

Abstract
Osteoarthritis (OA) is a highly prevalent, disabling joint disease with no existing therapies to slow or halt its progression. Cartilage degeneration hallmarks OA pathogenesis, and pannexin 3 (Panx3), a member of a novel family of channel proteins, is upregulated during this process. The function of Panx3 remains poorly understood, but we consistently observed a strong increase in Panx3 immunostaining in OA lesions in both mice and humans. Here, we developed and characterized the first global and conditional Panx3 knockout mice to investigate the role of Panx3 in OA. Interestingly, global Panx3 deletion produced no overt phenotype and had no obvious effect on early skeletal development. Mice lacking Panx3 specifically in the cartilage and global Panx3 knockout mice were markedly resistant to the development of OA following destabilization of medial meniscus surgery. These data indicate a specific catabolic role of Panx3 in articular cartilage and identify Panx3 as a potential therapeutic target for OA. Lastly, while Panx1 has been linked to over a dozen human pathologies, this is the first in vivo evidence for a role of Panx3 in disease.
Keywords: DMM, Cartilage, Mice, Pannexin 3, Osteoarthritis
Introduction
Osteoarthritis (OA) is one of the most disabling conditions worldwide, affecting an estimated 250 million people [1]. OA is associated with excess morbidity and mortality in affected patients and is, therefore, responsible for a tremendous socioeconomic burden [2–4]. Our ability to effectively treat this disease is hampered by a limited understanding of the molecular mechanisms that cause it, resulting in a lack of therapeutic strategies to modify progressive joint damage characteristic of OA. Current practices for OA treatment are limited to symptomatic management [5]. The end stage approach to unmanageable OA is joint replacement surgery, which is not a permanent solution, highlighting a need for more effective alternatives [6].
OA is hallmarked by a gradual destruction of the articular cartilage, but involves pathological changes in all joint tissues. Common features include subchondral sclerosis, synovial inflammation/hyperplasia, and osteophyte formation, which together cause severe pain leading to loss of joint function [7]. The onset of cartilage degeneration is triggered by an imbalance between anabolic and catabolic processes, resulting in gradual joint destruction [8]. Several factors, including small molecules like ATP and pro-inflammatory cytokines such as interleukin 1 beta (IL-1β), have been shown to activate catabolic processes in chondrocytes [9]. However, the factors stimulating their production and release in chondrocytes during the early stages of OA remain poorly understood. A previous study in our lab used microarray analysis of cartilage obtained in the early stages of surgically induced OA in rats to identify such mechanisms [10]. Panx3, a gene also upregulated in the prehypertrophic and hypertrophic zone of the murine growth plate, was observed to be increased fourfold in OA cartilage compared to controls [10, 11].
Panx3 encodes the protein pannexin 3 (Panx3), which, like the other two pannexin family members (Panx1 and Panx2), is a channel forming glycoprotein [12]. The pannexins were discovered in 2000 by Panchin and colleagues [13] and have since been implicated in a diverse array of normal and pathophysiological processes [14–16]. In chondrocytes, in vitro studies demonstrated that Panx3-mediated ATP release accelerated hypertrophic differentiation, a process critical for skeletal development [17]. Normally restricted to the transient cartilage of the growth plate, this catabolic process is recapitulated in OA and is likely an important factor stimulating cartilage breakdown [18, 19]. Chondrocyte hypertrophy drives cartilage resorption in preparation for bone deposition and is, therefore, paired with increased catabolic enzyme expression, most notably MMP13 [20]. Additionally, Panx3 is a target of the master hypertrophic regulator RUNX2, a transcription factor that also drives increased MMP13 expression [21]. Combined, this evidence suggests a role for Panx3 in driving ectopic chondrocyte hypertrophy in OA. Thus, we hypothesized that loss of Panx3 will delay the onset or progression of this disease.
In this study, we examined Panx3 expression and localization in both murine and human OA and explored the in vivo role of Panx3 by creating the first Panx3 knockout mice. We used these novel mouse lines to investigate the effects of global (Panx3−/−) and cartilage-specific (Panx3fl/fl:Col2cre) Panx3 deletion on skeletal development and osteoarthritis.
Materials and Methods
Generation of Panx3 Knockout Mice
The Panx3fl/fl strain used in this study was generated from an embryonic cell line (JM8A3.N1 Agouti (A/a), C57BL/6N), obtained from the NCRR-NIH supported KOMP Repository (www.komp.org) and generated by the Wellcome Trust Sanger Institute and the Mouse Biology Program (www.mousebiology.org) at the University of California Davis. Clone EPD0670_4_C10 for allele Panx3tm1a (KOMP)Wtsi of targeting project CSD24494, was engineered as a “Knockout First” (promoter driven) cassette using targeting vector PG00057_Y_A09_2.
Panx3fl/fl mice were generated through blastocyst injection into a C57BL/6N pseudo-pregnant female by the London Regional Transgenic and Gene Targeting Facility at the University of Western Ontario. Chimeric founders were mated with C57BL/6N mice and offspring showing germline transmission of the targeting allele were mated to C57BL/6J FLP deleter mice (B6(C3)-Tg(Pgk1-FLPo)10Sykr/J; Jaxmice #011065 (Jackson Laboratories, Bar Harbour, ME, USA) for removal of the FRT flanked selection cassette. Litters of the floxed Panx3 (Panx3fl/fl) mice were first crossed with C57BL/6J Cre deleter mice (B6.C-Tg(CMV-cre)1Cgn/J; Jaxmice # 006054) for germline deletion of the targeted exon. Offspring mice were PCR genotyped to confirm the hemizygous deletion of Panx3 exon 2. Mice hemizygous for the deleted exon were further crossed to produce homozygous null mice. Mating Panx3fl/fl mice with CMV-cre mice generated global Panx3−/− mice. Cre recombinase was out-bred following the generation of the Panx3−/− line. Chondrocyte specific Panx3-deficient mice were generated through mating Panx3fl/fl mice with C57BL/6J Col2a1-Cre mice [22–25].
Validation of Panx3 Ablation
PCR genotyping was performed to verify the lack of a Panx3 exon2 amplicon in Panx3 null mice (Panx3−/−) using an upper primer annealing to the intron just upstream of exon 2 in the floxed allele and the lower primer annealing to an exon 2 sequence. RT-PCR was performed on total RNA from mouse cartilage (extracted with a RNeasy kit; QIAGEN, Venlo, Limburg, Netherlands), with primers designed to amplify a product bridging the intron between exons 3 and 4 of Panx3, to verify that these exons are also not transcribed in the null mice. Western blots of protein lysates from 3-week-old wild type and null mice cartilage (from outer ears or tail tips) were performed as described earlier [26, 27] and probed with the anti-Panx3 antibody (CT-379), also previously described [27]. β-tubulin was used as loading control.
