

Abstract
Poly(ADP-ribose) polymerase 3 (PARP3) is a member of the PARP family enzymes which catalyze the ADP-ribosylation of proteins. PARP3 plays an important role in DNA damage repair and mitotic progression. In this study, we identified, using mass spectrometric techniques, two novel post-translational modification sites in PARP3, α-N-methylation and phosphorylation of serine 461 (S461). We found that the N-terminal α-amino group of PARP3 is heavily methylated in human cells, and N-terminal RCC1 methyltransferase (NRMT) is a key enzyme required for this methylation. We also observed that the phosphorylation level of S461 in PARP3 could be reduced in human cells upon treatment with flavopiridol, a cyclin-dependent kinase inhibitor. Moreover, we demonstrated that S461 phosphorylation, but not α-N-methylation of PARP3, may be involved in the cellular response toward DNA double-strand breaks. These findings provide novel insights into the post-translational regulation of PARP3.

Keywords: PARP3, post-translational modification, LC–MS/MS, N-terminal methylation, NRMT, phosphorylation, DNA double-strand break
Introduction
Poly(ADP-ribosyl)ation is an important post-translational modification (PTM) which regulates many cellular processes, including DNA damage repair, transcription, apoptosis, and mitosis.1 Poly(ADP-ribosyl)ation is catalyzed by poly(ADP-ribose) polymerases (PARPs), which transfer the ADP ribose unit from NAD+ to monomeric ADP-ribose, or short, long, or even branched poly(ADP-ribose) polymers to substrate pro-teins.2 Human PARPs comprise 17 members sharing a conserved catalytic domain.3 PARP1 is the best-studied member which can be activated rapidly in response to the formation of DNA strand breaks and facilitates the recruitment of DNA repair proteins to DNA damage sites.4–6 In addition to PARP1, PARP2, PARP3, and tankyrase-1 (a.k.a. PARP-5a) have also been implicated in DNA repair and/or the maintenance of genome stability.7,8
PARP3 was first discovered by screening an EST library using the catalytic domain of PARP1.9 The crystal structure of the catalytic domain of PARP3 is highly similar to those of PARP1 and PARP2.10–12 Apart from the catalytic domain, PARP3 contains an N-terminal region (NTR) and a WGR domain which is enriched with tryptophan, glycine, and arginine. Two isoforms of PARP3, differing by seven amino acids at the N terminus, are expressed in humans, whereas only the short isoform is present in mice.13 It remains unknown whether the two isoforms possess different functions.
PARP3 is an ADP-ribosyltransferase that can modify itself and other proteins in the presence of DNA breaks.14,15 Stimulation of PARP3 results in the transfer of ADP-ribose monomers or short poly(ADP-ribose) chains onto the target protein.14,16 A previous proteomic study revealed that PARP3 interacts with several proteins involved in base excision repair and nonhomologous end-joining (NHEJ) pathways.17 Subsequent studies indeed showed that the ADP-ribosyltransferase activity of PARP3 could be stimulated by DNA double-strand breaks (DSBs),14,15 and PARP3 functions together with APLF to promote NHEJ.14,18 Additionally, PARP3 was shown to be a critical player in the stabilization of the mitotic spindle and in telomere integrity by regulating mitotic components NuMA and tankyrase 1.19 Moreover, PARP3 affects the choice of homologous recombination (HR) and NHEJ pathways by limiting DNA end resection.20
PTMs are among the most efficient mechanisms for expanding proteins' structure, interactions, localization, and/or function.21 Recently, mass spectrometry has been widely used for assessing protein PTMs, which provides unambiguous identification of the sites and types of modifications,22 and allows for quantitative analysis.23 It has been reported that PARP3 is able to ADP-ribosylate some proteins.24 However, relatively little is known about the PTMs of PARP3 or their biological functions. In this study, we conducted a systematic identification of PTMs in PARP3 using mass spectrometric techniques, and our results expanded the functional studies of PARP3 at the post-translational level.
Materials and Methods
Cell Culture
Human HEK293T and U2OS cells were grown at 37 °C in Dulbecco's Modified Eagle medium (DMEM, ATCC) supplemented with 10% fetal bovine serum (Invitrogen) and 100 IU/mL penicillin/streptomycin (ATCC). Human A549 cells were grown at 37 °C in DMEM supplemented with 15% fetal calf serum, 50 IU/mL penicillin/streptomycin, and 2 mM l-glutamine (Invitrogen).
