

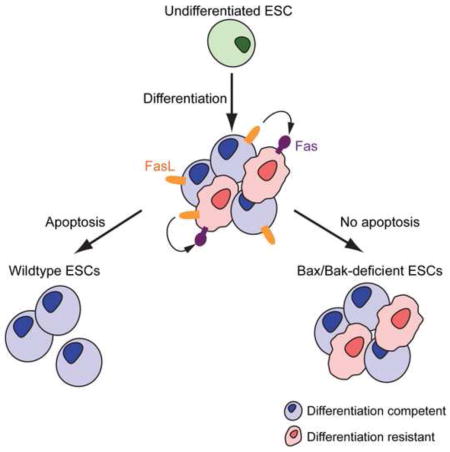
SUMMARY
The intrinsic (mitochondrial) apoptotic pathway is a conserved cell death program crucial for eliminating superfluous, damaged, or incorrectly specified cells [1], and the multi-domain pro-death BCL-2 family proteins BAX and BAK are required for its activation [2, 3]. In response to internal damage or developmental signals, BAX and/or BAK permeabilize the mitochondrial outer membrane [4, 5], resulting in cytochrome c release and activation of effector caspases such as Caspase-3 (Casp3) [6]. While the mitochondrial apoptotic pathway plays a critical role during late embryonic development in mammals [3], its role during early development remains controversial [7, 8]. Here, we show that Bax−/−Bak−/− murine embryonic stem cells (ESCs) display defects during the exit from pluripotency, both in culture and during teratoma formation. Specifically, we find that when ESCs are stimulated to differentiate, a subpopulation fails to do so and instead upregulates FAS in a p53-dependent manner to trigger Bax/Bak-dependent apoptosis. Blocking this apoptotic pathway prevents the removal of these poorly-differentiated cells, resulting in the retention of cells that have not exited pluripotency. Taken together, our results provide further evidence for heterogeneity in the potential of ESCs to successfully differentiate, and reveal a novel role for apoptosis in promoting efficient ESC differentiation by culling cells that are slow to exit pluripotency.
Graphical Abstract
RESULTS
Bax/Bak-dependent mitochondrial apoptosis is critical for proper development during late embryogenesis, as most Bax−/−Bak−/− mice die around embryonic day 17 due to defects in neural tube closure. The rare viable Bax−/−Bak−/− mice only survive a few weeks and possess many physical abnormalities [3]. Mice genetically deficient for other key players of this apoptotic pathway, such as Apaf-1 and Caspase-9, exhibit phenotypic defects similar to, but less dramatic than, those of Bax−/−Bak−/− mice [9–12]. However, the role of mitochondrial apoptosis during early embryogenesis remains poorly defined. A previous study reported that Casp3 cleaves the pluripotency factor Nanog to promote murine embryonic stem cell (mESC) differentiation without inducing apoptosis [13]. Conversely, others have reported that ESCs undergo apoptosis after withdrawal of the pluripotency-promoting cytokine leukemia inhibitory factor (LIF) [7] or during differentiation into cardiomyocytes [8]. Therefore, it remains unclear whether apoptosis has critical functions during ESC differentiation.
To study the role of the intrinsic apoptotic pathway during ESC differentiation, we generated ESCs deficient for Bax and Bak, which together are required for mitochondrial apoptosis upstream of Casp3. As germline-deficient Bax−/−Bak−/− mice generally die in utero [2], we isolated three different ESC lines (Figure S1A) from C57BL/6 Baxf/fBak−/− mice [14]; these were transiently transfected with Cre recombinase to obtain three Bax/Bak-deficient (double knockout, DKO) ESCs lines (Figures S1B, C). Subsequently, two lines (DKO#17 and DKO#18) were found to lack stable integration of Cre and were chosen for further analysis (Figure S1D); unless otherwise indicated, data shown are from line DKO#17. Deletion of Bax/Bak had no observable effects on size, shape, growth rate, or expression of the pluripotency factors Oct4 or Nanog [15–17] in undifferentiated ESCs (Figure S1E, F). Furthermore, DKO#17 and DKO#18 had normal karyotypes (40, XY) (Figure S1G).
