

Abstract
Dysregulated ether lipid metabolism is an important hallmark of cancer cells. Previous studies have reported that lowering ether lipid levels by genetic ablation of the ether lipid-generating enzyme alkyl-glycerone phosphate synthase (AGPS) lowers key structural and oncogenic ether lipid levels and alters fatty acid, glycerophospholipid, and eicosanoid metabolism to impair cancer pathogenicity, indicating that AGPS may be a potential therapeutic target for cancer. In this study, we have performed a small-molecule screen to identify candidate AGPS inhibitors. We have identified several lead AGPS inhibitors and have structurally characterized their interactions with the enzyme and show that these inhibitors bind to distinct portions of the active site. We further show that the lead AGPS inhibitor 1a selectively lowers ether lipid levels in several types of human cancer cells and impairs their cellular survival and migration. We provide here the first report of in situ-effective pharmacological tools for inhibiting AGPS, which may provide chemical scaffolds for future AGPS inhibitor development for cancer therapy.
Introduction
Cancer cells possess fundamentally altered lipid metabolism that underlies cancer pathogenicity, including heightened de novo lipogenesis and lipolysis as well as aberrant incorporation of exogenous lipids. This altered lipid metabolism is required both for cell proliferation, tumor growth, invasiveness, and metastasis. Lipids have diverse roles in driving cancer pathogenicity by contributing to cell membrane structure, formation of lipid rafts for oncogenic signaling, lipid signaling molecules that promote proliferation and tumor growth, and lipid-mediated post-translational modification of proteins [1].
Tumors also possess heightened levels of a particular lipid class, known as ether lipids, compared to normal tissues, and ether lipid levels have been correlated with proliferative capacity and tumorigenic potential of cancer cells [2–5]. One or more ether linkages, rather than an ester linkage, on the glycerol backbone characterize ether lipids. While the precise roles of intracellular and circulating ether lipids is not yet clear, their particular physicochemical properties contribute to their biological importance in cellular structure, membrane fusion and vesicle formation, free radicals scavenging, storage of lipid second messengers, and lipid signaling molecules. Ether lipid synthesis occurs in peroxisomes and begins with the esterification of dihydroxyacetone phosphate (DHAP) with a long-chain fatty acyl-CoA ester by the enzyme DHAP acyl-transferase (DHAPAT) and subsequent replacement of the fatty acyl chain by a fatty alcohol to form alkyl-DHAP by alkyl-glycerone phosphate synthase (AGPS) (Figure 1) [6–8].
Figure 1. AGPS functional role and inhibition of AGPS activity by lead inhibitors.

(A) AGPS catalyzes the formation of alkyl-DHAP from displacement of the acyl group by a fatty alcohol from the substrate acyl-DHAP. The enzyme is located in the peroxisomes. (B) Inhibition of Cavia porcellus AGPS activity was assessed by a radioactivity assay using 100 μM palmitoyl-DHAP, 100 μM [1-14C]hexadecanol, 180μM inhibitor and detecting the formation of [1-14C]hexadecanyl-DHAP as function of time. The controls were performed using AGPS alone and the catalytically inactive AGPS mutant Thr578Phe. Measurements were performed at least in triplicate [8].
We recently demonstrated that the critical AGPS enzyme is heightened in aggressive cancer cells and primary human breast tumors and that its genetic ablation significantly impairs cancer aggressiveness and tumorigenesis. Metabolomic profiling revealed that AGPS knockdown in breast cancer cells lowers the levels of several ether lipid species, arachidonic acid, and arachidonic acid-derived prostaglandins. Quite intriguingly, the pathogenic impairments conferred by AGPS knockdown in cancer cells are due to the specific depletion of the oncogenic signaling lipid lysophosphatidic acid ether (LPAe) and prostaglandins. These studies indicated that AGPS may serve as an attractive therapeutic target for combatting malignant human cancers, through altering the landscape of oncogenic signaling lipids that drive cancer aggressiveness.
Here, we have performed a small-molecule screen to identify AGPS inhibitors. We have identified several lead compounds whose inhibitory properties were investigated by biochemical and structural studies. One of the inhibitors is demonstrated to lower ether lipids and impair cancer pathogenicity in multiple different types of human cancer cells. We put forth the discovery of the first AGPS inhibitors, which we hope will open the door for developing a new therapeutic strategy for targeting aggressive and metastatic tumors.