Animals and Surgery
All animal experiments were approved by the Animal Use Subcommittee at the University of Western Ontario and conducted in accordance with guidelines from the Canadian Council on Animal Care. Experimental OA was induced at 20 weeks of age by performing DMM surgery on the left hind limb of male Panx3fl/fl (control) or Panx3fl/fl:Col2cre+/− mice as described [28–30]. Sham-operated animals were used as controls. 18 total mice were used for the surgical trial, nine Panx3fl/fl (control; 5 DMM and 4 Sham) and nine Panx3fl/fl:-Col2cre (5 DMM and 4 Sham). Littermates were used within each treatment group (DMM or Sham) to control for any variability associated with differing genetic backgrounds. All mice were sacrificed 8 weeks post-surgery by CO2 asphyxiation, and both operated and unoperated (right) limbs were harvested. The same protocol was used to assess OA progression following DMM surgery in control (WT) and globally deficient Panx3−/− mice (N=3).
Analysis of Skeletal Development
Newborn (P0) and 21 day old (P21) Panx3−/− and control mice were sacrificed using intraperitoneal Euthanyl (pentobarbital) injections (Animal Health Inc. Cambridge, ON, Canada). Alcian Blue/Alizarin Red was used to stain whole skeletons of 21-day-old mice as described [22, 23] (Sigma, St. Louis, MO, USA). Long bones (tibia, femur, and humerus) were isolated at P0 and P21, fixed in 4 % paraformaldehyde (PFA), decalcified in ethylenediaminetetraacetic acid (EDTA; Bioshop, Burlington, ON, Canada), and embedded in paraffin wax. Safranin-O/Fast Green was used to stain paraffin sections cut down the length of the long bones. Additionally, P0 tibia and femur growth plates and P21 tibia, humerus, and femur lengths were measured. Three wild-type and 3 Panx3 null mice from each time point (P0 or P21) were used for each trial described above.
Histopathology of the Knee
Knees harvested 8 weeks following surgeries were fixed in 4 % PFA (Sigma), decalcified in EDTA, and embedded in paraffin wax. Frontal sections were cut by the Molecular Pathology Facility and stained with Safranin-O/Fast Green (Sigma/VWR, Radnor, PA, USA) as previously described [31]. Ten sections through the entire joint were scored according to the OARSI histopathology scoring system by three blinded observers [32]. Scores from 0 to 6 were assigned to the medial and lateral tibia plateaus and femoral condyles based on depth and width of lesion. A score of 0 corresponds to healthy cartilage and is what is commonly seen in the joints of sham-operated animals. Scores ranging from 0.5 to 6 reflect increasing severity of cartilage degeneration. Focal aggrecan loss, surface fibrillations, and cracks without cartilage loss are assigned scores of 0.5, 1, or 2, respectively. Varying degrees of cartilage erosion ranging from less than 25 % to greater than 75 % are represented by scores from 3 to 6. Individual scores are averaged across observers and summed for each sample. These are then used to determine quadrant, compartment, and whole joint scores. These scores are than compared between Panx3fl/fl and Panx3fl/fl:Col2-cre+/− mice (N=5/4 (DMM/sham)) or WT and Panx3−/− mice (N=3).
Immunohistochemistry
Frontal sections of paraffin embedded knees and long bones were used for immunohistochemical analysis. Sections were dewaxed and re-hydrated as previously described [25, 31, 30]. Custom-made site-directed anti-Panx1 (CT-395) and anti-Panx3 (CT-379) antibodies (reported earlier by [27]) targeting mouse pannexin sequences and commercial anti-MMP13 (Protein Tech, Chicago, IL, USA) and anti-Collagen II (Fitzgerald, Acton, MA, USA) primary antibodies were used for immunolabeling. HRP-conjugated goat anti-rabbit secondary antibodies (Biorad, Hercules, CA, USA) were used with diaminobenzidine substrate (Sigma) to reveal immunopositive staining. Methyl Green (Sigma) was used to counterstain sections.
Human Cartilage
Knee OA cartilage samples were obtained from two patients diagnosed with advanced OA (patients were both female, 60 and 62 years of age) and undergoing knee replacement surgery under the guidelines of the Institutional Review Board of New York University (NYU) School of Medicine protocol #H9018 for use of surgically discarded human tissues. Regions of the tibial plateau and femoral condyle showing signs of cartilage degeneration along with regions showing no macroscopic signs of damage as internal controls was extracted from two female patients aged 60 and 62. Tissues were decalcified for 48 h in Formical (Decal Chemical Corp, Lodi, CA, USA) and subsequently embedded in Paraffin wax. Sections of tissue were stained with Safranin-O/Fast green, to assess proteoglycan content and immunostained with anti-Panx3 (CT379) antibody. Representative sections from lesion and non-lesion control tissue are presented; N=2.
Measurement of Osteophyte Size
To determine the area of osteophytes observed in DMM-operated control animals, three sections at similar depths spanning the medial anterior compartment of the joint were selected from each animal and imaged at ×200 magnification. Using the Leica Application Suite software, the margins of the osteophyte were traced and the area was determined using the software represented in square microns (mm2).
Statistical Analysis
For comparisons of bone lengths, growth plates and OARSI scores between control and Panx3−/− DMM-operated animals, two-tailed student t tests were employed. Two-way analysis of variance (ANOVA) was used to compare all four groups from cartilage specific Panx3 deficient in vivo surgical trial with Bonferroni post hoc analysis. All statistical analyses were performed using GraphPad Prism (Graphpad Software Inc. La Jolla, CA, USA) v.6.0. N≥3 for all animal experiments.
Results
Generation of Panx3fl/fl mice for global Panx3 ablation
Using Panx3-targeted embryonic cells (ES) with a C57BL/6N origin from the Knockout Mouse Project repository (Fig. 1a), we generated a genetically-modified mouse line using the Cre/loxP recombinase system for constitutive and tissue-specific deletion of Panx3. We first generated ubiquitous KO mice by breeding Panx3fl/fl mice to a CMV-Cre driver strain [33]. Homozygous null mice were identified by PCR genotyping (Fig. 1b) and later confirmed by RT-PCR (Fig. 1c) and Western blotting (Fig. 1d) to lack Panx3 expression in tissues rich in cartilage or bone, such as the outer ear and the tail, where Panx3 is abundant in the wild type control. Panx3−/− mice were found to be viable and fertile. However, litter sizes of Panx3+/− × Panx3+/− crosses as well as Panx3−/− × Panx3−/− crosses were significantly smaller than those of Panx3+/+ × Panx3+/+controls (Fig. 1e). Furthermore, Panx3+/− × Panx3+/− crosses failed to generate the predicted number of Panx3−/− offspring based on a normal Mendelian ratio (~25 %), and instead, produced only 8 % of Panx3−/− pups (Fig. 1f). Body weights of viable Panx3−/− males at 12 weeks of age did not show any significant differences compared to wild-type controls (Fig. 1G), and the overall appearance of 4-week-old and 8-month-old Panx3−/− mice was similar to the controls with no overt phenotypes (Fig. 1h, i).
Fig. 1.
