

Abstract
With the hope of discovering effective analgesics with fewer side effects, attention has recently shifted to allosteric modulators of the opioid receptors. In the past two years, the first chemotypes of positive or silent allosteric modulators (PAMs or SAMs, respectively) of μ- and δ-opioid receptor types have been reported in the literature. During a structure-guided lead optimization campaign with μ-PAMs BMS-986121 and BMS-986122 as starting compounds, we discovered a new chemotype that was confirmed to display μ-PAM or μ-SAM activity depending on the specific substitutions as assessed by endomorphin-1-stimulated β-arrestin2 recruitment assays in Chinese Hamster Ovary (CHO)-μ PathHunter cells. The most active μ-PAM of this series was analyzed further in competition binding and G-protein activation assays to understand its effects on ligand binding and to investigate the nature of its probe dependence.
Graphical abstract

INTRODUCTION
A prominent member of the G-protein-coupled receptor (GPCR) superfamily, the μ-opioid receptor is the main pharmacological target for both acute and chronic pain, as well as a target for the treatment of alcohol abuse, drug abuse, and addiction disorders.1,2 Although μ-opioid medications such as morphine and its derivatives remain the “gold-standard” for pain management, clinicians are rightly conservative in the administration of these drugs, owing to their dangerous adverse effects (e.g., respiratory depression, nausea, tolerance, dependence, and constipation), as well as social and legal issues. As a result, the development of new opioid analgesics that are free from side effects represents a critically important research objective for 21st century medicine.
Recent high-resolution structural information on the μ-opioid receptor,3 as well as novel paradigms of biased agonism (or functional selectivity) and allosterism at this receptor,4,5 may offer unprecedented opportunities for the discovery of opioid therapeutics with reduced adverse effects. Allosteric ligands are defined as binding to regions that are distinct from the site where the endogenous ligand binds (defined as the orthosteric binding site). Depending on whether they enhance or reduce the affinity and/or efficacy of the orthosteric ligand, they can be classified as positive allosteric modulators (PAMs) or negative allosteric modulators (NAMs), respectively. In principle, these ligands have several theoretical advantages over traditional orthosteric agonists and antagonists. First, because allosteric regions of GPCRs tend to be less evolutionarily conserved than orthosteric binding sites, allosteric ligands can attain improved receptor type selectivity, which can limit the occurrence of off-target effects (although this does not eliminate the possibility of on-target effects). In the case of opioid receptors, development of selective opioid drugs has not been a major impediment, and most of the untoward side effects of opioid agonists are target-mediated; therefore, this specific advantage of allosteric modulators may have limited applicability to opioid receptors. Another important advantage of allosteric modulators is that their effect is limited by their cooperativity, and therefore allosteric ligands may hold great potential as safer drugs with fewer on-target overdosing risks. This feature may be more important in the case of μ-opioid receptors, where safety risks associated with drug overdose are a very significant problem. Finally, another major theoretical advantage of PAMs compared to orthosteric agonists is that PAMs are likely to maintain the temporal and spatial fidelity of signaling in vivo as they only act in the presence of the endogenous ligand. Therefore, PAMs might be expected to produce significantly less desensitization and tolerance than direct-acting agonists, which continuously activate the receptor until the drug is cleared. Because tolerance and dependence produced by direct opioid agonists remain major issues limiting their therapeutic utility, this feature of PAMs may have great importance in the case of μ-opioid receptors specifically. Similarly, opioid PAMs may be able to avoid some of the ontarget side effects produced by opioid agonists by virtue of acting only in tissues where native opioid signaling is occurring. For a more thorough review on the potential advantages of opioid PAMs, see Burford et al.4 It is important to note that at present these potential advantages of opioid PAMs remain purely theoretical, as in vivo effects of opioid PAMs have not yet been reported.
Although several GPCR allosteric modulators have shown preclinical promise in neurodegenerative, psychiatric, or neurobehavioral diseases,6 the development and validation of drug-like allosteric modulators of the opioid receptors lags behind. The first opioid allosteric modulators were identified for the μ-opioid receptor from a recent high throughput screening campaign using a β-arrestin2 recruitment assay.7 Specifically, this screen identified two PAMs and two silent allosteric modulators (SAMs) of the μ-opioid receptor. While the μ-SAMs exhibited neutral cooperativity with orthosteric ligands in spite of their competitive binding at the allosteric site, the two μ-PAMs BMS-986121 and BMS-986122 potentiated the effects of endomorphin-1, DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin), and morphine in β-arrestin2 recruitment, G-protein activation, and adenylyl cyclase (AC) inhibition. Although a few PAMs of the δ-opioid receptor have also been recently identified,8,9 additional pharmacological tools are needed to investigate further the effect of allosterism on μ-opioid receptor signaling, and to test whether μ-opioid receptor PAMs will in fact provide the potential therapeutic advantages described above.
Here, we report the discovery of a new chemotype that, depending on the specific substitutions, exhibits μ-PAM or μ-SAM activity as assessed by endomorphin-1-stimulated β-arrestin2 recruitment assays in Chinese Hamster Ovary (CHO)-μ PathHunter cells. Further radioligand binding and G-protein activation assays were performed on the most active μ-PAM of this series (MS1) to understand both its effects on ligand binding and the nature of its probe dependence.