Results and Discussion
Identification of AGPS Inhibitors by ThermoFAD-Based Library Screening
AGPS is a flavoenzyme that catalyzes the formation of alkyl-glycerone phosphate using fatty alcohol and fatty acyl fatty acyl DHAP as substrates. The flavin of AGPS enables the acyl/alkyl exchange by covalently reacting with DHAP through an unusual non-redox catalytic mechanism [7–9].
Protein thermal stabilization assay is a well-established medium-throughput method to screen for strongly binding ligands. A variant of the classic ThermoFluor assay, ThermoFAD, measures the unfolding temperature of the protein by monitoring the increase in cofactor flavin adenine dinucleotide (FAD) fluorescence upon release from the protein [10]. In this way, artifacts caused by usage of fluorescent dyes are bypassed. We chose AGPS from C. porcellus, as a suitable system for inhibitor screening because of its stability and suitability for crystallographic studies [8]. We screened an initial set of 1360 small molecules from the Prestwick Chemical Library® that encompasses 1280 approved drugs and a subset of the Zinc database [11], at 180 μM against purified AGPS (5 μM protein). We identified lead compounds that affected the thermal stability of AGPS, increasing the melting temperature of the protein by 4° C (Table 1). They included (3R)-3-(2-fluorophenyl)-N-[(1R)-1-(2-oxo-1,3-dihydrobenzimidazol-5-yl)ethyl]butanamide (ZINC-69435460), and the anti-fungal agent Antimycin A (from Prestwick Chemical Library) [12].
Table 1.
Structure and effect on AGPS thermal stability of the identified inhibitors.
| ΔTm | ||
|---|---|---|
| ZINC-69435460 |
|
+3.5°C |
| 1a |
|
+3.5°C |
| 1e |
|
+2.0°C |
| Antimycin Aa |
|
+4°C |
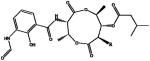
Commercial Antimycin A from Sigma-Aldrich is a mixture of Antimycin A1 (R = C6H13), A2 (R = C5H11), A3 (R = C4H9) and A4 (R = C3H7)
We next wanted to confirm whether these lead compounds inhibited AGPS activity. We used a radioactivity-based enzymatic assay, using palmitoyl-DHAP and [1-14C]hexadecanol as substrates and detecting the formation of [1-14C]hexadecanyl-DHAP as function of time in presence of an excess of inhibitor (180 μM) (Figure 1B). We were unable to calculate Ki values using this approach due to the insolubility of the lipid substrates and the low sensitivity of radioactive detection, but our data show that the three lead compounds inhibit AGPS enzymatic activity. We also attempted to measure the binding of the inhibitors to AGPS using surface plasmon resonance and isothermal calorimetry, but were unsuccessful due to insolubility of the compounds. Nonetheless, we estimated the affinity constants of the identified compounds as 200–700 nM for both Antimycin A and ZINC-69435460 through thermal shift assay according to procedures established by Matulis et al [13].
Three-Dimensional Structure of AGPS in Complex with the inhibitors ZINC-69435460 and Antimycin A
To explore the binding mechanisms between AGPS and the identified inhibitors, the crystal structures of AGPS in complex with Zinc-69435460 and Antimycin A were determined by X-ray crystallography at 2.1–2.2 Å resolution, enabling a detailed view of the inhibitor binding (Figure 2, S1A). In both cases, inhibitors reside in the V-shaped active site, whose vertex is the putative substrate cavity, in front of the FAD. Most importantly, both inhibitors establish specific interactions with the protein residues but their binding mode and location in the active site are significantly different (Figure 3). The two molecules can therefore be considered as distinct lead compounds with regard to their mode of binding and locate two, only partly overlapping, druggable sites within the protein.
Figure 2. Structural studies on Cavia porcellus AGPS in complex with the inhibitors 1a and Antimycin A.