Construction of Vectors
Three tandem repeats of the FLAG epitope tag (DYKD-DDDK) were inserted into BamHI and EcoRI sites of pRK7 vector to produce the pRK7-3×FLAG vector. The short isoform of PARP3 was amplified from HEK293T cells by reverse transcription PCR to introduce a 5′ XbaI site and a 3′ BamHI site, and subcloned into pRK7-3×FLAG vector. The pSUPER-PARP3 and pCD2E-PARP3 vectors were described previously.14 The PARP3 K4Q, PARP3 S461A, and PARP3 S461D mutant constructs were amplified by site-directed muta-genesis using primers containing designed mutations. The primers used are listed in Table S1, Supporting Information.
Protein Expression and Mass Spectrometric Analysis
HEK293T cells were grown to 70–80% confluency in a six-well plate and transfected with 1.5 μg of FLAG-tagged PARP3 plasmids using Lipofectamine 2000 (Invitrogen). After a 48-h incubation, the protein was isolated and purified using anti-FLAG M2 beads (Sigma). The purified proteins were digested by trypsin (Roche) or Glu-C (Roche), and subjected to LC–MS/MS analysis.
LC–MS/MS experiments were performed as described previously.25 Briefly, the peptides were separated on an EASY-nLC II and analyzed by an LTQ Orbitrap Velos mass spectrometer equipped with a nanoelectrospray ionization source (Thermo, San Jose, CA). The trapping column (150 μm × 50 mm) and separation column (75 μm × 120 mm) were both packed with ReproSil-Pur C18-AQ resin (3 μm in particle size, Dr. Maisch HPLC GmbH, Germany). The peptide samples were first loaded onto the trapping column in CH3CN/H2O (2:98, v/v) at a flow rate of 4.0 μL/min and resolved on the separation column with a 120 min linear gradient of 2–40% acetonitrile in 0.1% formic acid and at a flow rate of 300 nL/min. The LTQ-Orbitrap Velos mass spectrometer was operated in the positive-ion mode, and the spray voltage was 1.8 kV. The full-scan mass spectra (m/z 300–2000) were acquired with a resolution of 60 000 at m/z 400 after accumulation to a target value of 500 000 in the linear ion trap. MS/MS data were obtained in a data-dependent scan mode where one full MS scan was followed with 20 MS/MS scans. Dynamic exclusion parameters for data-dependent scan are listed in Table S2, Supporting Information. The relative collision energy was 35, and the mass window for precursor ion selection was 3 m/z units. For quantitative analysis, MS/MS experiments were conducted in selected-ion monitoring mode, where the peak areas found in the selected-ion chromatograms for monitoring the formation of a specific fragment ion from the unmodified and the corresponding modified peptides were employed for estimating the levels of modifications. In particular, the y2 ion at m/z 249.1 and the y112+ ion at m/z 578.3 were used to estimate the levels of α-N-methylation in the Glu-C-produced N-terminal peptide and the level of S461 phosphorylation in the tryptic peptide harboring residues 449–472, respectively. In this regard, we realize that the α-N-methylation and phosphorylation may alter the ionization efficiencies of the corresponding peptides; therefore, the levels of modifications estimated from the above measurements may differ from the actual modification levels. Nevertheless, the relative levels of modifications determined by the method under different cellular conditions should be valid.
In Vitro Methylation Assay
NRMT-His6 plasmid, a gift from Prof. Ian G. Macara,26 was expressed by inducing transformed Escherichia coli Rosetta (DE3) pLysS with 0.5 mM isopropyl 1-thio-β-d-galactopyranoside at room temperature overnight when the culture reached OD600 ≈ 0.8. Subsequently, the NRMT-His6 protein was purified by using Talon affinity resin (Clontech) following the manufacturer's recommended procedures.
In vitro methylation assay was performed as described previously.25,26 Briefly, 200 ng of unmodified peptide, i.e., APKPKPWVQTEG (Genemed Synthesis Inc.), was mixed with 100 μM S-adenosyl-l-methionine (S-AdoMet, Sigma) and 1 μg of NRMT to a final volume of 50 μL in the methyltransferase buffer (50 mM Tris, 50 mM potassium acetate, pH 8.0). The reaction mixture was incubated at 30 °C for 1 h and subjected to LC–MS/MS analysis.