MATERIALS AND METHODS
Dataset Generation and Clustering
The eMolecules database10 of 6 million compounds was used to search for chemical analogs of BMS-986121 and BMS-986122, including compounds with a significantly different chemotype. An eMolecules similarity cutoff of 0.6 to either parent compound led to the retrieval of 661 analogs of BMS-986121 and 675 analogs of BMS-986122 on January 31, 2014. A hierarchical clustering of the resulting 1336 molecules (Table S1) was performed using a Tanimoto distance matrix generated from linear chemical fingerprints with Canvas version 2.2 of the Schrödinger Suite 2014-4.11 Using default options such as the cluster linkage method and the Kelly criterion, molecules were grouped into 7 clusters separated by a Tanimoto distance of 0.9. The three most populated of these clusters included derivatives of BMS-986121 and BMS-986122, as well as a significantly different chemical scaffold herein termed “MS” (Figure 1). An additional 33 molecules with high chemical similarity (≥0.9) to MS1 were retrieved from both the eMolecules10 and ZINC12 (over 35 million purchasable compounds) databases. Twenty-eight of such compounds were purchased for experimental testing.
Figure 1.
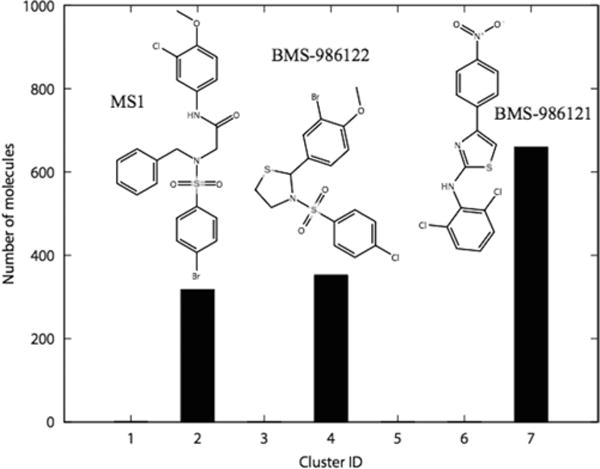
Clustering results of the 1336 analogs of BMS-986121 and BMS-986122 extracted from eMolecules. The three most populated clusters 2, 4, and 7 included analogs of MS1, BMS-986122, and BMS-986121, respectively.
Cell Lines
CHO PathHunter cells expressing enzyme acceptor (EA)-tagged β-arrestin2 PK-tagged human μ-opioid receptor (CHO-μ) were from DiscoveRx (Fremont, CA). PathHunter is a trademark of DiscoveRx.13,14 Cells were grown in F-12 media (Invitrogen 11765), containing Hyclone FBS 10%, Hygromycin 300 μg/mL (Invitrogen 10687), G418 800 μg/mL (Invitrogen 10131), and maintained at 37 °C in a humidified incubator containing 5% CO2. These cells were used for the β-arrestin recruitment assays. C6 rat glioma cells stably expressing the rat μ-opioid receptor (C6-μ) were generated as described previously.15 Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin with G418 to maintain μ-opioid receptor expression. Membranes prepared from these cells were used for the radioligand binding and guanosine 5′-O-[γ-thio]triphosphate (GTPγ35S) assays as described below.
Membrane Preparation
Confluent C6 rat glioma cells stably expressing rat μ-opioid receptor (C6-μ) were rinsed with phosphate buffered saline and then detached using harvesting buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.4, 0.68 mM ethylenediaminetetraacetic acid (EDTA), and 150 mM NaCl). Cells were pelleted following centrifugation at 300g for 3 min at room temperature. The supernatant was discarded and the pellet was resuspended in ice-cold 50 mM Trizma buffer, pH 7.4. The pellet was homogenized using a Tissue Tearor and then centrifuged at 20000g for 20 min at 4 °C. The supernatant was discarded and the pellet was resuspended, homogenized, and centrifuged once more. The final pellet was homogenized in 50 mM Trizma, pH 7.4 using a glass dounce homogenizer and aliquots were flash frozen and stored at −80 °C until use in assays. Concentration of protein was determined using a bicinchoninic acid assay (BCA) protein assay with bovine serum albumin as the standard.
Materials
PathHunter detection reagents were from DiscoveRx (Fremont, CA). Cell culture media and supplements were from Life Technologies (Carlsbad, CA). All other chemicals, unless otherwise specified, were purchased from Sigma (St. Louis, MO).
PathHunter β-Arrestin Assay
Confluent flasks of CHO-μ PathHunter cells were harvested with TrypLE Express, and resuspended in F-12 media supplemented with 10% FBS and 25 mM HEPES, at a density of 6.67e5 cells/mL and plated (3 μL/well) into white solid tissue culture (TC)-treated 1536-well plates (3727BC, Corning, NY). Plates were incubated over night at 37 °C in a humidified incubator containing 5% CO2. The next day, compounds (40 nL of 100 × final concentration in 100% DMSO) were added to cell plates by acoustic dispense using an Echo-550 (Labcyte, Sunnyvale, CA) from Echo-qualified 1536-well source plates (Labcyte). Next, 1 μL of either: assay buffer (Hanks Balanced Salt Solution (HBSS) with 25 mM HEPES, and 0.05% bovine serum albumin, pH 7.4) (agonist mode); or assay buffer containing a low concentration (~4 × EC20) of the orthosteric agonist endomorphin-1 (PAM mode); or a higher concentration (~4 × EC50) of endomorphin-1 (NAM mode); or a low concentration of endomorphin-1 (~4 × EC20) plus 40 μM BMS-986122 (10 μM final) (SAM mode) were added to assay plates. Plates were covered with a lid and incubated at room temperature for 90 min. Incubations were terminated by the addition of 2 μL PathHunter Reagent (DiscoveRx). One hour later, luminescence was detected using a Viewlux imaging plate reader (PerkinElmer, Cambridge, MA).