(A) Overall view of the dimeric enzyme bound to 1a (blue spheres). The inhibitor is extended along the hydrophobic substrate tunnel, delimited by the “gating helix” (residues 126–137). AGPS is known be associated to the lumenal side of the peroxisomal membrane as outlined in the figure [9]. FAD is shown in yellow. The overall rms deviation between the unbound protein (PDB entry 4bby) and the inhibitor complex is 0.36 Å. (B) Weighted 2Fo-Fc final electron density map contoured at 1.2 σ level for 1a in the same orientation of the left subunit shown in Figure 2A. Binding involves specific H-bonds (black dashed lines) with the catalytic residues Tyr580, His616, and His617. The position and the distance of the inhibitor suggest also π interactions with the cofactor isoalloxazine ring. A few side chains change their conformation with respect to the unbound enzyme (light orange). (C) Final weighted 2Fo-Fc map (1.2 σ contour level) for Antimycin A-enzyme complex. The electron density is fully consistent with Antimycin A1 (6-carbon alkyl moiety) being bound to the protein. Inhibitor binding causes the rearrangement of several residues around the active site (in orange superposition with native AGPS residues).
Figure 3. Electrostatic potential surfaces of AGPS in complex with the inhibitors 1a (A) and Antimycin A (B).

Negatively and positively charged regions are shown in red and blue, respectively. The orientation highlights the tripartite organization of the active site: the catalytic core close to the flavin, the positively charged entrance tunnel, and the more hydrophobic tunnel which is proposed to accommodate the alkyl moiety of the substrate [8]. The two inhibitors bind in only partly overlapping active-site regions. The aliphatic portion of Antimycin A is mainly extended along the lipophilic tunnel whereas the formamide-substituted aromatic ring interacts with the positively charged patch of the entrance tunnel. Conversely, 1a binds deeper inside active site as compared to Antimycin A. The dihydrobenzimidazole moiety stacks in front of FAD whereas the benzene ring extends along the lipophilic tunnel, as for the Antimycin A alkyl chain. In both cases, the ligands are not solvent accessible and far from the surface.
ZINC-69435460 (Table 1) makes multiple specific interactions with residues in the AGPS active site. The dihydrobenzimidazole moiety occupies the hydrophilic DHAP site, making π-stacking interactions on the FAD isoalloxazine ring and preventing the access to the reactive site N5 atom of the flavin (Figure S1A, 2B). On the other hand, the more hydrophobic amidic substituent points with the fluorophenyl ring toward the aliphatic tunnel that hosts the acyl chain of the substrate. Binding is mediated by hydrophobic interactions in the substrate acyl-chain tunnel and by H-bonds with several residues, including His616 and His617, which belong to an active-site loop, directly involved in catalysis (so-called “HHH loop) [8–9]. ZINC-69435460 binding causes localized rearrangements in the side chains of residues in the active site to enable inhibitor binding (Figure S1A).
Antimycin A shows a different mode of binding, in that this compound resides on an outer segment of the active site without any direct contact with the flavin (Figure 2C). Antimycin A consists of three portions: the dilactone ring in the middle, the 3-formylamino salicylic acid group, that is linked to the dilactone ring through an amide bond, and the hydrophobic alkyl and a 1-methyl butanoate tails on the other side of the dilactone ring. Commercial Antimycin A is a mixture of four compounds, differing in the length of the alkyl chain: Antimycin A1 (R = C6H13), A2 (R = C5H11), A3 (R = C4H9) and A4 (R = C3H7) (Table 1). From the crystal structure in complex with AGPS, it is evident that Antimycin A1 is binding to the enzyme as its bulky six-carbon side chain nicely fits the electron density. The dilactone ring is sitting in hydrophobic pocket in the central segment of the active-site tunnel with the alkyl chain pointing towards (but not in direct contact with) the flavin. Conversely, the butanoate substituent of the inhibitor is extended along the substrate tunnel, in the direction of the so-called “gating-helix” (Figure 2A, 2C, 3B). Finally, the 3-formylamino salicylic acid portion interacts with a positive charged region at the entrance of the active site, interlacing H-bonds with residues surrounding the catalytic site. The binding does not cause dramatic changes in the protein conformation, but as for ZINC-69435460, small changes in side chains orientation do occur, especially with regard to two Arg (Arg419 and Arg566) residues that H-bond to the inhibitor (Figure 2C). In essence, ZINC-69435460 binds deeper into the active site in direct contact with FAD, whereas Antimycin A acts as a plug that obstructs access to the hydrophobic active-site tunnel (Figure 3).