SiRNA Knockdown of NRMT and Real-Time Quantitative RT-PCR
NRMT knockdown was carried out as described previously. Briefly, HEK293T cells were cultured in six-well plates and transfected with 100 pmol of siRNAs using Lipofectamine 2000. After a 48-h incubation, 1.5 μg of PARP3 expression plasmids were cotransfected into the cells together with another aliquot of siRNA for another 48 h.
At 48 h following transfection, total RNA was extracted from the cells using the Total RNA Kit I (Omega), and cDNA was generated by using M-MLV reverse transcriptase (Promega) and an oligo(dT)16 primer. The knockdown efficiency of NMRT was evaluated by quantitative real-time RT-PCR using iQ SYBR Green Supermix kit (Bio-Rad) and the GAPDH gene was used as an internal control as described previously. Primers used in real-time PCR are listed in Table S1, Supporting Information.
Drug Treatments
FLAG-tagged PARP3 plasmids were transfected into HEK293T cells using Lipofectamine 2000. After a 48-h incubation, the cells were treated with 1 μM flavopiridol (Santa Cruz) for 6 h or 1 μM neocarzinostatin (NCS, Sigma) for 1 h. The FLAG-tagged PARP3 was subsequently purified, digested by trypsin or Glu-C, and analyzed by LC–MS/MS as described above.
GFP Reporter Assay for Monitoring NHEJ Level
U2OS cells with a chromosomally integrated copy of EJ5-GFP plasmid (a gift from Prof. Jeremy M. Stark) were transfected with the I-SceI expression vector pCBASce.27 The U2OS-GFP cells without I-SceI transfection were used as negative control. At 3 h after transfection, the medium was replaced with fresh medium containing 1 μM flavopiridol. Three days after transfection, the cells were washed with PBS and analyzed by flow cytometry (BD Biosciences FACSAria I).
RNA Interference of PARP3 and Western Blot
Transient PARP3 knockdown and complementation were performed as described previously.14 Briefly, A549 cells were cotransfected with a mixture (1:2 molar ratio) of pSUPER-PARP3 and empty pCD2E or shRNA target-resistant versions of pCD2E-PARP3 WT, pCD2E-PARP3 S461A, or pCD2E-PARP3 S461D construct. After a 24-h incubation, cells were placed into selection medium containing 1.5 mg/mL G418 for 6 days. The expression level of PARP3 was evaluated by Western blot as described previously.14 The antibodies that specifically recognized human PARP3 (rabbit polyclonal anti-PARP3, a gift from Francoise Dantzer) and α-actin (Sigma) were used at 1:1000 and 1:5000 dilutions, respectively. Antimouse or antirabbit HRP-conjugated secondary antibodies (Dako Cytomation) were employed at 1:5000 dilution.
Immunofluorescence
Immunofluorescence experiments were carried out as described previously.14 Briefly, A549 cells were γ irradiated (2 Gy) and cultured at 37 °C for the indicated periods of time. The cells were then rinsed in PBS and fixed in 4% paraformaldehyde for 5 min. Cells were subsequently permeabilized in 0.2% Triton X-100 for 2 min, blocked with 5% bovine serum albumin for 30 min, and incubated with mouse anti-phospho-histone γH2AX (Ser139) antibody (Millipore, 1:1000 dilution) and rabbit anti-CENPF (Abcam, 1:500) at 4 °C overnight. Cells were washed three times with PBS containing 0.1% Tween 20 and 0.02% SDS and incubated with Alexa Fluor 488 goat antimouse and Alexa Fluor 555 goat antirabbit secondary antibodies (Invitrogen, 1:500 dilution) at room temperature for 1 h. After washing the cells five times as described above, they were stained with DAPI (Sigma), mounted in Vectashield Mounting Medium (Vector Laboratories), and scored for γH2AX foci in G1 (CENPF-negative) cells using a Nikon Eclipse 50i microscope as described previously.14
Results
Identification of α-N-Methylation of PARP3
To identify the PTMs of PARP3, the short isoform of human PARP3 with C-terminal FLAG tag was expressed in HEK293T cells and purified using anti-FLAG M2 beads. The purified protein was digested with endoproteinase Glu-C or trypsin. The resulting peptides were subjected subsequently to LC–MS/MS analysis. A database search of the LC–MS/MS data led to the identification of a number of peptides derived from PARP3 with a sequence coverage of 89% (Supplementary Figure S1, Supporting Information). Our LC–MS/MS analysis also gave rise to the identification of two modification sites in PARP3, i.e., α-N-methylation and phosphorylation of serine 461.