Next, we tested whether loss of Bax and Bak affected ESC differentiation. DKO ESCs were differentiated with retinoic acid (RA) and compared to wildtype C57BL/6 ESCs (Bax+/+Bak+/+, WT), parental cells (Baxf/fBak−/−), and Casp3−/− ESCs. We first performed colony formation assays with RA-treated ESCs and found that both DKO and Casp3−/− ESCs inappropriately formed colonies expressing the pluripotency marker alkaline phosphatase (AP) [18] (Figures 1A, B), consistent with the previous Casp3 study [13]. Interestingly, DKO ESCs were more delayed in losing AP expression than Casp3−/− ESCs. To further assess this differentiation defect, we measured mRNA levels of Nanog and Oct4, and found significantly higher expression in DKO ESCs relative to WT cells after 1d and 2d of RA treatment (Figure 1C). Similarly, loss of Oct4 and Nanog proteins was delayed in both DKO ESC lines during differentiation (Figure 1D, S2A, B). We next examined DKO ESC differentiation during embryoid body (EB) formation and found elevated levels of Nanog and Oct4 protein in DKO relative to WT ESCs 4d after initiating EB formation (Figure 1E), indicating that the observed defect is not restricted to RA-induced differentiation. Thus, DKO ESCs exhibit a marked delay in exiting pluripotency in vitro.
Figure 1. Genetic Deletion of Bax and Bak Results in a Delay in ESC Differentiation.

(A) Representative phase and fluorescent images of each ESC line stained for AP after 2d of RA treatment. (B) Quantification of AP positive colonies during RA treatment; data plotted as mean ± SD. (C) Nanog and Oct4 mRNA levels from ESCs analyzed via qPCR; data plotted as mean ± SD. (D) Immunoblot of ESC lysates for Nanog and Oct4. (E) Immunoblot of ESC lysates for Nanog and Oct4 during EB formation. (F) Sections of teratomas stained with hematoxylin and eosin (H&E) (left panel) or for Oct4 expression (right panel), and quantification of Oct4+ nuclei. At least three independent biological samples were used for the AP colony-reforming assay, and Nanog and Oct4 qPCR. * significant at p < 0.05 (Student t test). See also Figures S1–S2.
To account for potential artifacts from Cre expression, we transiently expressed Cre in WT ESCs and selected individual clones without stable expression (Figure S1H), as was done to generate DKO cells. In two different clones, transient expression did not impair ESC differentiation (Figure S1I, J). Separately, to test whether differentiation-resistant DKO colonies could be stably propagated, we re-grew these cells in ESC media. Even after 4d of previous RA treatment, Oct4 and Nanog expression in these cells was similar to that of undifferentiated ESCs (Figure S1K), indicating that these cells were not stuck in a partially differentiated state. Furthermore, with similar efficiency to unmanipulated DKO ESCs, these ESCs re-differentiated upon treatment with RA (Figure S1L). Thus, the differentiation delay of some DKO ESCs may be a stochastic event and not an inherent defect of these cells.
Next, we tested WT, Baxf/fBak−/−, DKO, and Casp3−/− ESCs for their ability to differentiate in vivo during teratoma formation. Hematoxylin and eosin (H&E) stains of teratomas revealed that WT, Baxf/f Bak−/−, and to a lesser extent Casp3−/− lines were mainly comprised of differentiated cells representing all three germ layers (Figure 1F). In contrast, DKO teratomas largely consisted of sheets of undifferentiated cells. Moreover, Oct4 immunohistochemistry showed significantly more undifferentiated, Oct4-positive nuclei in DKO teratomas than those from the other lines. Taken together, our results demonstrate that the differentiation defect is more pronounced in Bax−/−Bak−/− than in Casp3−/− ESCs. Interestingly, this mirrors the greater resistance to apoptosis in Bax−/−Bak−/− cells compared to Casp3−/− cells [19].
Previously, Casp3 was reported to have a nonapoptotic role in promoting ESC differentiation [13]. To test this, we quantified apoptosis in ESCs during differentiation using AnnexinV and propidium iodide (PI). We found that levels of apoptotic cells were extremely low (~3%) in untreated ESCs, while ~12% were apoptotic after 2d of RA-induced differentiation (Figure 2A, S3A). In contrast, Casp3−/− and both DKO ESC lines lacked evidence of apoptosis under untreated and RA-treated conditions (Figure 2A, S2C). For confirmation, we performed TUNEL staining and found that while WT ESCs exhibited an increase in the proportion of TUNEL+ cells after 2d RA treatment, DKO cells were resistant (Figure 2B). Furthermore, Casp3 was only cleaved and activated after RA treatment in WT and Baxf/fBak−/− ESCs (Figure 2C, S2D). Therefore, Bax and Bak are required for Casp3 activation and differentiation-induced apoptosis.