Structure-Activity Relationships Surrounding ZINC-69435460 Derivatives
The investigation of Antimycin A was insightful with regard to its mode of binding and to uncover druggable sites in the protein. However, as it is known inhibitor of cytochrome C reductase, it is not an attractive lead compound for further investigations [14]. Along this line, we focused our efforts on the synthesis and evaluation of several structurally related analogues of ZINC-69435460. All molecules were tested in ThermoFAD and, in the case of a positive signal, also in inhibition assays. With only two exceptions (see below), all compounds showed no strong interactions as judged from the absence of a detectable shift in the melting temperature (Table S1). These results highlighted the specificity of inhibitor recognition by AGPS as all three inhibitor moieties, fluorophenyl, benzamide linker, and dihydrobenzimidazole ring, are involved in specific polar and van der Waals interactions with the residues lining the innermost segment of V-shaped active site tunnel (Figure 2B, 3A). For instance, any modification of the hydrogen-bonding groups on the dihydrobenzimidazole ring turned out to be detrimental for binding (Table S1). Among the tested ZINC-69435460 derivatives and analogues, only 1a and 1e featured detectable AGPS inhibition (Table 1, Figure 1B). 1e was synthesized with the idea that a double bond on the linker moiety would increase rigidity. As gathered from enzymatic and thermal shift assays, the compound demonstrated to retain good binding and inhibition although not as strong as the parent compound. Interestingly, the structural analysis revealed some alterations in the binding conformation with the dihydrobenzimidazole being flipped compared to the structure observed for ZINC-69435460, although the hydrogen-bonding interactions between inhibitor and the protein remain unaffected (Figure S1B). The more rigid double-bond containing linker of 1e possibly causes a suboptimal fit in the active site accounting for the observed reduction in binding affinity and inhibitory efficacy (ΔTm 2° smaller than that of ZINC-69435460; Table 1, Figure 1B). 1a differs from the parent compound simply for a methyl-to-hydrogen substitution on the linker portion of the inhibitor (Table 1). The molecule demonstrated to be as effective in AGPS inhibition as ZINC-69435460 (estimated Ki ~500 nM). Furthermore, the crystal structure showed that the binding was identical to that of the parent compound, the removal of the methyl substituent having no effect on the conformation of both inhibitor and interacting protein residues (Figure 2B, S1A). 1a it is more attractive as lead compound since it lacks a chiral center due to the elimination of a methyl-substituent. Therefore, given its strong binding affinity and inhibitory activity, we chose this molecule as lead compound for further biological evaluation.
AGPS Inhibitor 1a Lowers Ether Lipid Levels in Cancer Cells
We previously demonstrated that genetic knockdown of AGPS with RNA interference in malignant breast and melanoma cancer cells dramatically lowered both structural and oncogenic signaling ether lipid levels [15]. We thus next sought to determine whether the representative inhibitor 1a leads also lowers ether lipid levels in human cancer cells. We treated C8161 melanoma, 231MFP breast, and SKOV3 ovarian cancer cells with 1a (500 μM) and subsequently performed targeted single-reaction monitoring (SRM)-based lipidomic profiling of >120 lipid species, encompassing fatty acids, neutral lipids, phospholipids, sphingolipids, eicosanoids, and ether lipids. We show that 1a primarily lowers the levels of ether lipids across all three cancer cell lines, largely without affecting the levels of lipids from other classes (Figure 4A, 4B, 4C). More importantly, we show that the levels of the oncogenic signaling lipid LPAe, which we previously showed to be a driver of AGPS-mediated effects upon cancer pathogenicity, were also lowered upon 1a treatment (Figure 4). We did not observe ether lipid lowering upon treatment of cancer cells with a lower concentration of 1a (50 μM), possibly due to issues with cell (and/or peroxisome) penetrance (data not shown). Nonetheless, our results indicate that 1a inhibits AGPS in situ and rather selectively lowers the levels of ether lipids across multiple different types of human cancer cells.