Radioligand Binding Assay
Displacement of 3H-diprenorphine (DPN) (Kd = 0.15 nM) by increasing concentrations of orthosteric ligand was measured in the presence of vehicle (1% DMSO) or allosteric ligand. Briefly, C6-μ cell membranes (10 μg/well, prepared as described above) were incubated in a buffer (50 mM Trizma base pH 7.4, 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl) containing orthosteric ligand, allosteric ligand (or vehicle), 10 μM GTPγS, and 3H-DPN (0.2–0.3 nM) for 60–90 min shaking at room temperature. Nonspecific binding was assessed in the presence of 10 μM naloxone. Reactions were terminated by rapid filtration through glass microfiber GF/C filters (Whatman) using a Brandell harvester and washed three times using ice-cold 50 mM Trizma buffer pH 7.4. Filters were dried and radioactivity was measured using liquid scintillation counting with EcoLume liquid scintillation cocktail (MP Biomedicals) in a Wallac 1450 MicroBeta counter.
GTPγ35S Binding Assay
C6-μ cell membranes (10 μg/well; prepared as described above) were incubated for 1 h at 30 °C in a buffer (see composition above) containing 30 μM guanosine diphosphate (GDP), 0.10 nM GTPγ35S, orthosteric ligand, and allosteric ligand (or vehicle). Basal binding was measured in the absence of orthosteric ligand and maximal binding was determined using 10 μM DAMGO. The reaction was terminated and counted using filtration onto glass fiber filters and liquid scintillation counting as described above.
Data Analysis
Concentration response data were fit to a logistic eq 1 using nonlinear regression analysis to provide estimates of Ymin (bottom), Ymax (top), potency (EC50), and slope factor (Hill slope), using GraphPad Prism 5.01 (sigmoidal dose response with variable slope).
| (1) |
The β-arrestin curve shift assays in Figure 2 were analyzed using an allosteric ternary complex model (Graphpad Prism 5.01 Dose response-Special-Allosteric EC50 shift), to determine log Kb and the cooperativity factor (α) of the PAMs.
Figure 2.
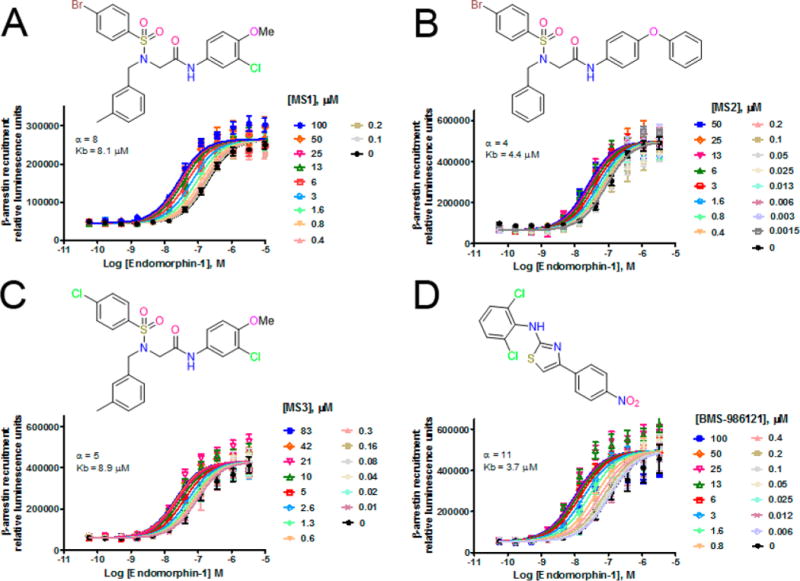
Allosteric EC50 shifts of endomorphin-1-stimulated β-arrestin recruitment in CHO-μ PathHunter cells in the presence of increasing concentrations of PAM: MS1 (A), MS2 (B), MS3 (C), and BMS-986121 (D). Kb = calculated binding affinity of PAM to free receptor; α = allosteric cooperativity factor.
Calculation and Statistical Analysis of Physicochemical Properties
The Schrödinger Suite 2014-4 was used to calculate the physicochemical properties of the 14 PAMs and 12 SAMs listed in Table 1. The two-dimensional (2D) sketcher tool of Maestro 10.0 was used to draw 2D structures of each of these 26 compounds, which were imported as 3D structures into LigPrep v3.2 with protonation states assigned at physiological pH using Epik v3.0. Fifty-two physicochemical properties were calculated using QikProp v4.2. Among them, 42 properties (see Tables S2 and S3) displayed nonzero variance across the 26 ligands calculated, and were therefore used for statistical analysis.
Table 1.