Figure 2. ESCs Undergo Fas-mediated, Bax/Bak-dependent Apoptosis During Differentiation.

(A) Quantification of percent apoptotic cells (AnnexinV positive/PI negative) during RA-mediated differentiation. (B) Representative images and quantification of TUNEL staining from untreated and 2d RA-treated WT and DKO ESCs. (C) Immunoblot for full-length (FL) and cleaved (CL) Casp3 from ESCs untreated or treated with RA for 2d. (D) Representative Q-PCR of Fas and Fas-L mRNA levels during ESC differentiation using two independent sets of primers for each target. (E) Schematic of Fas-mediated activation of Bax/Bak-dependent apoptosis. (F) Immunoblot for components of the Type II Fas death receptor pathway during ESC differentiation. (G) Fas and AnnexinV co-staining in ESCs after 2d of differentiation. (H) Immunoblot for Casp8 and Casp3 in ESCs sorted for Fas after 2d of differentiation. (I) Immunoblots for Fas and Casp3 from WT or DKO cells induced to form embryoid bodies by the hanging drop method. (J) Immunoblot of FAS and CASP3 from UCSF4 human ESCs differentiated with RA as indicated. Three independent biological samples were used for the AnnexinV assays. Data plotted as mean ± SD. * significant at p < 0.05 (Student t test). See also Figure S3.
We next searched for upstream factors that activate Bax/Bak during ESC differentiation. Based on a small microarray of approximately 100 cell death and survival genes, we found >30 fold upregulation of Fas mRNA after 4d of RA treatment; its cognate ligand Fas ligand (Fas-L) was upregulated ~3 fold as well (Table S1). We confirmed that during differentiation, Fas and Fas-L mRNA and protein were markedly upregulated (Figure 2D, F). A prototypical death receptor, Fas can trigger apoptosis through assembly of the death-inducing signaling complex (DISC) and subsequent activation of Caspase-8 (Casp8) [20]. In Type I cells, Fas-mediated activation of Casp8 is sufficient to activate the executioner Casp3; however, in Type II cells, Fas-mediated apoptosis requires Casp8-dependent activation of the BH3-only protein Bid and subsequent engagement of Bax and Bak [20] (Figure 2E).
As Bax and Bak are required for apoptosis during ESC differentiation, we asked whether ESCs are Type II cells. Indeed, we observed cleavage of Casp8 (to the active p18 fragment) and Bid during ESC differentiation (Figure 2F). Importantly, these events occurred upstream of Bax/Bak and in the absence of Casp3, suggesting that Casp8 is an initiator caspase rather than a substrate of downstream caspases. We then costained differentiating ESCs for Fas and AnnexinV and found significant enrichment of apoptotic cells in the Fas-positive subpopulation (Figure 2G, S3B). Furthermore, after sorting differentiating WT ESCs for Fas, we found elevated Casp8 and Casp3 activation in the Fas positive population (Figure 2H).
To determine the generality of Fas upregulation and Casp3 activation during ESC differentiation, we first analyzed EB formation and observed Fas upregulation in both WT and DKO ESCs, while cleaved Casp3 was only detected in WT ESCs (Figure 2I). Although Casp3 activation during EB formation could be critical for other developmental steps, such as cavitation, these data are consistent with our hypothesis that Fas signaling triggers apoptosis during ESC differentiation. We then analyzed RA-treated human ESCs (hESCs) and found transient FAS upregulation and CASP3 cleavage (Figure 2J). This is consistent with the mESCs phenotype and suggests that this apoptotic process is conserved across species.
To determine whether differentiated or undifferentiated ESCs are variably susceptible to apoptosis, we utilized Oct4-GFP reporter ESCs [21] to measure levels of AnnexinV staining after RA treatment. Strikingly, levels of apoptosis were significantly higher in the undifferentiated (GFPhigh) subpopulation (Figure 3A, B). We then sorted these cells for GFP and found that the GFPhigh subpopulation contained significantly more cleaved Casp3 than the GFPlow subpopulation (Figure 3C). Thus, a subgroup of poorly-differentiating ESCs preferentially initiates apoptosis through the Bax/Bak pathway.
Figure 3. Fas and Casp8 Promote the Removal of Poorly Differentiating cells during ESC Differentiation.