Figure 4. AGPS inhibitor 1a lowers ether lipid levels in cancer cells.

Shown are lipidomic profiling data from C8161 melanoma (A), 231MFP breast (B), and SKOV3 ovarian (C) cancer cells treated with vehicle (DMSO) or 1a (500 μM) for 24 h. Shown in heat maps are relative levels compared to control for each lipid measured by targeted SRM-based LC-MS/MS lipidomics. Darker and lighter blue compared to control indicates elevated or reduced levels, respectively, upon 1a treatment compared to controls. Bar graphs show lipid species that were significantly (p<0.05) changed >2-fold upon 1a treatment compared to vehicle-treated controls. Data shown are from n=5/group. Data in bar graphs are shown as average ± SEM.
AGPS Inhibitor 1a Impairs Cancer Pathogenicity
We previously showed that AGPS genetic knockdown in cancer cells impaired cell survival and migration. Having established that 1a treatment evidently inhibited AGPS in situ and was capable of lowering ether lipid levels in cancer cells, we next tested whether 1a and pharmacological blockade of AGPS also impairs cancer pathogenicity. Indeed, we show that 1a treatment impairs serum-free cell survival and migration in C8161 melanoma, 231MFP breast, and SKOV3 ovarian cancer cells (Figure 5A–5F). Our results overall recapitulate the pathogenic impairments observed previously with genetic knockdown of AGPS in cancer cells.
Figure 5. AGPS inhibitor impairs cancer cell pathogenicity.

Shown are effects of 1a upon cell survival and migration in C8161 melanoma (A, B), 231MFP breast (C, D), and SKOV3 ovarian (E, F) cancer cells. Vehicle (DMSO) or 1a were pre-incubated with cancer cells before seeding cells into serum-free survival or migration assays. Data shown as average ±SEM, n=3–4/group. Significance is represented as *p<0.05 in relation to control groups.
Conclusion
In this study, we put forth the first report of an AGPS inhibitor that directly binds to the enzyme active site, inhibits enzymatic activity, lowers ether lipid levels, and impairs pathogenicity in several human cancer cells. We also present a medium-throughput pipeline to screen for AGPS inhibitors, which can be used in subsequent studies, coupled with insights from our structural analysis of inhibitor-AGPS interactions, to optimize the potency, selectivity, and in vivo efficacy of future AGPS inhibitors.
Targeting dysregulated lipid metabolism has arisen as an attractive strategy towards combatting human cancer. Prominent examples include targeting de novo fatty acid synthesis through fatty acid synthase inhibition, targeting dysregulated lipolysis and fatty acid release by inhibiting monoacylglycerol lipase, manipulating tumor-suppressing lipids by inhibiting platelet activating factor acetylhydrolase 1B2 and 1B3 (PAFAH1B2 and PAFAH1B3), or targeting specific aspects of ether lipid metabolism by inhibiting the 2-acetylmonoalkylglycerol ether hydrolase KIAA1363 [16–17]. We recently characterized AGPS as a dysregulated enzyme in cancer that drives the heightened ether lipid metabolism that has been historically correlated with highly proliferative human tumors. We showed that genetic knockdown of AGPS by RNA interference impaired cellular survival, motility, and invasiveness through lowering key oncogenic signaling lipids and altering the overall landscape of fatty acid metabolism to impair other protumorigenic lipid signaling molecules that subserve cancer aggressiveness. We also showed that AGPS knockdown had dramatic effects upon in vivo tumor xenograft growth in mice, indicating that AGPS inhibitors may have therapeutic benefit in cancer treatment. We report here 1a as a lead AGPS inhibitor which shows in vitro inhibition of AGPS activity and apparent in situ AGPS inhibition as demonstrated by the lowering of ether lipid levels and impaired cancer pathogenicity that recapitulate the effects observed with genetic knockdown of this enzyme. While the inhibitors reported here are unlikely to be in vivo-active, we hope that the lead inhibitors reported and characterized here can serve as scaffolds for future structure-activity relationship studies to optimize potency, selectivity, and pharmacokinetic parameters so that AGPS inhibitors can be tested for their safety and efficacy in vivo in preclinical and clinical settings. While loss-of-function mutations in AGPS in humans have been shown to cause neurodevelopmental and neurobehavioral deficits through impairing peroxisomal function [18], whether pharmacological inhibition of AGPS in adulthood would cause toxicity remains unknown. Thus, the development of in vivo efficacious AGPS inhibitors will be crucial in assessing the future promise of AGPS inhibitors for cancer therapy. They will also be very valuable tools for further investigations of the biology of ether phospholipids.