LC–MS/MS results showed that the initiating methionine of PARP3 was cleaved and the N-terminal α-amino group of the protein was heavily methylated (Figure 1A). Unmodified N-terminal peptide was hardly detectable (Supplementary Figure S2, Supporting Information). Fragmentation of the methylated peptide produced a tandem mass spectrum revealing unambiguously the α-N-methylation of PARP3 (Figure 1B–D).
Figure 1.
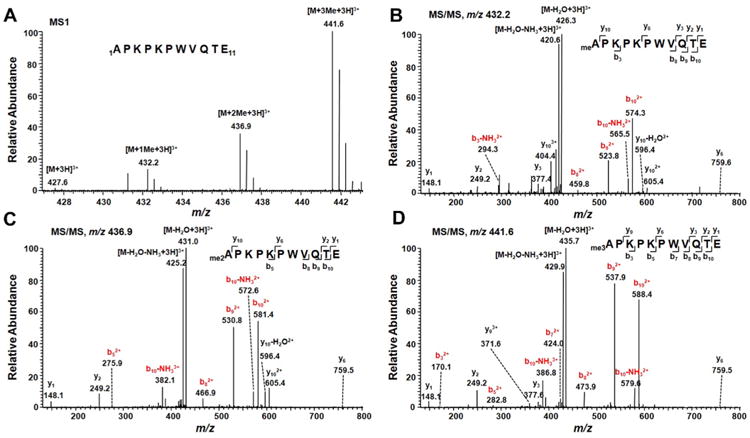
Identification of α-N-methylation of PARP3. (A) ESI-MS of the N-terminal peptide APKPKPWVQTE from the Glu-C digestion of C-terminally FLAG-tagged human PARP3. (B–D) MS/MS of mono-, di-, and trimethylated N-terminal peptide of PARP3. Those fragment ions carrying the methylated N-terminus are highlighted in red.
Most known N-terminally methylated proteins harbor a common N-terminal sequence motif of XPK (“X” represents alanine, proline, or serine),28 though methylation in other N-terminal sequence contexts was also found.29,30 To address the importance of APK motif in N-terminal methylation of PARP3, we mutated lysine 4 to glutamine and analyzed the N-terminal methylation pattern of the K4Q mutant of PARP3 by LC–MS/MS. Our result showed that the K4Q mutation abolished the N-terminal methylation (Supplementary Figure S3, Supporting Information), indicating that lysine 4 is essential for the N-terminal methylation of PARP3.
NRMT Is Involved in the α-N-methylation of PARP3
NRMT can catalyze the α-N-methylation of a number of human proteins carrying the conserved N-terminal XPK motif.26 Thus, we reasoned that NRMT may also catalyze the α-N-methylation of PARP3, which possesses an N-terminal APK motif. To test this, we incubated the N-terminal peptide of PARP3, i.e., APKPKPWVQTEG, with NRMT and S-adenosyl-l-methionine, which served as the methyl group donor. The results showed that the N-terminal peptide of PARP3 can be mono-, di-, and trimethylated by NRMT, as supported by LC–MS/MS (Figure 2A). However, we can only find unmodified peptide in the samples without the addition of NRMT (Figure 2B). Together, these results demonstrated that NRMT can catalyze the α-N-methylation of the N-terminal peptide of PARP3 in vitro.
Figure 2.

NRMT can catalyze α-N-methylation of PARP3. (A and B) ESI-MS for monitoring the in vitro methylation of the N-terminal peptides of PARP3 in the presence (A) or absence (B) of NRMT. (C) Relative levels of different methylation forms of N-terminal peptide of PARP3 isolated from HEK293T cells treated with control or NRMT siRNA, as estimated from quantitative LC–MS/MS analysis.
To assess whether NRMT is required for the α-N-methylation of PARP3 in cells, we knocked down NRMT in HEK293T cells by using siRNA and transfected the cells with the plasmid expressing FLAG-tagged PARP3. After protein purification and Glu-C digestion, the resulting peptide mixture was subjected to LC–MS/MS analysis in the selected-ion monitoring mode. We then estimated the relative level of α-N-methylation based on the relative abundances of fragment ions formed from the unmodified and α-N-methylated peptides. The results showed that the level of α-N-methylation of PARP3 was substantially decreased upon NRMT knockdown (Figure 2C, Supplementary Figure S4, Supporting Information), suggesting that NRMT is required for the α-N-methylation of PARP3 in human cells.