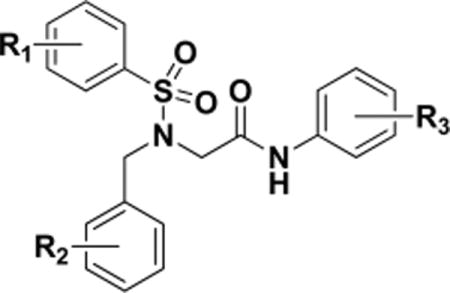
Structure Activity Relationship of a New μ-PAM/SAM Chemotype in a PathHunter β-Arrestin Recruitment Assay
| ||||||
|---|---|---|---|---|---|---|
| sample ID | R1 | R2 | R3 | EC50, μM (PAM/SAM mode)a | PAM mode Ymax % | activity |
| MS1 | 4-Br | H | 3-Cl,4-MeO | 6.5 | 85.1 | PAM |
| MS2 | 4-Br | H | 4-PhO | 6.2 | 109.2 | PAM |
| MS3 | 4-Cl | 3-Me | 3-Cl,4-MeO | 5.7 | 44.6 | PAM |
| MS4 | 4-Br | H | 3-Cl | 3.9 | 32.4 | PAM |
| MS5 | 3-Me,4-MeO | H | 3-Cl,4-MeO | 4.7 | 21.0 | PAM |
| MS6 | 4-Me | H | 3-Cl,4-MeO | 5.3 | 21.8 | PAM |
| MS7 | 4-Br | H | 4-MeO | 5.4 | 22.1 | PAM |
| MS8 | 4-Cl | H | 4-EtO | 6.4 | 31.8 | PAM |
| MS9 | 4-F | H | 3-Cl,4-MeO | 7.0 | 24.0 | PAM |
| MS10 | 4-Cl | H | 4-MeO | 8.9 | 26.4 | PAM |
| MS11 | 4-OMe | H | 3-Br | 14.5 | 33.0 | PAM |
| MS12 | 4-Me | 4-Me | 3-Cl,4-MeO | 21.9 | 10.3 | PAM |
| MS13 | H | H | 3-Br | > 30 | 20 | PAM |
| MS14 | H | H | 3-Cl,4-MeO | 76.1 | 28.4 | PAM |
| MS15 | H | 2,4-di-Cl | 3-Cl,4-MeO | 6a | NAb | SAM |
| MS16 | 4-Cl | H | 3-MeO | 6.5a | NAb | SAM |
| MS17 | H | 4-F | 3-Cl,4-MeO | 6.9a | NAb | SAM |
| MS18 | 4-Me | H | 4-MeO | 14.4a | NAb | SAM |
| MS19 | H | 4-Me | 3-Cl | 16.2a | NAb | SAM |
| MS20 | H | 4-Cl | 4-MeO | 16.6a | NAb | SAM |
| MS21 | H | 4-Cl | 3-Me | 21.2a | NAb | SAM |
| MS22 | H | 4-Br | 4-MeO | 22.7a | NAb | SAM |
| MS23 | 4-Cl | H | 3-Cl,6-MeO | 23.5a | NAb | SAM |
| MS24 | H | 4-Cl | 3-Cl,4-MeO | 24.2a | NAb | SAM |
| MS25 | H | 4-Cl | 2-MeO,5-Me | 27.7a | NAb | SAM |
| MS26 | 4-Cl | H | 2-MeO,5-Me | >30a | NAb | SAM |
| MS27 | H | 4-Me | 3-Cl,4-MeO | NAb | inactive | |
| MS28 | 4-MeO | H | 2-CF3 | NAb | inactive | |
SAM compounds were detected by incubating a serial dilution of the compound with cells in the presence of an EC20 of endomorphin-1 plus an EC80 of BMS-986121 PAM. Under these conditions, the SAM compound acts as an antagonist of PAM binding, reducing PAM activity. Control PAMs: BMS-986121 (EC50 2.2 μM, Ymax 86%) and BMS-986122 (EC50 16.2 μM, Ymax 108%).
NA = not active in PAM mode.
The bayesglm function in the “arm” R package was used to build a Bayesian logistic regression model for the classification of the PAM versus SAM activities at the μ-opioid receptor (see Table 1) based on their calculated physicochemical properties (see Table S2). Specifically, PAMs and SAMs were assigned effective values of 1 and 0, respectively, and the logistic model was built to predict numerical effective values for these compounds, ideally 0.5–1.0 for PAMs and 0–0.5 for SAMs. Binomial distribution was selected as the error distribution model. All models containing combinations of up to five properties (without interaction terms) were estimated, and the best linear model was selected based on the Akaike Information Criterion (AIC).
Ligand-Based 3D Pharmacophore Modeling
Ligand-based 3D pharmacophore modeling was performed using the PHASE workflow within the Schrödinger Suite 2014-4 and the same set of PAMs and SAMs reported in Table 1. First, thorough conformational sampling of these ligands was performed using the ConfGen search method, which produced about 50 conformations for each ligand. Classical pharmacophoric features were identified in the ligand dataset, specifically: hydrogen bond acceptors (A), hydrogen bond donors (D), hydrophobic groups (Hyd), and aromatic rings (R). Common 3D pharmacophore hypotheses were then generated by PHASE version 4.1 using all PAMs in Table 1, and requesting an exact match for the top three most active PAMs. These common pharmacophoric hypotheses were then scored based on optimal alignment between the PAMs of Table 1 and the pharmacophoric features. The default active survival scoring function that corresponds to 1 × vector score + 1 × site score + 1 × volume score + 1.1 × (number of matches − 1) was used for this evaluation. The hypotheses were further filtered so that the root-mean-square deviation (RMSD) of PAMs to the pharmacophore features was no more than 1.2 Å. SAMs were also aligned to each qualifying pharmacophore hypothesis model to derive their alignment fitness scores (as the sum of vector, site, and volume scores).