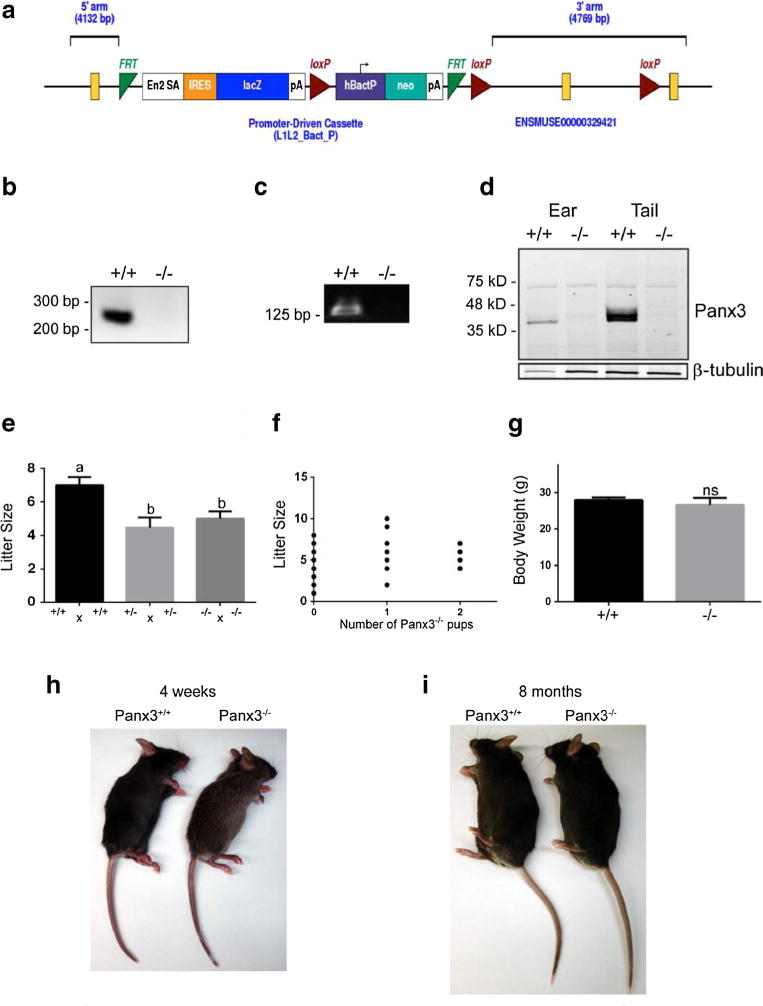
Generation of Panx3−/− mice. a Panx3 targeting vector from KOMP project ID CSD24494. The “Knockout-First” reporter tagged insertion promoter-driven cassette targets the second exon of Panx3, creating a truncated protein product. Source: www.knockoutmouse.org. b PCR genotyping showing lack of Panx3 exon2 amplicon in Panx3 null mice (Panx3−/−). Upper primer is in the intron upstream of exon 2 in the floxed allele; the lower primer is in exon 2. PCR produces a 232-bp in the wild type (Panx3+/+), but not in the null. c RT-PCR of mRNA from the mouse cartilage, using primers designed to amplify a 128-bp product bridging the intron between exons 3 and 4 of Panx3, revealed no amplification in the null mouse. d Western blots of protein lysates from wild type and Panx3−/− mouse cartilage confirmed the complete absence of Panx3 protein in the Panx3−/− mice. β-tubulin was used as loading control. e Litter size was significantly reduced in Panx3+/− × Panx3+/− crosses as well as Panx3−/− × Panx3−/− crosses compared to Panx3+/+ × Panx3+/+ crosses (N=13 litters, P<0.05; one-way ANOVA followed by Tukey test). weight of a 3-month-old Panx3−/− male mice was not significantly different (ns) to Panx3+/+ controls (N=10, t test, P>0.05). A 4-week-old (h) and an 8-month-old males examined (i) showed no overt morphological phenotypes when compared to Panx3+/+ controls.
Global Loss of Panx3 Has no Major Effect on Skeletal Development
Current evidence shows that Panx3 is an important regulator of hypertrophic differentiation of chondrocytes [17]. Due to the importance of this process during skeletal development, we investigated the effects of global Panx3 loss on skeletal development. Growth plates of Panx3−/− mice were compared to wild-type (WT) C57BL/6 mice at P0 and P21. There were no major abnormalities in growth plate morphology and organization in Panx3−/− mice compared to WT (Fig. 2a). This is complemented by the absence of differences in the length of the growth plate and its individual zones (Fig. 2b, c). At P0 and P21, Panx3 immunostaining appears concentrated in the pre-hypertrophic zone in WT mice, but this staining is absent in Panx3 null growth plates, confirming its deletion (Fig. 2a). Whole skeletal preparations of P21 WT and Panx3−/− mice stained with Alizarin Red and Alcian Blue revealed no gross skeletal abnormalities in Panx3−/− mice (Fig. 2d). Measurements of humerus, femur, and tibia lengths confirmed that there were no differences in size between WT and Panx3−/− mice at this stage (Fig. 2d). Skeletal morphology (data not shown) and long bone lengths (Fig. 2d) remain similar between WT and null mice at 6 weeks of age.
Fig. 2.
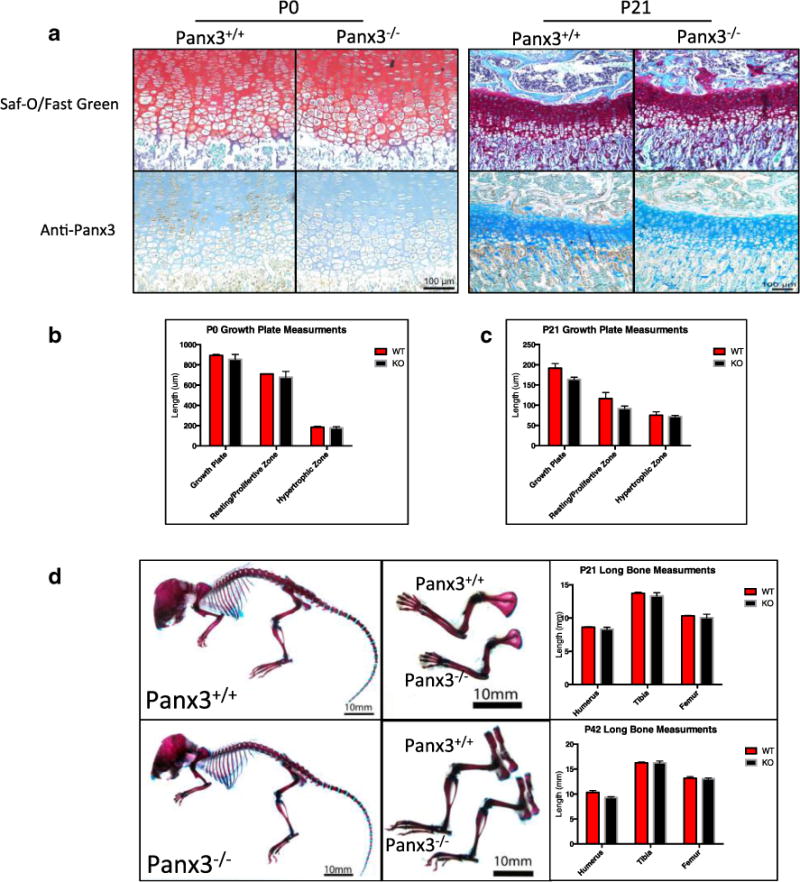
Global loss of Panx3 has no effect on the development of the murine skeleton. Newborn (P0), 21 day old (P21), and 42 day old (P42) wild-type (WT) and Panx3 null mice were examined for signs of abnormal skeletal development. a Representative images of Safranin-O/Fast Green stained sections of WT and Panx3−/− tibia growth plates at P0 and P21. b, c Measurements of total growth plate, resting/proliferative zone, and hypertrophic zone length at P0 and P21, respectively, show no significant size differences (N=3 WT-null pairs, P<0.05; two-tailed t test). d Photographs of Alizarin Red/Alcian Blue-stained whole skeletons (left) and upper (top middle) and lower (bottom middle) limbs of 21-day-old mice. Long bone measurements of P21 (top right) and P42 (bottom right) long bones show no size differences between WT and Panx3 null mice (N=3 WT-null pairs for each time point, P<0.05; two-tailed t test). Results show mean ± SEM.