PARP3 Is Phosphorylated at S461
Our LC–MS/MS analysis also led to the identification of phosphorylation of serine 461 in PARP3. Trypsin digestion produced a 24-amino acid peptide spanning residues Glu449 and Arg472. The observed [M + 3H]3+ ion (m/z 903.4325) exhibited a m/z value that is 26.6555 higher than the calculated m/z of the corresponding unmodified peptide (m/z 876.7770), which is consistent with the monophosphorylation of this peptide. Figure 3A depicts the MS/MS of the [M + 3H]3+ ion of the peptide 449EHHINTDNPSLKpSPPPGFDSVIAR472 with one phosphorylated amino acid. The phosphorylation site could be readily determined from fragment ions found in the MS/MS, with the consideration of the mass shift introduced by phosphorylation (80 Da). Along this line, the existence of the y12 and b13 ions carrying a phosphorylated amino acid provides unambiguous evidence supporting the phosphorylation of S461 (Figure 3A).
Figure 3.

Identification of S461 phosphorylation of PARP3. (A) MS/MS of phosphorylated peptide 449EHHINTDNPSLKSPhoPPPGFDSVIAR472 of PARP3. (B) LC–MS/MS-based relative quantification showed that flavopiridol treatment could lead to a reduced phosphorylation level of S461. (C) The effect of flavopiridol treatment on DNA DSB repair via the NHEJ pathway. (D) Quantitative results revealed that NCS treatment could augment the S461 phosphorylation level.
Trypsin cleaves the amide bonds on the C-terminal sides of lysine and arginine. Thus, the phosphorylated peptide 449EHHINTDNPSLKSPhoPPPGFDSVIAR472 contains a miscleavage. We failed to observe the phosphorylated peptide without miscleavage, though the corresponding unmodified peptide, 461SPPPGFDSVIAR472, could be readily detected (Supplementary Figure S5, Supporting Information). The negatively charged Asp189 in the substrate-binding pocket of trypsin is responsible for binding positively charged lysine and arginine in substrate proteins.31 S461 phosphorylation in PARP3 introduces negative charges close to Lys460, which, similar to what was reported previously for other phosphorylated proteins,32 is expected to diminish the binding of trypsin to Lys460. Thus, the miscleavage of the amide bond on the N-terminal side of S461 provides another line of evidence to support its phosphorylation.
Cyclin-dependent kinases (CDKs) are a family of protein kinases regulating cell cycle progression, and some CDKs can phosphorylate protein substrates at serine or threonine residues preceding a proline.33–35 Considering that S461 of PARP3 also resides at a serine-proline site, we reason that CDKs might be involved in this phosphorylation. To test this, we used a CDK inhibitor, flavopiridol, to treat the HEK293T cells expressing FLAG-tagged PARP3 and measured the phosphorylation level of S461. Our results revealed that flavopiridol treatment resulted in a significant decline in the phosphorylation level of S461 (Figure 3B), suggesting the involvement of CDKs in this phosphorylation.
Role of S461 Phosphorylation of PARP3 in the Cellular Response toward DNA DSBs
Previous studies revealed that PARP3 is an important regulator of NHEJ pathway during DNA DSB repair.14 To assess whether the S461 phosphorylation of PARP3 might be involved in NHEJ pathway, we first employed flavopiridol to treat the U2OS cells with a chromosomally integrated EJ5-GFP reporter.36 EJ5-GFP carries a promoter that is separated from a GFP coding cassette by a puromycin-resistant gene flanked by two I-SceI sites; NHEJ-mediated repair of the I-SceI-induced DSBs results in the restoration of the GFP cassette.36 Flow cytometry-based measurement of GFP-positive U2OS cells after I-SceI transfection facilitated us to quantify the levels of NHEJ in control and flavopiridol-treated cells. Our results revealed that flavopiridol treatment led to significantly diminished repair of DSBs via the NHEJ pathways (Figure 3C).