MATERIALS AND METHODS
Protein production and crystallization
Protein expression, purification and crystallization were performed according to the methods previously described [8]. AGPS crystals obtained by sitting-drop vapor diffusion at 20° C mixing protein 8 mg/ml and 20–24% wt/vol PEG 1500 in Na/Hepes pH 7.5–100 mM were soaked with the cryo-protectant solution (30% wt/vol PEG 1500 in Na/Hepes pH 7.5–100 mM) containing 1 mM inhibitor o/n at 20°C and then washed in cryo-protectant solution without inhibitor before freezing.
ThermoFAD assays
ThermoFAD experiments were performed with cpAGPS 5 μM, inhibitors 180 uM, KPO4 buffer 50 mM pH 7.5, 50 mM NaCl, 5% glycerol, 20 μl final. Temperature gradient 25–70°C, fluorescence detection every 0.5°C at 485 ± 30 nm excitation and 625 ± 30 nm emission for 5 s (BioRad MiniOpticon Real-Time PCR System).
To estimate the binding of the identified compounds, the same ThermoFAD protocol was followed, using serial dilutions of the compounds from 500 μM to 10 nM, tested with 5 μM AGPS in KPO4 buffer 50 mM pH 7.5, 50 mM NaCl, 5% glycerol, 20 μl final. To estimate the affinity constant, ΔUHT of AGPS was calculated according to equation 6 [13] using ThermoFAD output, and ΔUCp was estimated according to AGPS size and structure [19]. The calculation of the estimated Kd was performed according to equation 14 [13], intentionally considering a range of uncertainty due to the reasonable assumption of ΔbHTm being 40 kJ/mol (relatively unimportant for the overall Kd calculation).
Enzymatic assays
Activity assays were performed in the same conditions previously described using radioactive [1-14C]hexadecanol [9, 20]. The following final concentrations were used: 500 nM AGPS, 50 mM Tris/HCl pH 8.2, 50 mM NaF, 0.1% (w/v) Triton X-100, 100 μM palmitoyl-DHAP [21], 96 μM [1-14C]hexadecanol (Sigma; specific radioactivity adjusted to 13000 dpm/nmol) in a total volume of 100 μl at 30° C. The palmitoyl-DHAP stock solution was sonicated before usage. Aliquots (10 μl) were withdrawn at different times and spotted on DEAE cellulose disks. After an extensive wash with ethanol, the disks were transferred to scintillation vials and counted by means of a Tri-Carb 2100TR (Packard) scintillation counter to measure the amount of the radioactive [1-14C]hexadecanyl-DHAP product. When appropriate, an inhibitor or the vehicle (DMSO) was added to the reaction mixture to final concentration of 180–200 μM after pre-incubation with the protein for 30 min on ice.
Targeted lipidomic analyses
Metabolite measurements were conducted using modified previous procedures [15]. Cancer cells were grown in serum-free media for 24 hr to minimize the contribution of serum-derived metabolites to the cellular profiles. Cells were treated with 1a (500 μM) for 24 h. Cancer cells (1 × 106 cells/6 cm dish or 2 × 106 cells/6cm dish) were washed twice with phosphate buffer saline (PBS), harvested by scraping, and isolated by centrifugation at 1400 × g at 4°C and cell pellets were flash frozen and stored at −80°C until metabolome extractions. Lipid metabolites were extracted in 4 ml of a 2:1:1 mixture of chloroform:methanol:Tris buffer with inclusion of internal standards C12:0 dodecylglycerol (10 nmol) and pentadecanoic acid (10 nmol). Organic and aqueous layers were separated by centrifugation at 1000 × g for 5 min and the organic layer was collected. The aqueous layer was acidified (for metabolites such as LPA) by adding 0.1 % formic acid, followed by the addition of 2 ml chloroform. The mixture was vortexed, and the organic layers were combined, dried down under N2 and dissolved in 120 μl chloroform, of which 10 μl was analyzed by both single-reaction monitoring (SRM)-based LC-MS/MS or untargeted LC-MS. LC separation was achieved with a Luna reverse-phase C5 column (50 mm × 4.6 mm with 5 μm diameter particles, Phenomenex). MS analysis was performed with an electrospray ionization (ESI) source on an Agilent 6430 QQQ LC-MS/MS. Representative metabolites were quantified by SRM of the transition from precursor to product ions at associated collision energies.