Panx3 and MMP13 Immunostaining is Increased in Areas of Cartilage Degeneration in Mice and Humans
Immunostaining for Panx3 and MMP13 was done on paraffin sections obtained from 12-week-old male WT C57BL/6 mice that either underwent destabilization of medial meniscus (DMM) surgery to induce OA [28, 29] or sham surgery. At 5 weeks post-surgery, mice displayed mild OA. Histologically, this appeared as loss of proteoglycan staining in Safranin-O/Fast Green staining and as delamination of superficial cartilage from the underlying calcified matrix (Fig. 3a). Strong Panx3 staining was localized in areas of mild cartilage degeneration. Similarly, immunostaining for MMP13 on serial sections of lesions revealed strong staining in the same areas of cartilage degeneration. Cartilage from sham-operated animals appeared healthy and did not stain strongly for either Panx3 or MMP13 (Fig. 3a).
Fig. 3.
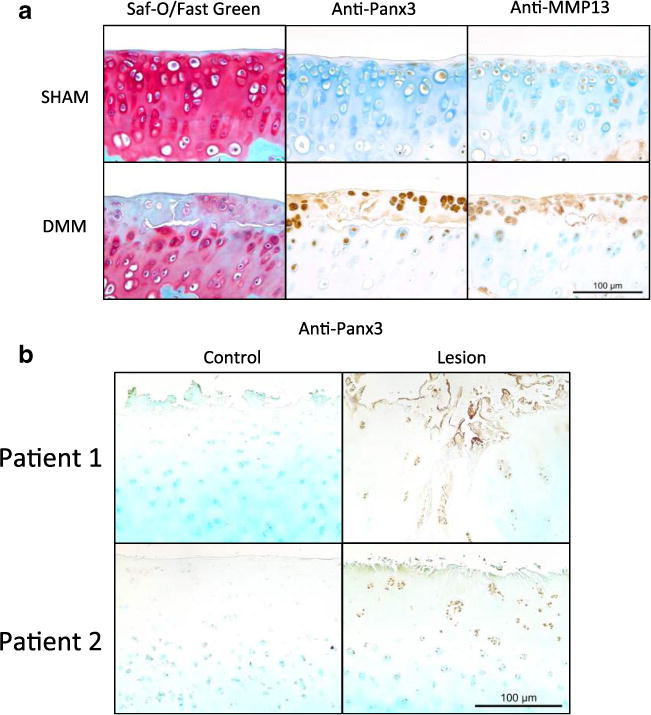
Panx3 and MMP13 are seen in chondrocyte clusters in early cartilage lesions after DMM surgery in mice and in human osteoarthritic cartilage. a Sham and DMM operated joints were stained with Safranin-O/Fast Green (left). Serial sections were immunostained using anti-Panx3 (middle) and anti-MMP13 (right) antibodies. b Cartilage isolated from lesioned and non-lesioned areas of human knee joints were immunostained for Panx3.
In addition to being localized to cartilage lesions in murine OA, Panx3 was also detected in human osteoarthritic cartilage (Fig. 3b). Tissue was obtained from both lesioned and non-weight-bearing (control) regions of the knee from patients undergoing joint replacement surgeries. In areas showing histological features of OA such as decreased proteoglycan staining, chondrocyte clustering, and cartilage surface degeneration, we observed an increased in Panx3 immunostaining compared to control regions.
Cartilage-Specific Deletion of Panx3 Protects Against Osteoarthritis Development In Vivo
To assess the importance of Panx3 in OA in vivo, we generated cartilage-specific Panx3 KO mice to specifically address its role in chondrocytes in a model of post-traumatic OA. In agreement with the data from Panx3−/− mice, these mice did not show any defects in skeletal development and growth (data not shown). We performed DMM surgery to induce OA in a 20-week-old control (Panx3fl/fl) and Panx3fl/fl:-Col2cre mice that are deficient in Panx3 specifically in chondrocytes. Paraffin sections of knees isolated 8 weeks post-surgery were stained with Safranin-O/Fast Green and scored according to the OARSI histopathology scoring system [32]. Control mice developed cartilage degeneration as expected. Histologically, advanced cartilage degeneration and osteophyte formation was seen in DMM-operated control but not in Panx3fl/fl:Col2-cre mice, which showed at worst minor proteoglycan loss (Fig. 4a, b). Whole joint scores confirmed significantly reduced OA severity in Panx3fl/fl:Col2-cre (Fig. 4e). The medial compartment is most severely affected in DMM surgery and scores from medial tibia plateaus and medial femoral condyles are significantly lower in Panx3fl/fl:-Col2-cre mice (Fig. 4a). Medial compartment (Fig. 4c, d) and whole joint scores (Fig. 4e) from sham-operated mice of either genotype revealed no damage. This was complemented by the absence of histological signs of cartilage degeneration in Safranin-O/Fast Green stained sections (Fig. 4a, b).
Fig. 4.
