonstrating distinct glial fibrillary acidic Protein (GFAP)-positive, reticulin-poor glial neoplastic areas and GFAP-negative, reticulin-rich sarcomatous areas [32]. Of the remaining 3, one had small cell features, one was fibrillary, and one was gemistocytic. Molecular and immunohistochemical analysis demonstrated that these tumors predominantly lacked EGFR amplification (0/4), strong p53 immunolabeling (1/5), mutant IDH1 (R132H) protein expression (0/5), or the alternative lengthening of telomeres phenotype (1/5). Median overall survival was 32 months (range, 10-118) (Tables 1 and 2).
Table 2. Clinical, pathological and molecular features of GBM group with NF1 loss.
| AGE | Tumor Type | Tumor Location | Histologic subtype | Ch17 | PODOPLANIN IHC | EGFR FISH | IDH1 (R132H) | p53 IHC | ALT | Overall survival (Mo) |
|---|---|---|---|---|---|---|---|---|---|---|
| 81 | Adult GBM | R temporal lobe | Gemistocytic | Monosomy | Pos | NA | Neg | Neg | Neg | 32 |
| 47 | Adult GBM | R frontal lobe | Fibrillary | Monosomy | Pos | No AMP | Neg | Neg | Neg | 118 |
| 14 | Peds GBM | Cerebellum | Small cell features | NF1 loss | Neg | No AMP | Neg | Neg | Pos | 42 |
| 59 | Adult GBM | L temporal lobe | Gliosarcoma | Monosomy | Pos | No AMP | Neg | Pos | Neg | 21 |
| 59 | Adult GBM | L parietooccipital | Gliosarcoma | NF1 loss | Pos | No AMP | Neg | Neg | Neg | 10 |
Abbreviations: R, right; L, left; Ch, chromosome; IHC, immunohistochemistry; Pos, positive; Neg, negative; AMP, amplification; NA, not available.
Given the strong association between NF1 loss and the mesenchymal phenotype in glioblastoma, we next tested the samples with an antibody against podoplanin, which is in broad clinical use and previously used as a marker of the mesenchymal subtype in paraffin tissues [33]. Podoplanin immunoreactivity when present was diffuse and varied primarily in staining intensity, therefore the latter represented the scoring parameter. Glioblastomas with NF1 loss expressed podoplanin more frequently (4/5) than other diffuse gliomas (45% [55/122]), although this was not statistically significant (P = .18), likely related to the small number of cases of interest (Fig. 4). Of note, almost all IDH1 (R132H) mutant tumors (except for two) were podoplanin negative.
Fig. 4.
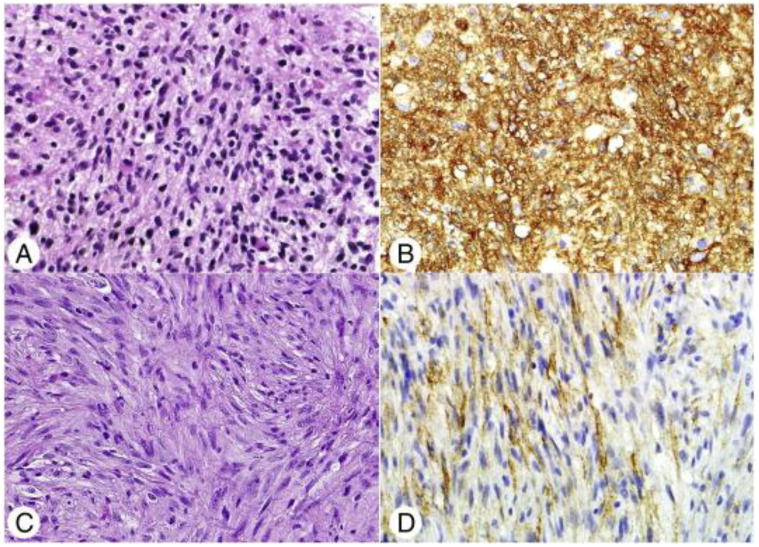
Histologic and immunohistochemical features of diffuse gliomas with NF1 loss. The histology of the 5 tumors with NF1 loss was variable, including fibrillary astrocytoma (H&E; original magnification ×400) (A) and 2 gliosarcomas The latter were characterized by neoplastic spindle cells (C), lacking GFAP expression and demonstrating pericellular reticulin staining (not shown). Matched podoplanin immunohistochemical stains demonstrating strong (B) and more modest (D) overexpression in these tumors (original magnification ×400).