We next used neocarzinostatin (NCS), a radiomimetic drug, to treat HEK293T cells expressing FLAG-tagged PARP3 and measured the phosphorylation level of S461. Our result showed that NCS treatment led to an increase in the phosphorylation level of S461 in PARP3 (Figure 3D). To gain further insights into the function of S461 phosphorylation, we produced plasmids for expressing wild-type PARP3 (PARP3 WT) and the corresponding phospho-defective (PARP3 S461A) and phospho-mimetic (PARP3 S461D) mutants. It is of note that PARP3 S461A and PARP3 S461D mutants were still predominantly nuclear (Figure 4). Subsequently, we compared the rates of γH2AX induction and removal following γ-ray exposure in A549 cells expressing PARP3 WT, PARP3 S461A, or PARP3 S461D. In this respect, A549 cells display low background levels of damage in unperturbed cells and have been established as a useful cell line for analyzing repair rates of DNA DSBs.14,37,38 The number of γH2AX foci in cells after γ-ray exposure reflects the number of chromosomal DSBs.39–41 Our results showed that the cellular sensitivity toward ionizing radiation arising from PARP3 deficiency could be restored to a similar extent by complementing the cells with wild-type PARP3 and PARP3 S461D, whereas PARP3 S461A displayed a significant delay in the removal of γH2AX foci (Figure 5A). Our result, therefore, indicates that the S461 phosphorylation of PARP3 may confer cellular resistance toward DSBs.
Figure 4.

Confocal microscopy results showed the nuclear localization of wild-type PARP3 (PARP3 WT), PARP3 S461A, and PARP3S461D mutants in U2OS cells.
Figure 5.

Role of S461 phosphorylation of PARP3 in the cellular response to DSBs. (A) Mock-depleted (A549) or transiently PARP3-depleted A549 cells (PARP3 KD) transfected with empty vector (+V) or shRNA target-resistant wild-type PARP3 (PARP3 WT), PARP3 S461A, or PARP3 S461D construct, were exposed to γ-rays and analyzed by γH2AX immunofluorescence. Right panels show the expression level of PARP3 in the indicated cells. (B) Normal A549 cells (Mock) or PARP3-deficient A549 cells (PARP3 KD) transiently transfected with empty vector (+V), targeting-resistant wild-type PARP3 or PARP3 K4Q vector, were γ-irradiated and analyzed by γH2X immunofluorescence. Right panels show Western blot results of PARP3 in these cells. Error bar represents the SEM (n = 3). *P < 0.05. **P < 0.01. The P value was calculated by using unpaired two-tailed Student's t-test.
We also examined the effect of NCS treatment on N-terminal methylation of PARP3. However, the level of α-N-methylation of PARP3 was not altered upon NCS treatment (Supplementary Figure S6, Supporting Information). Moreover, the compromised clearance of γH2AX foci induced by PARP3 knockdown can be restored PARP3 K4Q mutant to a similar level as the wild-type PARP3 (Figure 5B), suggesting that the N-terminal methylation of PARP3 may not be involved in DSB repair.
Discussion
Protein N-terminal α-methylation is a type of PTM that is conserved from E. coli to man. NRMT is a critical enzyme that catalyzes the α-N-methylation of a number of proteins in humans.26,43 Most substrates of NRMT possess a conserved N-terminal sequence motif of XPK (“X” represents alanine, proline, or serine) after the initiator methionine residue is cleaved. 28,29 Human PARP3 contains two isoforms differed by seven amino acids on the N-terminus, where the longer isoform lacks the conserved domain for N-terminal methylation. In contrast, the short isoform, which carries an N-terminal APK motif, was found to be N-terminally methylated in this study. It has been reported that the two isoforms of PARP3 may have different cellular localizations, where the long splicing isoform was considered as a core component of the centrosome and the short variant was a nuclear protein.13 Although it remains unknown whether these two isoforms have different biological functions, we speculate that the differences, if present, may be attributed, in part, to the α-N-methylation.
PARP3 plays diverse roles in many cellular processes, including DNA damage repair,14 mitotic progression, and maintenance of telomere stability.19 In this study, we found that S461 phosphorylation could be stimulated by a DNA-damaging agent (i.e., NCS), indicating that S461 phosphorylation may be required for PARP3's function in DNA damage repair. Additionally, we demonstrated that inhibition of CDK-mediated phosphorylation of PARP3 led to compromised NHEJ in human cells. Furthermore, we showed that S461A mutant exhibited slower γH2AX foci clearance than wild-type PARP3 and S461D mutant after γ-ray exposure. Together, we conclude that the S461 phosphorylation of PARP3 may be involved in the cellular response toward DNA DSBs. On the other hand, we found that N-terminal methylation of PARP3 is neither stimulated by NCS nor affects the γH2AX foci removal following γ-ray exposure, suggesting the lack of involvement of α-N-methylation in DNA DSB repair. It is possible that α-N-methylation is important for PARP3's role in other cellular processes.