(A) Representative FACS plots and quantification of Oct4-GFP ESCs stained for AnnexinV after 2d of RA treatment. (B) Quantification of apoptotic Oct4-GFP ESCs after 3d of RA treatment. (C) Immunoblots of lysates from 2d-treated Oct4-GFP ESCs after sorting for GFP expression. (D) Representative FACS plots and quantification of Oct4-GFP ESCs stained for Fas after 2d of RA treatment. RA-treated ESCs were used for isotype control staining. (E) Immunoblot for Oct4 from 2d-treated WT ESCs after sorting for Fas expression. (F) Q-PCR using two different primer sets for Fas-L mRNA from Oct4-GFP ESCs treated or untreated with RA for 2d. (G) Immunoblot of ESC lysates for Casp8 and Fas after siRNA knockdown and 2d RA treatment. (H) AnnexinV staining of ESCs quantified by FACS after indicated siRNA knockdown and 2d RA treatment. (I) Number AP+ colonies after indicated siRNA knockdown and 2d RA treatment. (J) Immunoblot of ESC lysates for Nanog after siRNA knockdown and 2d RA treatment. Three independent biological samples were used for the AnnexinV, qPCR, and AP assays. Data plotted as mean ± SD. * significant at p < 0.05 (Student t test).
We next determined the distribution of Fas and Fas-L among differentiating ESCs. Using Oct4-GFP ESCs, we found that Fas-expressing cells were predominantly undifferentiated (GFPhigh) (Figure 3D). Likewise, after sorting differentiating WT ESCs for Fas, we found that Fashigh cells had markedly higher levels of Oct4 than Faslow cells (Figure 3E). To determine the source of Fas-L, we isolated mRNA from GFP-sorted Oct4-GFP ESCs and found equivalent upregulation of Fas-L mRNA in undifferentiated and differentiated cells (Figure 3F), suggesting that poorly-differentiated cells acquire this signal through both autocrine and paracrine sources.
To test the roles of Fas and Casp8 in ESC differentiation, we performed siRNA knockdowns (Figure 3G). In comparison to cells transfected with nontargeting control siRNA, knockdown of Fas or Casp8 significantly reduced the levels of apoptotic cells (Figure 3H). Furthermore, knockdown of Fas or Casp8 increased the number of AP-positive colonies and delayed the loss of Nanog after RA treatment (Figures 3I, J). Taken together, our data strongly suggest that engagement of Fas is a critical upstream activator of the intrinsic apoptotic pathway during ESC differentiation, and that deletion of Bax and Bak prevents the elimination of poorly differentiating cells.
Next, we tested known transcriptional regulators of Fas and discovered that its expression was largely dependent on p53 (Figure 4A–C). p53−/− ESCs could not efficiently upregulate Fas, and these cells were protected against apoptosis and Casp3 cleavage (Figure 4D, E).
Figure 4. p53 is De-repressed During ESC Differentiation to Initiate Apoptosis in Poorly Differentiating Cells.

(A) Immunoblot of p53 from p53+/+ and p53−/− ESCs. (B) mRNA and (C) protein levels of Fas from RA-treated p53+/+ and p53−/− ESCs. (D) AnnexinV staining of p53+/+ and p53−/− ESCs after 2d RA treatment. (E) Immunoblot of CASP3 from p53+/+ and p53−/− ESCs treated with RA. (F) Immunoblot for p53 from WT and DKO ESCs during RA treatment. (G) mRNA levels of p53 from WT and DKO ESCs during RA treatment. Immunoblot for AurA from (H) WT and DKO or (I) p53+/+ and p53−/− ESCs during RA treatment. (J) Immunoblot for Fas from p53+/+ and p53−/− ESCs treated with MLN8237 for 24 hr. (K) Immunoblot for Fas from p53+/+ and p53−/− ESCs transfected with control siRNA or siRNA against AurA. AnnexinV staining of p53+/+ and p53−/− ESCs after treatment with (L) MLN8237 for 24 hr or (M) knockdown with control or AurA-specific siRNA. (N) Immunoblot for γ-H2AX from WT and DKO ESCs treated with RA. (O) Immunoblot for AurA, phospho-serine15-p53, total p53, and γ-H2AX from Oct4-GFP ESCs sorted for GFP expression after 2d RA treatment. (P) Representative immunofluorescence images and quantification of γ-H2AX+ nuclei from WT and DKO ESCs untreated or treated with RA for 4d. (Q) Schematic of Fas-L-mediated activation of Fas to induce apoptosis in undifferentiated ESCs. Three independent biological samples were used for the AnnexinV and qPCR assays. Data plotted as mean ± SD. * significant at p < 0.05 (Student t test). See also Figure S4.