RESULTS
Discovery and Structure–Activity Relationship of a New μ-PAM/SAM Chemotype
In the hunt for more active allosteric modulators of the μ-opioid receptor, we searched the eMolecules database for analogs of the recently identified μ-PAMs BMS-986121 and BMS-986122,7 including significantly different chemical scaffolds. Clustering of the resulting 1336 molecules (Table S1) led to their grouping into 7 clusters (Figure 1). While cluster 1 contained 2 elements and clusters 3, 5, and 6 contained 1 element only, clusters 2, 4, and 7 were highly populated (see Figure 1). Specifically, clusters 4 and 7 contained 353 and 660 close analogs of BMS-986122 and BMS-986121, respectively, whereas the 318 molecules of cluster 2 corresponded to a significantly different chemotype (e.g., compare MS1 to BMS-986122 and BMS-986121 in Figure 1). Additional analogs of this new chemotype were retrieved through a chemical similarity search to MS1 in both the eMolecules and ZINC databases (see Methods for details).
Twenty-eight of these compounds were purchased for experimental testing, and the results of a primary screen based on a PathHunter β-arrestin recruitment assay are shown in Table 1. While none of these compounds displayed agonist activity alone, all of them but two (i.e, MS27 and MS28) displayed PAM or SAM activity in the presence of low concentration of endomorphin-1, a μ-opioid receptor agonist. No NAM activity (inhibition of an EC50 concentration of endomorphin-1) was detected (data not shown). As shown in Table 1, most PAMs had low potencies in the single and low double digit μM range with efficacy (Ymax) values below 40% compared to endomorphin-1 maximal stimulation. The exceptions were MS1, MS2, and MS3, which displayed a Ymax value larger than 44% in PAM mode with EC50 values in the single digit μM range. The Ymax activity gives an indication of the degree of cooperativity exhibited by these compounds suggesting that MS1, MS2, and MS3 have greater cooperativity compared to the other MS compounds tested. The remaining 12 compounds are either SAMs or weak PAMs judging from their reduced PAM activity. As expected, SAMs behave as competitive antagonists at the allosteric site, having little to no allosteric efficacy themselves but inhibiting the binding of a higher efficacy PAM to the allosteric binding site.
For the most efficacious MS1–MS3 compounds, we assessed the Kb and α values of cooperativity by performing full concentration–response curves of the orthosteric ligand endomorphin-1 in the presence of increasing concentrations of the allosteric compounds in the β-arrestin recruitment assay (see Figure 2). The results confirm the ability of these molecules to act as PAMs with Kb and α cooperativity values slightly weaker, but comparable, to those observed for the previously reported BMS-986121.7
The allosteric compound with the highest α value of cooperativity, MS1, was analyzed further in competition binding and G-protein activation assays to understand its effects on ligand binding and to investigate the nature of its probe dependence. Saturation binding using the neutral antagonist 3H-diprenorphine (3H-DPN) was performed in cell membranes prepared from C6 glioma cells stably expressing rat μ-opioid receptor (Figure 3). The Kd of 3H-DPN was unchanged in the presence of 10 μM MS1 (Kd with veh = 0.25 ± 0.10 nM; Kd with MS1 = 0.35 ± 0.15 nM, data not shown). In contrast, MS1 was able to enhance the affinity of the agonist L-methadone to bind μ-opioid receptor (Figure 3A). The Ki of L-methadone in the absence or presence of 10 μM MS1 was enhanced by 7-fold (Ki in the presence of vehicle = 1177 ± 329 nM, Ki in the presence of MS1 = 161 ± 38 nM; p = 0.04). Notably, MS1 exhibited strong probe dependence in that it failed to alter the affinity of the agonists DAMGO, endomorphin-1, and morphine to bind μ-opioid receptor (Figure 3B–D).
Figure 3.
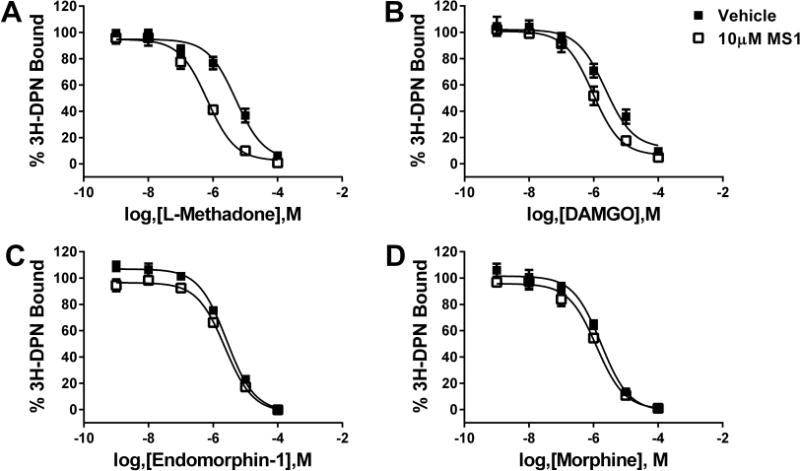
Effects of MS1 on the binding of various orthosteric μ-opioid receptor agonists. Displacement of 3H-DPN by L-methadone (A), DAMGO (B), endomorphin-1 (C), and morphine (D) was measured in the presence of vehicle (■) or 10 μM MS1 (□) using C6-μ cell membranes.