In conclusion, we identified, for the first time, two novel PTMs, i.e., α-N-methylation and S461 phosphorylation, of PARP3 in human cells. Our results suggested that the S461 phosphorylation, but not the α-N-methylation, may be required for DNA DSB repair in human cells. Thus, the results from the present study expanded the function of PARP3 at the post-translational level, which serves as the foundation for further study of PARP3.
Supplementary Material
Figure S1: The sequence coverage of PARP3 obtained from LC–MS/MS analysis.
Figure S2: MS/MS of the [M + 3H]3+ ion of the unmodified N-terminal peptide of PARP3.
Figure S3: PARP3-K4Q mutant abrogates α-N-methylation in human cells.
Figure S4: Relative mRNA level of NRMT by real-time PCR using GAPDH as control.
Figure S5: MS/MS of the [M + 2H]2+ ion of the unmodified peptide containing residues 461–462 of PARP3.
Figure S6: Effect of NCS treatment on the level of α-N-methylation of PARP3.
Table S1: The primers used in this study.
Table S2: The parameters for dynamic exclusion analysis.
Acknowledgments
The authors would like to thank Prof. Ian G. Macara for the NRMT-His6 vector, Prof. Jeremy M. Stark for U2OS-EJ5-GFP cells, Prof. Françoise Dantzer for the PARP3 antibody, and Dr. Luis Polo for the helpful comments. This work was supported by the National Institutes of Health (R01 ES019873 to Y.W.) and grants from Cancer Research UK (C6563/A1307 and C6563/A16771 to K.W.C.).
Footnotes
Notes: The authors declare no competing financial interest.
Supporting Information: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Burzio LO, Riquelme PT, Koide SS. ADP ribosylation of rat liver nucleosomal core histones. J Biol Chem. 1979;254:3029–3037. [PubMed] [Google Scholar]
- 3.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 4.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 5.Allinson SL, Dianova II, Dianov GL. Poly(ADP-ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta Biochim Polym. 2003;50:169–179. [PubMed] [Google Scholar]
- 6.Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson MK, Jacobson EL. Discovering new ADP-ribose polymer cycles: protecting the genome and more. Trends Biochem Sci. 1999;24:415–417. doi: 10.1016/s0968-0004(99)01481-4. [DOI] [PubMed] [Google Scholar]
- 8.Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18–25. doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Johansson M. A human poly(ADP-ribose) polymerase gene family (ADPRTL): cDNA cloning of two novel poly(ADP-ribose) polymerase homologues. Genomics. 1999;57:442–445. doi: 10.1006/geno.1999.5799. [DOI] [PubMed] [Google Scholar]
- 10.Lehtio L, Jemth AS, Collins R, Loseva O, Johansson A, Markova N, Hammarstrom M, Flores A, Holmberg-Schiavone L, Weigelt J, Helleday T, Schuler H, Karlberg T. Structural basis forinhibitor specificity in human poly(ADP-ribose) polymerase-3. J Med Chem. 2009;52:3108–3111. doi: 10.1021/jm900052j. [DOI] [PubMed] [Google Scholar]
- 11.Ruf A, Mennissier de Murcia J, de Murcia G, Schulz GE. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc Natl Acad Sci USA. 1996;93:7481–7485. doi: 10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver AW, Ame JC, Roe SM, Good V, de Murcia G, Pearl LH. Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res. 2004;32:456–464. doi: 10.1093/nar/gkh215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC, Apiou F, Vonesch JL, Kock M, Bornens M, De Murcia G. PARP-3 localizes preferentially to thedaughter centriole and interferes with the G1/S cell cycle progression. J Cell Sci. 2003;116:1551–1562. doi: 10.1242/jcs.00341. [DOI] [PubMed] [Google Scholar]
- 14.Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–7775. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouleau M, McDonald D, Gagne P, Ouellet ME, Droit A, Hunter JM, Dutertre S, Prigent C, Hendzel MJ, Poirier GG. PARP-3 associates with polycomb group bodies and with components of the DNA damage repair machinery. J Cell Biochem. 2007;100:385–401. doi: 10.1002/jcb.21051. [DOI] [PubMed] [Google Scholar]
- 18.Fenton AL, Shirodkar P, Macrae CJ, Meng L, Koch CA. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res. 2013;41:4080–4092. doi: 10.1093/nar/gkt134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boehler C, Gauthier LR, Mortusewicz O, Biard DS, Saliou JM, Bresson A, Sanglier-Cianferani S, Smith S, Schreiber V, Boussin F, Dantzer F. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci USA. 2011;108:2783–2788. doi: 10.1073/pnas.1016574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck C, Boehler C, Guirouilh Barbat J, Bonnet ME, Illuzzi G, Ronde P, Gauthier LR, Magroun N, Rajendran A, Lopez BS, Scully R, Boussin FD, Schreiber V, Dantzer F. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014;42:5616–5632. doi: 10.1093/nar/gku174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 22.Zhang K, Yau PM, Chandrasekhar B, New R, Kondrat R, Imai BS, Bradbury ME. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: an application for determining lysine 9 acetylation and methylation of histone H3. Proteomics. 2004;4:1–10. doi: 10.1002/pmic.200300503. [DOI] [PubMed] [Google Scholar]