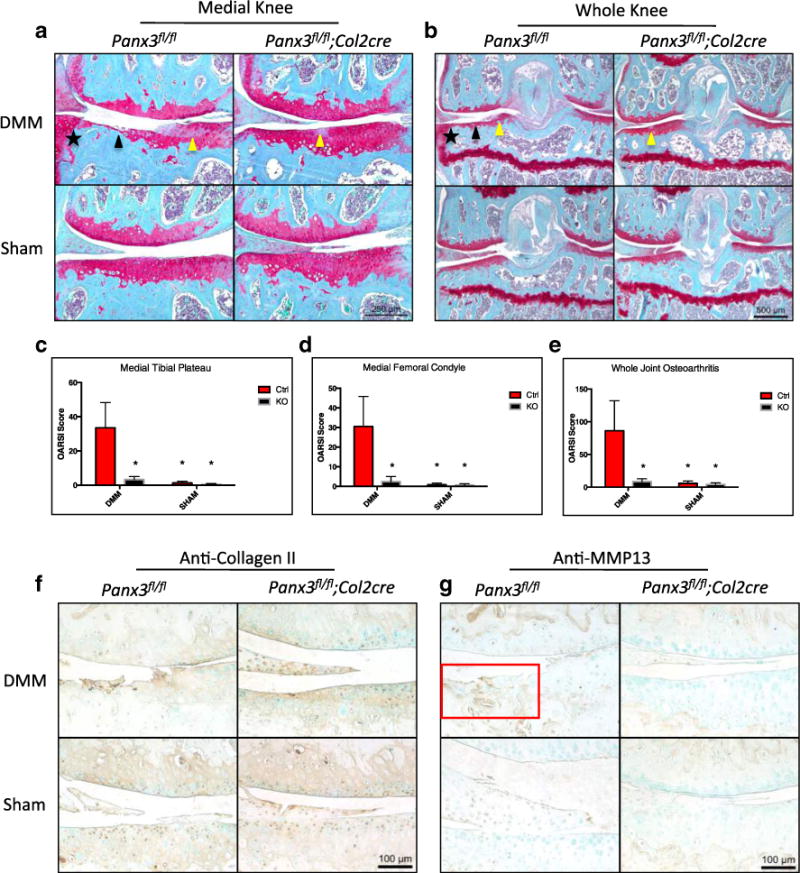
Chondrocyte specific loss of Panx3 protects against the development of surgically induced OA in mice. Representative images of Safranin-O/Fast Green-stained sections of Panx3fl/fl and Panx3fl/fl; Col2cre+/− DMM and Sham operated knees 8 weeks post-surgery (a, b). Black arrows indicate cartilage erosion. Yellow arrows indicate proteoglycan loss. Black stars indicate osteophyte formation. Medial knee and whole joint images were acquired. Cumulative OARSI scores of the medial tibia plateau (c), medial femoral condyle (d), and entire joint (e) are shown. Asterisk indicate means significantly different from Ctrl DMM scores (N=5, 4 DMM, sham per genotype; P<0.05, two-way ANOVA followed by Tukey’s test. Expressed as mean ± SEM. Serial sections from sham and DMM-operated joints harvested from Panx3f/f and Panx3f/f:Col2cre mice were immunostained with anti-collagen II and anti-MMP13 antibodies (f, g). Counter staining was done using methyl green. The red box highlights increased MMP13 immunolabeling within degenerated cartilage.
Type II Collagen Staining is Decreased, While MMP13 Staining is Increased in DMM-Operated Panx3f/f Mice
Type II collagen is a critical matrix molecule in articular cartilage and its destruction marks the onset of terminal OA. As expected, matrix and pericellular type II collagen staining appears reduced in DMM-operated Panx3−/− mice. In contrast, strong matrix and pericellular type II collagen staining is apparent in DMM-operated Panx3f/f:Col2cre mice and in sham-operated controls from each genotype (Fig. 4f). Immunostaining of MMP13, the main collagenase associated with type II collagen destruction in OA, is increased in the remaining cartilage in DMM-operated Panx3f/f mice (Fig. 4g). In contrast, DMM-operated Panx3f/f:Col2cre and sham-operated controls of either genotype reveal minimal MMP13 staining below the calcified cartilage, but none in the articular cartilage (Fig. 4g).
Global Panx3 Deletion Protects Against the Development of Surgically Induced OA
As our data show, cartilage-specific Panx3 deletion protects against cartilage destruction and OA following DMM surgery. Because of the ubiquitous expression of Panx3 in joint tissues (most notably cartilage and bone), we next examined the effects of global Panx3 deletion on the OA development following DMM surgery. We performed DMM surgery on 20-week-old control (WT) or Panx3−/− mice. Paraffin sections of knees isolated 8 weeks post-surgery were stained with Safranin-O/Fast Green and scored according to the OARSI histopathology scoring system. Significant joint damage was detected in DMM-operated WT control mice similar to that seen in DMM-operated Panx3f/f mice. In contrast, some proteoglycan loss and minimal cartilage degeneration was observed in DMM-operated Panx3−/− mice (Fig. 5a, b). Medial tibia and whole joint scores reveal statistically significant differences in the magnitude of cartilage damage seen in DMM-operated WT compared to Panx3−/− mice (Fig. 5c, e). Although there are noticeable differences in cartilage degeneration on the femoral condyles of DMM-operated WT and Panx3−/− mice, these are not significantly different (Fig. 5d).
Fig. 5.
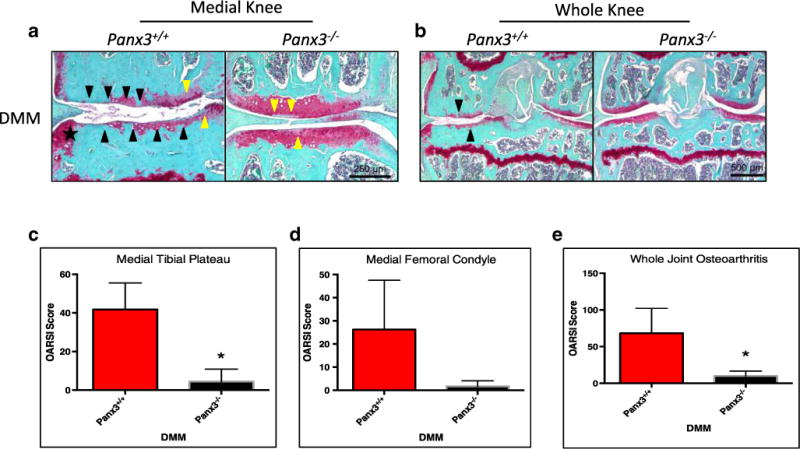
Global loss of Panx3 protects against the development of surgically induced OA. Representative images of Safranin-O/Fast Green stained sections of WT and Panx3−/− DMM-operated knees 8 weeks post-surgery (a, b). Black arrows indicate cartilage erosion, while yellow arrows denote proteoglycan loss. The black star indicates the location of an osteophyte. Cumulative OARSI scores of the medial tibia plateau (c), medial femoral condyle (d), and entire joint (e) are shown. Asterisk indicates significantly different from WT DMM scores (N=3, P<0.05; two-tailed t test). Expressed as mean ± SEM.