In addition to binding, the ability of MS1 to alter the activity of μ-opioid receptor agonists was investigated using GTPγ35S binding in C6-μ cell membranes. Although MS1 (up to 30 μM) failed to have any activity alone (data not shown), the presence of 10 μM MS1 enhanced the potency of L-methadone to activate G-protein by over 4-fold (Figure 4A) but had no effect on the degree of maximal stimulation. Again, MS1 showed strong probe dependence and failed to alter the potency or maximal stimulation of DAMGO and endomorphin-1 to activate G-protein (Figure 4B,C, respectively). However, MS1 did enhance the maximal activation by morphine to that of a full agonist while having no effect of morphine’s potency (Figure 4D). The lack of effect of MS1 on endormorphin-1 stimulation of GTPγ35S binding was unexpected in view of the enhancement of endomorphin-1 recruitment of β-arrestin, but is in line with reports that the endomorphins are arrestin-biased agonists.16,17
Figure 4.
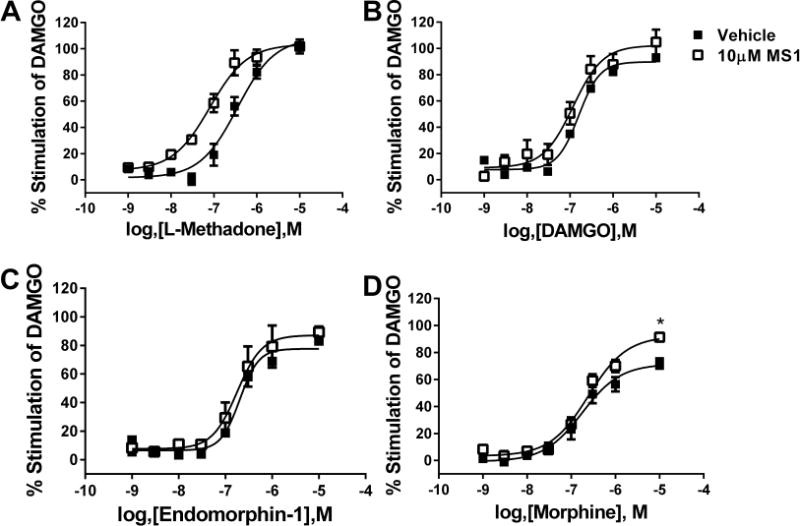
Effects of MS1 on the potency of various orthosteric μ-opioid receptor agonists to activate G-protein. Agonist-stimulated GTPγ35S binding was measured for L-methadone (A), DAMGO (B), endomorphin-1 (C), and morphine (D) in the presence of vehicle (■) or 10 μM MS1 (□) using C6-μ cell membranes.
Molecular Descriptors of μ-PAMs/SAM and Their Statistical Analysis
We calculated fifty-two physicochemical properties for each of the 14 μ-PAMs and 12 μ-SAMs reported in Table 1. The numerical values of those descriptors that displayed nonzero variance across these twenty-six ligands (Table S2) are reported in Table S3. Considering combinations of up to 5 descriptors, Bayesian logistical regression analysis identified four descriptors that could best separate between μ-PAMs and μ-SAMs as assessed experimentally. Specifically, the best linear model according to AIC (Table S4) resulted from using the following four properties: the predicted central nervous system activity (CNS), the conformation-independent predicted aqueous solubility (CIQPlogS), the Parameterized Model Number 3 (PM3)-calculated electron affinity (EA.eV.), and the van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms (PSA). Using this model, only four (MS12, MS13, MS14, and MS23) out of twenty-six compounds could not be confidently assigned the same μ-PAM or μ-SAM activity inferred from experiments owing to their predicted effect value below or above an arbitrary 0.5 cutoff, respectively (Table S5). The remaining 11 PAMs exhibited calculated average values of −0.82 ± 0.40, −7.12 ± 0.75, +1.07 ± 0.14, and +76.51 ± 3.62 for CNS, CIQPlogS, PM3, and EA.eV., respectively, whereas the remaining 11 SAMs had corresponding values of −0.91 ± 0.54, −6.54 ± 0.51, +0.94 ± 0.08, and +74.04 ± 2.78.
Common 3D-Pharmacophore of μ-PAMs
We built a ligand-based 3D pharmacophore model to elaborate further on the molecular and structural determinants that differentiate μ-PAMs from μ-SAMs. The best 3D pharmacophore model of this kind (Figure 5) includes: (i) two H-bond acceptors (i.e., the two oxygen atoms of the sulfur dioxide group) labeled A1 and A2 in the figure, (ii) one halogen substituent or hydrophobic group (i.e., R1 = Br, Cl, Me; see Table 1) labeled Halo/Hyd in the figure, and (iii) the three aromatic rings R1–R3 related by the distances and angles reported in Tables S6 and S7, respectively. Using this model, all μ-PAMs (but MS13) could be separated from all μ-SAMs (but MS16 and MS23) according to an arbitrary cutoff of 1.7 for the pharmacophore alignment fitness scores (see Table S8). In the case of MS13, an optimal alignment of this compound to the best pharmacophore model could not be found because of competition in the alignment between the hydrophobic substituent on ring 3 (R3) and that of ring 1 (R1). Although MS16 and MS23 could indeed be successfully aligned to the pharmacophore model, the R3 methoxy substitution at the ortho-position might interfere with the position of the ligand amide atoms although it is also possible that the R3 methoxy substitution at ortho- or meta- positions clashes with the receptor environment. More sophisticated strategies than simple docking are currently being tested in our lab to be eventually be able to support or dispute unambiguously this possibility.