- 23.Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, Grauslund M, Hansen AM, Jensen ON. Quantitative proteomic analysis of post-translational modifications of human histones. Mol Cell Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Loseva O, Jemth AS, Bryant HE, Schuler H, Lehtio L, Karlberg T, Helleday T. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J Biol Chem. 2010;285:8054–8060. doi: 10.1074/jbc.M109.077834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y. Identification of novel α-N-methylation of CENP-B thatregulates its binding to the centromeric DNA. J Proteome Res. 2013;12:4167–4175. doi: 10.1021/pr400498y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tooley CE, Petkowski JJ, Muratore-Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG. NRMT is an α-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature. 2010;466:1125–1128. doi: 10.1038/nature09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- 28.Chen T, Muratore TL, Schaner-Tooley CE, Shabanowitz J, Hunt DF, Macara IG. N-terminal α-methylation of RCC1 is necessary for stable chromatin association normal mitosis. Nat Cell Biol. 2007;9:596–603. doi: 10.1038/ncb1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petkowski JJ, Schaner Tooley CE, Anderson LC, Shumilin IA, Balsbaugh JL, Shabanowitz J, Hunt DF, Minor W, Macara IG. Substrate specificity of mammalian N-terminal α-amino methyltransferase NRMT. Biochemistry. 2012;51:5942–5950. doi: 10.1021/bi300278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai PJ, Bai DL, Russell DH, Macara IG, Shabanowitz J, Hunt DF, Black BE, Foltz DR. Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proc Natl Acad Sci USA. 2013;110:11827–11832. doi: 10.1073/pnas.1300325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graf L, Jancso A, Szilagyi L, Hegyi G, Pinter K, Naray-Szabo G, Hepp J, Medzihradszky K, Rutter WJ. Electrostatic complementarity within the substrate-binding pocket of trypsin. Proc Natl Acad Sci USA. 1988;85:4961–4965. doi: 10.1073/pnas.85.14.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Previs MJ, VanBuren P, Begin KJ, Vigoreaux JO, LeWinter MM, Matthews DE. Quantification of protein phosphorylation by liquid chromatography-mass spectrometry. Anal Chem. 2008;80:5864–5872. doi: 10.1021/ac800337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishidate T, Elewa A, Kim S, Mello CC, Shirayama M. Divide and differentiate: CDK/Cyclins and the art of development. Cell Cycle. 2014;13:1384–1391. doi: 10.4161/cc.28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–324. [PubMed] [Google Scholar]
- 35.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. Cyclin-dependent kinase 2 phosphorylates S/T-P sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol. 2012;86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- 39.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. γH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 40.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 41.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stock A, Clarke S, Clarke C, Stock J. N-terminal methylation of proteins: structure, function and specificity. FEBS Lett. 1987;220:8–14. doi: 10.1016/0014-5793(87)80866-9. [DOI] [PubMed] [Google Scholar]
- 43.Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE. NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem J. 2013;456:453–462. doi: 10.1042/BJ20131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: The sequence coverage of PARP3 obtained from LC–MS/MS analysis.
Figure S2: MS/MS of the [M + 3H]3+ ion of the unmodified N-terminal peptide of PARP3.
Figure S3: PARP3-K4Q mutant abrogates α-N-methylation in human cells.
Figure S4: Relative mRNA level of NRMT by real-time PCR using GAPDH as control.
Figure S5: MS/MS of the [M + 2H]2+ ion of the unmodified peptide containing residues 461–462 of PARP3.
Figure S6: Effect of NCS treatment on the level of α-N-methylation of PARP3.
Table S1: The primers used in this study.
Table S2: The parameters for dynamic exclusion analysis.