Cell survival and migration studies
Cell survival studies were performed as previously described [15, 22]. Cells were pre-incubated with 1a (500 μM) before seeding cells into survival and migration assays. For survival assays, cells were washed twice in PBS, harvested by trypsinization, washed in serum-free media and cells were seeded into 96-well plates (30,000 cells per well in a volume of 200 μl containing serum-free media) for 48 h prior to addition of WST-1 (10 μl) for 1 h at 37°C in 5 % CO2. Absorbance was then measured at 450 nm using a spectrophotometer. For 231MFP cells, rather than the addition of WST-1, cell viability was measured by first aspirating the media followed by the addition of 100 μl of fixation solution containing 5 μM Hoescht in formalin. The fixation solution was aspirated and the cells were washed with PBS. Absorbance was then measured using a fluorescent plate reader with an excitation of 350 nm and an emission of 461 nm. Migration assays were performed as previously described [15]. Migration assays were performed in Transwell chambers (Corning) coated with collagen. SKOV3, C8161, and 231MFP migration was measured at 5, 6, and 8 h, respectively.
Supplementary Material
Acknowledgments
This work was supported by grants from the Telethon (GGP12007 to A.Mattevi), Associazione Italiana Ricerca sul Cancro (IG-15208 to A.Mattevi), National Institutes of Health (R01CA172667 to D.K.N.), American Cancer Society Research Scholar Award (RSG14-242-01-TBE to D.K.N.), and DOD Breakthroughs Award (CDMRP W81XWH-15-1-0050 to D.K.N.), FIRB grant (RBFR10ZJQT to A.Mai), IIT-Sapienza Project (RF-2010-2318330 grant to A.Mai), and FP7 Project (BLUEPRINT/282510 to A.Mai). We acknowledge SLS and ESRF for provision of synchrotron radiation facilities and excellent supervision by their staff during data collection.
Footnotes
ACCESSION CODES
The atomic coordinates and structures have been deposited in the Protein Data Bank,www.pdb.org.
AUTHOR CONTRIBUTIONS
The manuscript was written through contributions of all authors.
The authors declare no competing financial interests.
Additional experimental, compound characterization, and assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nomura DK, Cravatt BF. Lipid metabolism in cancer. Biochim Biophys Acta, Mol Cell Biol Lipids. 2013;1831:1497–1498. doi: 10.1016/j.bbalip.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Snyder F, Blank ML, Morris HP. Occurrence and nature of O-alkyl and O-alk-1-enyl moieties of glycerol in lipids of morris transplanted hepatomas and normal rat liver. Biochim Biophys Acta, Mol Cell Biol Lipids. 1969;176:502–510. doi: 10.1016/0005-2760(69)90217-3. [DOI] [PubMed] [Google Scholar]
- 3.Howard BV, Morris HP, Bailey JM. Ether-lipids, -glycerol phosphate dehydrogenase, and growth rate in tumors and cultured cells. Cancer Res. 1972;32:1533–1538. [PubMed] [Google Scholar]
- 4.Albert DH, Anderson CE. Ether-linked glycerolipids in human brain tumors. Lipids. 1977;12:188–192. doi: 10.1007/BF02533292. [DOI] [PubMed] [Google Scholar]
- 5.RooS DS, Choppin PW. Tumorigenicity of cell lines with altered lipid composition. Proc Natl Acad Sci USA. 1984;81:7622–7626. doi: 10.1073/pnas.81.23.7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis PA, Hajra AK. Stereochemical specificity of the biosynthesis of the alkyl ether bond in alkyl ether lipids. J Biol Chem. 1979;254:4760–4763. [PubMed] [Google Scholar]
- 7.de Vet EC, Hilkes YH, Fraaije MW, van den Bosch H. Alkyl-dihydroxyacetonephosphate synthase. Presence and role of flavin adenine dinucleotide. J Biol Chem. 2000;275:6276–6283. doi: 10.1074/jbc.275.9.6276. [DOI] [PubMed] [Google Scholar]