Interestingly, we found that p53 mRNA and protein levels decline during WT ESC differentiation (Figure 4F, G), a counterintuitive result consistent with previous reports [22–24]. We reasoned that p53 activity could increase despite a decrease in its levels because its activity is restrained in undifferentiated ESCs, and differentiation could lift this signaling brake. In fact, a previous study demonstrated that Aurora Kinase A (AurA), an important mitotic kinase, phosphorylates p53 in undifferentiated ESCs to inhibit its transcriptional activity [22]. Immunoblotting for AurA revealed significant reductions in its levels during differentiation, irrespective of Bax/Bak or p53 status (Figure 4H, I). However, the pluripotency factors Oct4, Nanog, and Klf4 are not responsible for regulating AurA levels in ESCs (Figure S4). Furthermore, either treatment of undifferentiated ESCs with the AurA-specific kinase inhibitor MLN8237 or siRNA knockdown of AurA was sufficient to induce p53-dependent upregulation of Fas and apoptosis (Figure 4J–M). Thus, AurA restrains p53 function in undifferentiated ESCs, and its downregulation during differentiation allows p53-mediated upregulation of Fas and initiation of Bax/Bak-dependent apoptosis.
As p53 guards the integrity of the genome, we analyzed levels of phosphorylated histone protein H2AX (γ-H2AX), a classical DNA damage marker, and saw a transient increase in WT ESCs but a steady accumulation in DKO ESCs during differentiation (Figure 4N). Staining for γ-H2AX foci confirmed that DKO ESCs retain significantly more γ-H2AX-positive cells than WT ESCs after 4d of RA treatment (Figure 4P). Interestingly, p53 protein levels did not significantly decline in DKO ESCs during differentiation (Figure 4F), suggesting that an inability to remove damaged ESCs precluded downregulation of p53 protein at the population level.
We next used Oct4-GFP ESCs to investigate why Fas upregulation and apoptosis primarily occur in poorly differentiating ESCs. Although we observed no significant difference in AurA or total p53 levels between GFPhigh and GFPlow cells, we found increased levels of γ-H2AX and phospho-Ser15-p53, an activating p53 modification, in undifferentiated ESCs (Figure 4O). Thus, our data suggest that loss of AurA-mediated inhibition licenses p53 activity in all differentiating cells, while activation of p53 primarily occurs in the differentiation-resistant ESC subpopulation (Figure 4Q).
DISCUSSION
Here, we show that upregulation and engagement of Fas on poorly differentiating ESCs selectively triggers their demise. The most parsimonious explanation for the differentiation delay in Bax−/−Bak−/− ESCs is a failure to remove differentiation-resistant cells. This defect is consistent with a previous report studying Casp3 [13]. However, in contrast to their conclusion that Casp3 does not induce apoptosis, our results support a pro-apoptotic role for Casp3. This discrepancy may be because they used DAPI staining rather than the more sensitive, yet specific, markers employed here. Although our data argue that Casp3 promotes apoptosis in undifferentiated cells during differentiation, Casp3 may have non-apoptotic functions in cells that survive and differentiate. In fact, Fas itself may have additional non-apoptotic functions during differentiation, as it can activate NF-κB signaling [25–27], which may be important for ESC differentiation [28]. Nevertheless, our results clearly identify a pro-apoptotic role for Fas during ESC differentiation.
In contrast to our findings, a previous study reported downregulation of FAS during hESC differentiation [29]. This study compared pluripotent hESCs to fetal and adult human tissues, and hESCs differentiated for 20d, while our study focused on early ESC differentiation. Thus, the difference may be partly due to the timing of analysis. However, in closer agreement with our findings, conversion of human fibroblasts or hepatocytes into induced pluripotent stem cells is associated with a reduction in FAS mRNA levels [30, 31], while differentiation of hESCs into pancreatic islet-like cells or mesenchymal stem cells is associated with increased FAS mRNA levels [32, 33]. Further studies will be required to resolve these differences.
The accumulation of γ-H2AX in DKO ESCs coupled with its efficient loss in WT ESCs during differentiation suggests that DNA damage may be crucial to activate p53. However, future studies are necessary to determine the potential form of damage and its sensor(s). Furthermore, questions such as whether some undifferentiated ESCs have already accumulated DNA damage, or whether an inability to differentiate itself causes a DNA damage response, remain to be addressed.