4. Discussion
The NF1 gene has emerged in recent years as an important tumor suppressor gene in gliomas, not only in those associated with the syndrome but also those that (more commonly) develop sporadically. Previous analyses of the TCGA dataset in glioblastoma uncovered a strong association between somatic NF1 mutation and the mesenchymal molecular subtype [18]. The mesenchymal subtype, as the name suggests, was recognized initially by a gene expression signature [19]. Subsequent work demonstrated that this phenotype is driven by a set of transcription factors (C/EBPβ and STAT3), which are able to reprogram neural stem cells along this phenotype [34]. Numerous markers have been proposed as being useful in characterizing the mesenchymal molecular subtype. We chose podoplanin in this study because it is commercially available and routinely used in many laboratories, and has been proposed by some groups as a marker useful in proteomic subclassification of glioblastoma [33], although it has also been found to label reactive glial cells in the context of gliomas and other pathologies [35]. One caveat is that glioblastoma and other diffuse gliomas are increasingly recognized as demonstrating genetic and phenotypic heterogeneity, and the small areas studied in TMAs may not be fully representative of the tumor expression patterns.
In this study, we evaluated a cohort of diffuse gliomas for copy number alterations involving the NF1 gene. Heterozygous deletion of the NF1 gene region/17q or whole chromosomal loss (monosomy 17) was actually restricted to a small subset of glioblastomas and were completely absent in a group of diffuse gliomas of various types and grades. Although these tumors have relatively distinct molecular genetic features, the histology was variable. Of interest, 2 of the cases with NF1/Ch17 loss were histologically gliosarcomas. Prior cytogenetic studies of gliosarcoma have demonstrated a similar genetic profile to glioblastoma, including 7,X,9q, and 20q gain, as well as 9p, Ch10, and 13q loss [36,37], although 17q loss does not appear to be particularly common.
Also of interest is that the survival of the patients with the NF1-deleted group was not necessarily poor, although this alteration was strictly limited to the glioblastoma group. Despite the expression of podoplanin by 4 (of 5) of these cases, the relatively long survival of some of these cases raises the possibility that these tumors are not all of the mesenchymal subtype. This subtype has been previously associated with a high frequency of somatic NF1 loss, although the alterations identifiable in the TCGA dataset include point mutations and small deletions. It is unclear if large deletions identifiable by FISH or whole chromosomal loss has the same significance but may identify a distinct subset of glioblastoma. Of interest, two of the tumors in our dataset with NF1 loss were gliosarcomas, and the survival of these 2 patients was shorter (10 and 21 months).
When looking at data from the TCGA, deletions involving the NF1 gene were relatively rare, although given the higher resolution of the platforms employed, only homozygous deletions were studied. They were present in both the glioblastoma and lower-grade glioma group. Although slightly more frequent in glioblastoma, the difference was not statistically significant. This contrasts with the absence of NF1 genetic loss in our cohort of lower grade diffuse gliomas using FISH. This discrepancy could be explained in part by the fact that large NF1 deletions identifiable by FISH reflect increased genomic instability in higher grade gliomas, that is, glioblastoma, and that smaller NF1 deletions present in lower-grade gliomas require platforms with higher resolution using frozen tissue, such as those used by the TCGA.
A clinically important finding when looking at the TCGA data is that NF1 loss (homozygous deletions or predicted inactivating mutations) was associated with worse survival in the lower-grade glioma group. Given the correlation of NF1 loss with the mesenchymal molecular subtype in glioblastoma, it is possible that sporadic lower-grade gliomas with NF1 loss represent a group more similar to primary glioblastoma than other lower-grade gliomas. The predominant histologic type in this group was diffuse astrocytoma (WHO grade II), which suggests that histologic type (but probably not grade) may account for some of these survival differences. In fact, only 3 pure oligodendrogliomas were present in this group, and CIC and FUBP1 mutations, typical of oligodendrogliomas [38], were absent in the latter. Of interest, IDH1/2 mutations occurred in approximately a third of these lower-grade gliomas with NF1 loss, but not in glioblastomas, albeit at a lower frequency than the lower-grade glioma group as a whole (∼80%). This suggests that the effects of NF1 loss on this group cannot be ascribed completely to early glioblastomas, at the pathologic or molecular level, but that it may have additional prognostic implications. However, characterization of the full clinical and biologic significance of NF1 genetic loss in diffuse gliomas, and its cooperation with other genetic alterations, will require further studies.