Figure 5.
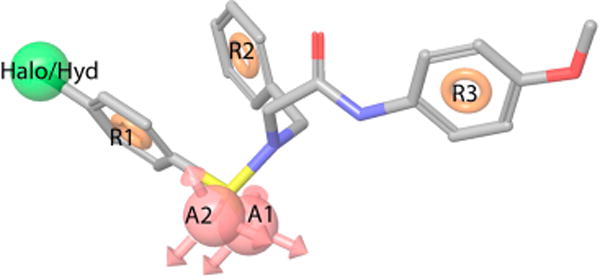
Ligand-based 3D pharmacophore model built from μ-PAMs shown in Table 1. The best 3D pharmacophore model that separates μ-PAMs and μ-SAMs includes two H-bond acceptors (i.e., the two oxygen atoms of the sulfur dioxide group), one halogen substituent or hydrophobic group (i.e., R1 = Br, Cl, Me; see Table 1), and three aromatic rings related by specific distances and angles listed in Tables S6 and S7.
DISCUSSION
Recently, attention has shifted to allosteric rather than orthosteric opioid ligands as a means of potentially providing effective pain relief that is free from debilitating adverse effects.5 These allosteric modulators are expected to be receptor type selective, and to act by enhancing the antinociceptive activity of endogenous opioid ligands. Therefore, μ-opioid receptor PAMs may have fewer on-target side effects and overdosing risks, and may produce less tolerance and dependence than currently used opioid agonists. It has been suggested that opioid ligands that bias receptor signaling toward the G-protein mediated pathway instead of β-arrestin2 may be therapeutically beneficial.18 Whether caused by the receptor conformational plasticity, allosterism, or dimerization/oligomerization, this G-protein-biased agonism has been suggested to remove the on-target side effects such as drug tolerance associated with the μ-opioid receptor internalization (e.g., see ref 5). The current findings with endomorphin-1 suggest that the μ-opioid receptor PAM MS1 may promote signaling bias in the opposite direction (favoring β-arrestin versus G-protein activation), at least with this peptide. Further studies are needed in order to understand more fully both the signaling bias and probe-dependence of this PAM, and to determine whether these properties can be altered through modifications to the chemical structure.
The only two known μ-PAMs at the time of this work, i.e., BMS-986121 and BMS-986122, are limited in their ease of synthesis. Not only is the new allosteric modulator chemotype we identified easier to derivatize by synthetic chemistry, and offering an additional point of diversity for structure–activity relationship studies compared to previously published compounds, but the new scaffold increases the chemical diversity of known ligands for the allosteric site of the μ-opioid receptor. However, undesirable “off-target” effects may still be present for this scaffold, and must be evaluated before further development.
In competition binding and G-protein activation assays, MS1 displayed marked probe dependence. Indeed, the largest effects of MS1 were seen with L-methadone. The prototype μ-PAM BMS-986122 also showed the highest levels of cooperativity with methadone and its isomers.19 Because MS1 is a new scaffold, this similar probe dependence may be reflective of a similar mechanism of action and/or mode of binding. In addition, MS1 enhanced the maximal activation of the partial agonist morphine to activate G-protein. This again fits with the probe dependence of BMS-986122 in which the efficacy of partial agonists was increased. The mechanism of BMS-986122 action was found to be through allosteric disruption of sodium ion binding19 and it would be interesting to determine if this new chemotype also functions in a similar manner.
Cheminformatics analysis of the set of newly identified μ-PAMs and μ-SAMs suggested that physicochemical properties such as the predicted CNS, the CIQPlogS, the EA.eV., and the PSA may be used as searching criteria to identify additional compounds with potential PAM activity at μ-opioid receptors. Specifically, our best statistical model shows that μ-PAMs have higher predicted values of central nervous system activity, PM3-calculated electron affinity, and van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms, but lower calculated values of conformation-independent predicted aqueous solubility, compared to μ-SAMs. However, it must be kept in mind that the dataset we used is limited in number and no thorough cross-validation of the presented statistical models could be performed. Although the same limitation exists for the predicted common 3D pharmacophore model of μ-PAMs vs μ-SAMs, the suggested model can be used as an initial criterion to either design more highly potent derivatives of the newly identified μ-PAM or to search for completely different chemotypes that retain the same pharmacophore features. These inferences can and will eventually be combined with structural studies using the crystal structure of the active μ-opioid receptor that has appeared in the literature during review of this paper.20 In the meantime, additional ligand-based studies are ongoing in our laboratories to optimize the newly identified chemotype and to explore the potential of this scaffold for the development of new therapeutics.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants DA026434 and DA034049 (to M.F.) and DA035316 (to J.T.). Computations were run on resources available through the Scientific Computing Facility at Mount Sinai and the Extreme Science and Engineering Discovery Environment (XSEDE) under MCB080109N, which is supported by National Science Foundation grant number OCI-1053575.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.5b00388.