Analysis of Osteophyte Size and Pannexin Staining in DMM-Operated Panx3f/f Mice
In addition to extensive cartilage loss, all five DMM-operated Panx3f/f mice showed extensive osteophyte formation in the medial margins of the joint. Using three slides from each animal, the average osteophyte area was calculated to be 0.138±0.0156 mm2. No osteophytes were detected in DMM-operated Panx3f/f/:Col2cre mice or sham-operated animals of either genotype, thus prohibiting further analysis or comparisons. Immunohistochemical analysis of Panx1 and Panx3 in osteophytes revealed strong immunoreactivity in osteophytes following DMM surgery (Fig. 6a, b). Immunostaining with MMP13 antibodies revealed similar localization in these osteophytes as well (Fig. 6c). Additionally, since Panx3fl/fl: Col2cre mice did not develop such osteophytes, it was not possible to examine whether Panx1 is upregulated in a compensatory manner. Additionally, because Col2cre-mediated Panx3 deletion is not 100 % penetrant, there is low intensity Panx3 immunoreactivity throughout the cartilage of Panx3-fl/fl:Col2cre mice. Regardless, it was apparent that Panx1 and the minimal amount of remaining Panx3 were unable to drive cartilage degeneration and OA in the surgically challenged Panx3fl/fl:Col2cre mice.
Fig. 6.
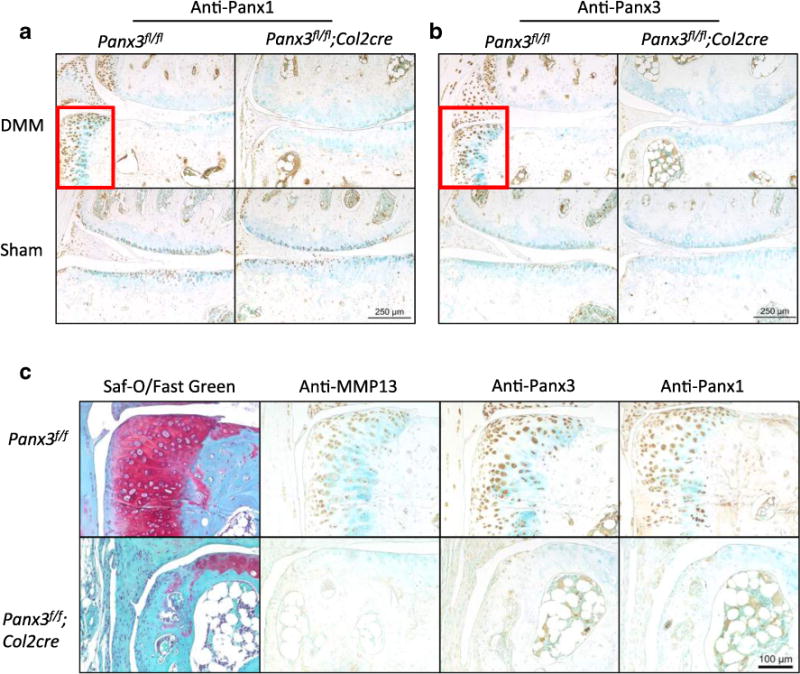
Extensive osteophyte formation is detected in all DMM operated Panx3f/f/ mice and is associated with increased MMP13, Panx1, and Panx3 staining. Serial sections from DMM-operated joints harvested from Panx3fl/f and Panx3fl/fl:Col2cre mice were immunostained with anti-Panx1 (a, c), anti-Panx3 (b, c), or anti-MMP13 (c) antibodies. Counterstaining was done using methyl green. Osteophyte formation was only detected in Panx3fl/f DMM-operated knees.
Discussion
The pannexins are a unique three-member family of channel-forming glycoproteins. The most extensively studied and ubiquitously expressed member, Panx1, has been implicated in a diverse array of physiological and pathological processes [34–36, 15]. Recently, there has been increasing interest in elucidating the roles of Panx2 and Panx3, both of which show more restricted expression patterns [12]. Despite newer evidence suggesting a more ubiquitous expression pattern for Panx2 than previously anticipated [37], it does not appear to be expressed in bone or cartilage. Panx3, however, is expressed in prehypertrophic chondrocytes, acts to accelerate their terminal differentiation through increased ATP release [11, 17], and is upregulated in OA [10]. With the aim to further examine these roles of Panx3 in vivo, we generated Panx3fl/fl mice and investigated the consequence of global and cartilage-specific deletion on skeletal development and OA.
Using the Cre/loxP recombinase system, we targeted the second exon of Panx3. Exon 2 contains 143 coding nucleotides that encode ~47 amino acids corresponding to the first extracellular loop of mouse Panx3. Removal of the second exon from the gene destabilizes and ablates the expression of the entire Panx3 mRNA and protein, as we confirmed by RT-PCR and Western blot, respectively. To our knowledge, this is the first Panx3 null mouse model to be reported in the literature. Panx3−/− mice were viable and fertile with no overt phenotypes to report. However, due to the less-than-Mendelian ratio of Panx3−/− offspring, we hypothesize a higher intra-uterine death rate in Panx3−/− mice. At this moment, the reason for the apparent embryonic death of KO mice is unknown.
In this study, we show that global deletion of Panx3 does not affect gross skeletal development at P0, P21, or P42. However, because we only examined gross skeletal morphology, detailed morphometric analyses are required to determine whether subtle changes in bone morphology occur in Panx3−/− mice. Growth plate morphology is unchanged in Panx3 null mice at P0 or P21. These findings are somewhat surprising given previous in vitro data which show that siRNA-mediated Panx3 knock down delays hypertrophic differentiation of chondrogenic ATDC5 cells [17]. We suspect that in Panx3−/− mice, this is likely due to the ubiquitous expression of Panx1 throughout the murine growth plate in both WT and Panx3 null long bones at P0, P21, and P42 (Supplemental Fig. 1). Panx1 and Panx3 share 59 % homology at the amino acid level, which is likely correlated to functionally similar proteins. Therefore, it is likely that these proteins have overlapping and/or redundant roles in cells co-expressing Panx1 and Panx3 such as chondrocytes [12].
However, because we see significant differences between DMM-operated control and Panx3-deficient mice (Panx3−/− and Panx3f/f:Col2cre) in OA progression, there is no evidence of a compensatory effect of Panx1 in articular cartilage under these conditions. In 20-week-old mice that had undergone DMM surgery, animals lacking Panx3 globally or specifically in cartilage showed significant protection against OA development at 8 weeks post-surgery. The substantial cartilage degeneration and joint pathology observed in DMM-operated control mice was contrasted with overall healthy joints in Panx3f/f:Col2cre and Panx3−/− mice, which showed only minor proteoglycan loss and early degenerative changes. Our data, therefore, suggest that Panx3 is an important player in driving OA pathogenesis following joint destabilizing surgery.
Nevertheless, we do see increased Panx1 (and Panx3) staining in osteophytes, which were seen in all five DMM-operated control animals, but not Panx3-deficient animals (Fig. 6). Osteophytes are bony projections extending from the margins of the joint and are commonly seen in OA. A number of different factors ultimately drive their growth and development, including mechanical instability and aberrant cytokine signaling [38]. Patients presenting with these outgrowths commonly experience significant pain and joint dysfunction. Interestingly, the process driving the development and growth of these structures is highly similar to endochondral ossification, and therefore, it was not surprising that both Panx1 and Panx3 were found in abundance in these regions.