In summary, NF1/Ch17 gains occur in a subset of diffuse gliomas, irrespective of grade and pathologic subtype. Conversely, NF1/Ch17 loss identifiable by FISH is restricted to a small GBM subset, a finding that deserves further exploration. In TCGA datasets NF1 alterations occur in both GBM and lower-grade diffuse gliomas and may be associated with worse outcome in the latter group, which has prognostic implications.
Supplementary Material
Acknowledgments
The authors thank Norman Barker and Sharon Blackburn for assistance with illustrations. The authors also thank the Cytogenetic Shared Resource at Mayo Clinic for assistance with FISH experiments. This work was supported in part by the Childhood Brain Tumor Foundation (F.J.R.) and The Children's Cancer Foundation, Inc (C.G.E., F.J.R.).
Footnotes
The authors have no conflict of interest to disclose.
Supplementary data: Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.humpath.2015.05.014.
References
- 1.Cawthon RM, Weiss R, Xu GF, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 2.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 3.Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. 2000;403:895–8. doi: 10.1038/35002593. [DOI] [PubMed] [Google Scholar]
- 4.Carroll SL. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012;123:321–48. doi: 10.1007/s00401-011-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutmann DH, James CD, Poyhonen M, et al. Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology. 2003;61:1397–400. doi: 10.1212/wnl.61.10.1397. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–9. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kluwe L, Hagel C, Tatagiba M, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol. 2001;60:917–20. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- 8.Sievert AJ, Jackson EM, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19:449–58. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–49. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–87. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- 12.Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–12. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Guan X, Vengoechea J, Zheng S, et al. Molecular subtypes of glioblastoma are relevant to lower grade glioma. PLoS One. 2014;9:e91216. doi: 10.1371/journal.pone.0091216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messiaen LM, Callens T, Mortier G, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–55. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 22.Wimmer K, Roca X, Beiglbock H, et al. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5′ splice-site disruption. Hum Mutat. 2007;28:599–612. doi: 10.1002/humu.20493. [DOI] [PubMed] [Google Scholar]
- 23.Mautner VF, Kluwe L, Friedrich RE, et al. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J Med Genet. 2010;47:623–30. doi: 10.1136/jmg.2009.075937. [DOI] [PubMed] [Google Scholar]
- 24.Wimmer K, Yao S, Claes K, et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–76. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- 25.Kluwe L, Nguyen R, Vogt J, et al. Internal tumor burden in neurofibromatosis Type I patients with large NF1 deletions. Genes Chromosomes Cancer. 2012;51:447–51. doi: 10.1002/gcc.21931. [DOI] [PubMed] [Google Scholar]
- 26.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odia Y, Orr BA, Bell WR, Eberhart CG, Rodriguez FJ. cMYC expression in infiltrating gliomas: associations with IDH1 mutations, clinicopathologic features and outcome. J Neurooncol. 2013;115:249–59. doi: 10.1007/s11060-013-1221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez FJ, Scheithauer BW, Giannini C, Bryant SC, Jenkins RB. Epithelial and pseudoepithelial differentiation in glioblastoma and gliosarcoma: a comparative morphologic and molecular genetic study. Cancer. 2008;113:2779–89. doi: 10.1002/cncr.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleihues P, Burger PC, Aldape K, et al. Glioblastoma (gliosarcoma) In: Louis DN, Ohgaki H, Wiestler OD, editors. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Motomura K, Natsume A, Watanabe R, et al. Immunohistochemical analysis-based proteomic subclassification of newly diagnosed glioblastomas. Cancer Sci. 2012;103:1871–9. doi: 10.1111/j.1349-7006.2012.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolar K, Freitas-Andrade M, Bechberger JF, et al. Podoplanin: a marker for reactive gliosis in gliomas and brain injury. J Neuropathol Exp Neurol. 2014;74:64–74. doi: 10.1097/NEN.0000000000000150. [DOI] [PubMed] [Google Scholar]
- 36.Actor B, Cobbers JM, Buschges R, et al. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer. 2002;34:416–27. doi: 10.1002/gcc.10087. [DOI] [PubMed] [Google Scholar]
- 37.Boerman RH, Anderl K, Herath J, et al. The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. J Neuropathol Exp Neurol. 1996;55:973–81. doi: 10.1097/00005072-199609000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–22. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.