- 8.Nenci S, Piano V, Rosati S, Aliverti A, Pandini V, Fraaije MW, Heck AJR, Edmondson DE, Mattevi A. Precursor of ether phospholipids is synthesized by a flavoenzyme through covalent catalysis. Proc Natl Acad Sci USA. 2012;109:18791–18796. doi: 10.1073/pnas.1215128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razeto A, Mattiroli F, Carpanelli E, Aliverti A, Pandini V, Coda A, Mattevi A. The crucial step in ether phospholipid biosynthesis: structural basis of a noncanonical reaction associated with a peroxisomal disorder. Structure. 2007;15:683–692. doi: 10.1016/j.str.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Forneris F, Orrù R, Bonivento D, Chiarelli LR, Mattevi A. ThermoFAD, a Thermofluor((R))-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J. 2009;276:2833–2840. doi: 10.1111/j.1742-4658.2009.07006.x. [DOI] [PubMed] [Google Scholar]
- 11.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slater EC. The mechanism of action of the respiratory inhibitor, antimycin. Biochim Biophys Acta, Bioenerg. 1973;301:129–154. doi: 10.1016/0304-4173(73)90002-5. [DOI] [PubMed] [Google Scholar]
- 13.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005;44:5258–5266. doi: 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- 14.Huang LS, Cobessi D, Tung EY, Berry EA. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J Mol Biol. 2005;351:573–597. doi: 10.1016/j.jmb.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamin DI, Cozzo A, Ji X, Roberts LS, Louie SM, Mulvihill MM, Luo K, Nomura DK. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc Natl Acad Sci USA. 2013;110:14912–14917. doi: 10.1073/pnas.1310894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang JW, Zuhl AM, Speers AE, Niessen S, Brown SJ, Mulvihill MM, Fan YC, Spicer TP, Southern M, Scampavia L, Fernandez-Vega V, Dix MM, Cameron MD, Hodder PS, Rosen H, Nomura DK, Kwon O, Hsu KL, Cravatt BF. Selective inhibitor of platelet-activating factor acetylhydrolases 1b2 and 1b3 that impairs cancer cell survival. ACS Chem Biol. 2015;10:925–932. doi: 10.1021/cb500893q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulvihill MM, Benjamin DI, Ji X, Le Scolan E, Louie SM, Shieh A, Green M, Narasimhalu T, Morris PJ, Luo K, Nomura DK. Metabolic profiling reveals PAFAH1B3 as a critical driver of breast cancer pathogenicity. Chemistry & Biology. 2014;21:831–840. doi: 10.1016/j.chembiol.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wanders RJA, Waterham HR. Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta, Mol Cell Res. 2006;1763:1707–1720. doi: 10.1016/j.bbamcr.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Robertson DE, Giangiacomo KM, de Vries S, Moser CC, Dutton PL. Two distinct quinone-modulated modes of antimycin-sensitive cytochrome b reduction in the cytochrome bc1 complex. FEBS Letters. 1984;178:343–350. doi: 10.1016/0014-5793(84)80630-4. [DOI] [PubMed] [Google Scholar]
- 20.Zomer A, Deweerd W, Langeveld J, Vandenbosch H. Ether lipid-synthesis - purification and identification of alkyl-dihydroxyacetone phosphate synthase from guinea-pig liver. Biochim Biophys Acta. 1993;1170:189–196. doi: 10.1016/0005-2760(93)90070-p. [DOI] [PubMed] [Google Scholar]
- 21.Hajra AK, Agranoff BW. Acyl dihydroxyacetone phosphate. A rapidly labeled lipid in guinea pig liver mitochondria. J Biol Chem. 1967;242:1074–1075. [PubMed] [Google Scholar]
- 22.Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, Nomura DK. Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00240. Epub April, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.