Current evidence suggests that this pathological role of Panx3 in OA could be the result of Panx3-mediated hypertrophic-like changes in articular chondrocytes in a mechanically unstable joint, possibly through ATP release. Normally, ATP is released by chondrocytes through multiple mechanisms (likely including Panx1 and Panx3) in response to physiologic mechanical stimulation [39, 40, 34, 41]. This leads to the induction of anabolic genes encoding proteins such as lubricin, aggrecan, and type II collagen, which maintain healthy cartilage and joint function [42]. In contrast, abnormal mechanical loading results in increased ATP release, which has been linked to the induction of catabolic genes such as Mmp13 that are critical in the pathogenesis of OA [43, 44]. Both ATP release and mechanical loading have also shown to increase levels of Runt-related transcription factor 2 (RUNX2), a potent inducer of chondrocyte hypertrophy [45, 46]. Interestingly, putative Runx2 binding sites have been identified in Panx3 promoter sequences, indicating that Runx2 is a potential transcriptional regulator of Panx3 expression [21].
Based on these data we hypothesize that the altered mechanical environment following joint injury leads to the Runx2-mediated induction of Panx3, enhancing ATP release from articular chondrocytes. Combined with the ATP released by other mechanisms, local extracellular ATP concentrations rise. This increase in extracellular ATP can lead to activation of purine receptors (P2Y and P2X) and downstream effectors including ERK1/2. ERK1/2, together with altered mechanical loading, further activates RUNX2 [45], leading to enhanced Panx3 and Mmp13 expression and cartilage matrix breakdown. This begins the vicious cycle that ultimately leads to the complete destruction of the articular cartilage and joint.
Critical to OA progression is the induction of Mmp13 gene expression and increased MMP13 activity. Although other pathways lead to increases in MMP13, our data suggest that increases in Panx3 are correlated with increases in MMP13. DMM surgery in 12-week-old WT mice caused mild cartilage damage 5 weeks post-surgery. Increased Panx3 immunostaining was observed in these cartilage lesions, with overlapping increases in MMP13 immunostaining. Our data also show that chondrocyte-specific Panx3 deletion is associated with reduced MMP13 staining following DMM surgery. Furthermore, this is also associated with increased type II collagen in the articular cartilage. While loss of Panx3 potentially affects other catabolic factors as well (e.g. aggrecanases such as ADAMTS4 or 5), MMP13 is a critical catabolic enzyme in cartilage because it cleaves type II collagen, an essential structural component of the articular cartilage [47, 48]. Ultimately, genetic loss of Panx3, specifically in chondrocytes, protects against the development of surgically induced OA by reducing the chondrocytes ability to pathologically respond to altered mechanical loading. Equally important, loss of Panx3 has no overt consequence on joint development and health. It is likely that Panx1 alone is sufficient to mediate relatively normal skeletal and joint development, and in permanent adult cartilage, Panx3 is not normally found in significant levels, therefore, having minimal effect on normal joint health.
In conclusion, we have successfully generated Panx3 global and conditional mouse lines and used them to investigate the role of Panx3 in OA. This is the first study to show that genetic deletion of Panx3 prevents the development of OA in a mouse model of joint instability-induced OA. We have thus identified Panx3, a novel channel forming glycoprotein found in human osteoarthritic tissue, as a potential target for novel OA therapies.
Supplementary Material
Key message.
Panx3 is localized to cartilage lesions in mice and humans.
Global Panx3 deletion does not result in any developmental abnormalities.
Mice lacking Panx3 are resistant to the development of osteoarthritis.
Panx3 is a novel therapeutic target for the treatment of osteoarthritis.
Acknowledgments
This study was funded by a Canadian Institutes of Health Research operating grant (MOP130530) to DWL, FB, and SP. DWL and FB are supported by Canada Research Chair Awards. SBA and AM are supported in part by U.S. National Institutes of Health grant R01-AR054817.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00109-015-1311-1) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare that they have no competing interest.
Contributor Information
Dale W. Laird, Email: Dale.Laird@schulich.uwo.ca.
Frank Beier, Email: fbeier@uwo.ca.
References
- 1.Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. 2014;10(7):437–441. doi: 10.1038/nrrheum.2014.44. [DOI] [PubMed] [Google Scholar]
- 2.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum. 2008;58(1):15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 3.Nuesch E, Dieppe P, Reichenbach S, Williams S, Iff S, Juni P. All cause and disease specific mortality in patients with knee or hip osteoarthritis: population based cohort study. BMJ. 2011;342:d1165. doi: 10.1136/bmj.d1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II Arthritis Rheum. 2008;58(1):26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J, Towheed T, Welch V, Wells G, Tugwell P, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res. 2012;64(4):465–474. doi: 10.1002/acr.21596. [DOI] [PubMed] [Google Scholar]
- 6.Weinstein AM, Rome BN, Reichmann WM, Collins JE, Burbine SA, Thornhill TS, Wright J, Katz JN, Losina E. Estimating the burden of total knee replacement in the United States. J Bone Joint Surg Am. 2013;95(5):385–392. doi: 10.2106/JBJS.L.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roach HI, Aigner T, Soder S, Haag J, Welkerling H. Pathobiology of osteoarthritis: pathomechanisms and potential therapeutic targets. Curr Drug Targets. 2007;8(2):271–282. doi: 10.2174/138945007779940160. [DOI] [PubMed] [Google Scholar]
- 9.Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: pathobiology-targets and ways for therapeutic intervention. Adv Drug Deliv Rev. 2006;58(2):128–149. doi: 10.1016/j.addr.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007;56(6):1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- 11.James CG, Stanton LA, Agoston H, Ulici V, Underhill TM, Beier F. Genome-wide analyses of gene expression during mouse endochondral ossification. PLoS One. 2010;5(1):e8693. doi: 10.1371/journal.pone.0008693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penuela S, Gehi R, Laird DW. The biochemistry and function of pannexin channels. Biochim Biophys Acta. 2013;1828(1):15–22. doi: 10.1016/j.bbamem.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Panchin Y, Kelmanson I, Matz M, Lukyanov K, Usman N, Lukyanov S. A ubiquitous family of putative gap junction molecules. Curr Biol. 2000;10(13):R473–474. doi: 10.1016/s0960-9822(00)00576-5. [DOI] [PubMed] [Google Scholar]
- 14.Bennett MV, Garre JM, Orellana JA, Bukauskas FF, Nedergaard M, Saez JC. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 2012;1487:3–15. doi: 10.1016/j.brainres.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penuela S, Harland L, Simek J, Laird DW. Pannexin channels and their links to human disease. Biochem J. 2014;461(3):371–381. doi: 10.1042/BJ20140447. [DOI] [PubMed] [Google Scholar]