Table S1 provides a list of the 1336 molecules identified in eMolecules as close analogs of BMS-986121 and BMS-986122. Table S2 provides a list of the physicochemical properties displaying nonzero variance across the twenty-six ligands shown to have either a PAM or a SAM activity at the μ-opioid receptor. Table S3 reports calculated values of the physicochemical properties displaying nonzero variance across the twenty-six ligands with a μ-PAM or μ-SAM activity. Table S4 reports the statistics of the best predictive model for PAM/SAM differentiation based on Bayesian logistic regression analysis of combinations of up to 5 descriptors. Table S5 reports the predicted effect values of MS1-MS26 based on Bayesian logistic regression analysis of combinations of up to five descriptors. Table S6 and S7 provide distance and angle measurements for the best pharmacophore model, respectively. Table S8 reports values of the best pharmacophore alignment fitness (PDF).
Notes
The authors declare no competing financial interest.
References
- 1.Pasternak GW. Opiate Pharmacology and Relief of Pain. J Clin Oncol. 2014;32:1655–1661. doi: 10.1200/JCO.2013.53.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spetea M, Asim MF, Wolber G, Schmidhammer H. The mu Opioid Receptor and Ligands Acting at the mu Opioid Receptor, as Therapeutics and Potential Therapeutics. Curr Pharm Des. 2014;19:7415–7434. doi: 10.2174/13816128113199990362. [DOI] [PubMed] [Google Scholar]
- 3.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal Structure of the mu-Opioid Receptor Bound to a Morphinan Antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burford NT, Traynor JR, Alt A. Positive Allosteric Modulators of the mu-Opioid Receptor: A Novel Approach for Future Pain Medications. Br J Pharmacol. 2015;172:277–286. doi: 10.1111/bph.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson GL, Kelly E, Christopoulos A, Canals M. Novel GPCR Paradigms at the mu-Opioid Receptor. Br J Pharmacol. 2015;172:287–296. doi: 10.1111/bph.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickols HH, Conn PJ. Development of Allosteric Modulators of GPCRs for Treatment of CNS Disorders. Neurobiol Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O’Connell J, Traynor JR, Alt A. Discovery of Positive Allosteric Modulators and Silent Allosteric Modulators of the μ-Opioid Receptor. Proc Natl Acad Sci U S A. 2013;110:10830–10835. doi: 10.1073/pnas.1300393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burford NT, Livingston K, Canals M, Ryan M, Budenholzer L, Han Y, Shang Y, Herbst JJ, O’Connell J, Banks M, Zhang L, Filizola M, Bassoni D, Wehrman TS, Christopoulos A, Traynor JR, Gerritz SW, Alt A. Discovery, Synthesis and Molecular Pharmacology of Selective Positive Allosteric Modulators of the delta-Opioid Receptor. J Med Chem. 2015;58:4220–4229. doi: 10.1021/acs.jmedchem.5b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burford NT, Wehrman T, Bassoni D, O’Connell J, Banks M, Zhang L, Alt A. Identification of Selective Agonists and Positive Allosteric Modulators for mu- and delta-Opioid Receptors from a Single High-Throughput Screen. J Biomol Screening. 2014;19:1255–1265. doi: 10.1177/1087057114542975. [DOI] [PubMed] [Google Scholar]
- 10.eMolecules. eMolecules, Inc.; La Jolla, CA: http://www.emolecules.com/ [Google Scholar]
- 11.Small-Molecule Drug Discovery Suite 2014-4. Schrödinger; New York, NY: 2014. [Google Scholar]
- 12.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bassoni DL, Raab WJ, Achacoso PL, Loh CY, Wehrman TS. Measurements of Beta-Arrestin Recruitment to Activated Seven Transmembrane Receptors Using Enzyme Complementation. Methods Mol Biol. 2012;897:181–203. doi: 10.1007/978-1-61779-909-9_9. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Jones A, Olson KR, Peng K, Wehrman T, Park A, Mallari R, Nebalasca D, Young SW, Xiao SH. A Homogeneous Enzyme Fragment Complementation-Based Beta-Arrestin Translocation Assay for High-Throughput Screening of G-protein-Coupled Receptors. J Biomol Screening. 2008;13:737–747. doi: 10.1177/1087057108321531. [DOI] [PubMed] [Google Scholar]
- 15.Lee KO, Akil H, Woods JH, Traynor JR. Differential Binding Properties of Oripavines at Cloned mu- and delta-Opioid Receptors. Eur J Pharmacol. 1999;378:323–330. doi: 10.1016/s0014-2999(99)00460-4. [DOI] [PubMed] [Google Scholar]
- 16.McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. mu-Opioid Receptors: Correlation of Agonist Efficacy for Signalling with Ability to Activate Internalization. Mol Pharmacol. 2010;78:756–766. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, Rosethorne EM, Charlton SJ, Krasel C, Bailey CP, Henderson G, Kelly E. Endomorphin-2: A Biased Agonist at the mu-Opioid Receptor. Mol Pharmacol. 2012;82:178–188. doi: 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional Selectivity at the mu-Opioid Receptor: Implications for Understanding Opioid Analgesia and Tolerance. Pharmacol Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livingston KE, Traynor JR. Disruption of the Na+ Ion Binding Site as a Mechanism for Positive Allosteric Modulation of the mu-Opioid Receptor. Proc Natl Acad Sci U S A. 2014;111:18369–18374. doi: 10.1073/pnas.1415013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK. Structural insights into μ-opioid receptor activation. Nature. 2015;524:315–21. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.