- 16.Dahl G, Keane RW. Pannexin: from discovery to bedside in 11+/−4 years? Brain Res. 2012;1487:150–159. doi: 10.1016/j.brainres.2012.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwamoto T, Nakamura T, Doyle A, Ishikawa M, de Vega S, Fukumoto S, Yamada Y. Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J Biol Chem. 2010;285(24):18948–18958. doi: 10.1074/jbc.M110.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis and cartilage/OARS. Osteoarthr Res Soc. 2012;20(3):223–232. doi: 10.1016/j.joca.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Pitsillides AA, Beier F. Cartilage biology in osteoarthritis–lessons from developmental biology. Nat Rev Rheumatol. 2011;7(11):654–663. doi: 10.1038/nrrheum.2011.129. [DOI] [PubMed] [Google Scholar]
- 20.D’Angelo M, Yan Z, Nooreyazdan M, Pacifici M, Sarment DS, Billings PC, Leboy PS. MMP-13 is induced during chondrocyte hypertrophy. J Cell Biochem. 2000;77(4):678–693. [PubMed] [Google Scholar]
- 21.Bond SR, Lau A, Penuela S, Sampaio AV, Underhill TM, Laird DW, Naus CC. Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J Bone Miner Resh Off J Am Soc Bone Miner Res. 2011;26(12):2911–2922. doi: 10.1002/jbmr.509. [DOI] [PubMed] [Google Scholar]
- 22.Wang G, Woods A, Agoston H, Ulici V, Glogauer M, Beier F. Genetic ablation of Rac1 in cartilage results in chondrodysplasia. Dev Biol. 2007;306(2):612–623. doi: 10.1016/j.ydbio.2007.03.520. [DOI] [PubMed] [Google Scholar]
- 23.Solomon LA, Li JR, Berube NG, Beier F. Loss of ATRX in chondrocytes has minimal effects on skeletal development. PLoS One. 2009;4(9):e7106. doi: 10.1371/journal.pone.0007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillespie JR, Ulici V, Dupuis H, Higgs A, Dimattia A, Patel S, Woodgett JR, Beier F. Deletion of glycogen synthase kinase-3beta in cartilage results in up-regulation of glycogen synthase kinase-3alpha protein expression. Endocrinology. 2011;152(5):1755–1766. doi: 10.1210/en.2010-1412. [DOI] [PubMed] [Google Scholar]
- 25.Pest MA, Russell BA, Zhang YW, Jeong JW, Beier F. Disturbed cartilage and joint homeostasis resulting from a loss of mitogen-inducible gene 6 in a mouse model of joint dysfunction. Arthritis Rheumatol. 2014;66(10):2816–2827. doi: 10.1002/art.38758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penuela S, Bhalla R, Nag K, Laird DW. Glycosylation regulates pannexin intermixing and cellular localization. Mol Biol Cell. 2009;20(20):4313–4323. doi: 10.1091/mbc.E09-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penuela S, Bhalla R, Gong XQ, Cowan KN, Celetti SJ, Cowan BJ, Bai D, Shao Q, Laird DW. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J Cell Sci. 2007;120(Pt 21):3772–3783. doi: 10.1242/jcs.009514. [DOI] [PubMed] [Google Scholar]
- 28.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr Cartil/OARS Osteoarthr Res Soc. 2007;15(9):1061–1069. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Welch ID, Cowan MF, Beier F, Underhill TM. The retinoic acid binding protein CRABP2 is increased in murine models of degenerative joint disease. Arthritis Res Ther. 2009;11(1):R14. doi: 10.1186/ar2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ratneswaran A, LeBlanc EA, Walser E, Welch I, Mort JS, Borradaile N, Beier F. Peroxisome proliferator-activated receptor delta promotes the progression of posttraumatic osteoarthritis in a mouse model. Arthritis Rheum. 2015;67(2):454–464. doi: 10.1002/art.38915. [DOI] [PubMed] [Google Scholar]
- 31.Usmani SE, Pest MA, Kim G, Ohora SN, Qin L, Beier F. Transforming growth factor alpha controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone. 2012;51(1):131–141. doi: 10.1016/j.bone.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil/OARS Osteoarthr Res Soc. 2010;18(Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 33.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23(24):5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467(7317):863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Billaud M, Sandilos JK, Isakson BE. Pannexin 1 in the regulation of vascular tone. Trends Cardiovasc Med. 2012;22(3):68–72. doi: 10.1016/j.tcm.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poon IK, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature. 2014;507(7492):329–334. doi: 10.1038/nature13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Vasseur M, Lelowski J, Bechberger JF, Sin WC, Naus CC. Pannexin 2 protein expression is not restricted to the CNS. Front Cell Neurosci. 2014;8:392. doi: 10.3389/fncel.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Kraan PM, van den Berg WB. Osteophytes: relevance and biology. Osteoarthr Cartil. 2007;15(3):237–244. doi: 10.1016/j.joca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Garcia M, Knight MM. Cyclic loading opens hemichannels to release ATP as part of a chondrocyte mechanotransduction pathway. J Orthop Res Off Pub Orthop Res Soc. 2010;28(4):510–515. doi: 10.1002/jor.21025. [DOI] [PubMed] [Google Scholar]
- 40.Graff RD, Lazarowski ER, Banes AJ, Lee GM. ATP release by mechanically loaded porcine chondrons in pellet culture. Arthritis Rheum. 2000;43(7):1571–1579. doi: 10.1002/1529-0131(200007)43:7<1571::AID-ANR22>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 41.Ishikawa M, Iwamoto T, Nakamura T, Doyle A, Fukumoto S, Yamada Y. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J Cell Biol. 2011;193(7):1257–1274. doi: 10.1083/jcb.201101050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogawa H, Kozhemyakina E, Hung HH, Grodzinsky AJ, Lassar AB. Mechanical motion promotes expression of Prg4 in articular cartilage via multiple CREB-dependent, fluid flow shear stress-induced signaling pathways. Genes Dev. 2014;28(2):127–139. doi: 10.1101/gad.231969.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waldman SD, Usprech J, Flynn LE, Khan AA. Harnessing the purinergic receptor pathway to develop functional engineered cartilage constructs. Osteoarthr Cartil/OARS Osteoarthr Res Soc. 2010;18(6):864–872. doi: 10.1016/j.joca.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Leong WS, Russell RG, Caswell AM. Stimulation of cartilage resorption by extracellular ATP acting at P2-purinoceptors. Biochim Biophys Acta. 1994;1201(2):298–304. doi: 10.1016/0304-4165(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 45.Papachristou DJ, Pirttiniemi P, Kantomaa T, Papavassiliou AG, Basdra EK. JNK/ERK-AP-1/Runx2 induction “paves the way” to cartilage load-ignited chondroblastic differentiation. Histochem Cell Biol. 2005;124(3–4):215–223. doi: 10.1007/s00418-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 46.Costessi A, Pines A, D’Andrea P, Romanello M, Damante G, Cesaratto L, Quadrifoglio F, Moro L, Tell G. Extracellular nucleotides activate Runx2 in the osteoblast-like HOBIT cell line: a possible molecular link between mechanical stress and osteoblasts’ response. Bone. 2005;36(3):418–432. doi: 10.1016/j.bone.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 47.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H, et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest. 1997;99(7):1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544–1550. doi: 10.1074/jbc.271.3.1544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.