Based on the dramatic differentiation defects in apoptosis-deficient (p53−/−, Bax−/−Bak−/−, and Casp3−/−) ESCs observed here and previously [13, 23], one might expect obvious problems in early embryogenesis in mice genetically deficient for these genes. However, p53-deficient mice are born at normal Mendelian ratios without gross developmental defects [34]. Bax−/−Bak−/− and Casp3−/− mice also progress through early embryogenesis without obvious problems [3, 12]. We speculate that this may be due to compensation for the loss of these genes, as has been seen in other circumstances [3, 35–38]. Nevertheless, these mutant mice, and p53 haploinsufficent humans (Li-Fraumeni syndrome), have a predisposition to cancer [34, 39, 40]. Future studies will be required to test whether this elevated risk of cancer is partly due to an inability to delete differentiation-defective cells during early embryogenesis.
In summary, our findings show that the mitochondrial apoptotic pathway is critical for removing poorly-differentiated ESCs during their exit from pluripotency. These results highlight the heterogeneity of isogenic ESCs and reveal the importance of apoptosis in controlling cell fate decisions at the earliest stages of ESC differentiation.
EXPERIMENTAL PROCEDURES
ESC Cultures
Mouse ESC lines were maintained in ESC Media: DMEM containing high glucose (Sigma) with HEPES hemisodium salt (MP Biomedical), 1% L-glutamine and 1% non-essential amino acids (UCSF Cell Culture Facility), 15% ESC-qualified fetal bovine serum and 100 mM β-mercaptoethanol (Gibco), and LIF (made in-house). Cells were maintained on irradiated mouse embryonic fibroblasts (MEFs). For differentiation experiments, ESCs were plated away from MEFs onto gelatinized dishes (0.2% gelatin from porcine skin, Sigma) and treated with 1 μM retinoic acid (Sigma) in ESC media without LIF as indicated. Casp3−/− ESCs were a generous gift from T. Zwaka (Mt. Sinai). Embryoid bodies were formed using the hanging drop method, with 400 cells plated in 20 μl drops of ESC media without LIF.
For additional methods, see Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We would like to thank Raga Krishnakumar for her assistance in performing the karyotyping analysis. This work was supported an NSF Fellowship (E.S.W.), F31NS083323 (E.S.W.), F31NS626272 (N.A.R.), R01CA136717 (A.G.), Atwater Family Foundation (A.G.), R01GM101180 (R.B.), R01CA136577 (S.A.O.), R01DK095306 (S.A.O.), HHMI Physician-Scientist Early Career Award (S.A.O.), American Cancer Society Research Scholar Award (S.A.O.), Juvenile Diabetes Research Foundation (S.A.O.), Harrington Discovery Institute Scholar-Innovator Award (S.A.O.).
Footnotes
Conflict of Interest: None to report
AUTHOR CONTRIBUTIONS
N.A.R. and C.M. derived the DKO ESCs and did initial characterization of apoptotic and differentiation phenotypes. E.S.W. helped characterize the DKO ESCs, performed experiments to determine the mechanism of the phenotype, and wrote the manuscript with input from R.B. and S.A.O. N.E.H. performed the microarray experiment. O.M. performed the hESC experiments. A.G., R.B., and S.A.O. provided input into the conception of the project and the design of experiments. R.B. and S.A.O. reviewed and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews Molecular cell biology. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 2.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science (New York, N Y. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochimica et biophysica acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 5.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annual review of biochemistry. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 6.Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harbor perspectives in biology. 2013;5 doi: 10.1101/cshperspect.a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duval D, Malaise M, Reinhardt B, Kedinger C, Boeuf H. A p38 inhibitor allows to dissociate differentiation and apoptotic processes triggered upon LIF withdrawal in mouse embryonic stem cells. Cell death and differentiation. 2004;11:331–341. doi: 10.1038/sj.cdd.4401337. [DOI] [PubMed] [Google Scholar]
- 8.Akbari-Birgani S, Hosseinkhani S, Mollamohamadi S, Baharvand H. Delay in Apoptosome Formation Attenuates Apoptosis in mouse Embryonic Stem Cell Differentiation. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M113.536730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Maria R, Testa U, Luchetti L, Zeuner A, Stassi G, Pelosi E, Riccioni R, Felli N, Samoggia P, Peschle C. Apoptotic role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood. 1999;93:796–803. [PubMed] [Google Scholar]
- 10.Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J. Adult Apaf-1-deficient mice exhibit male infertility. Developmental biology. 2000;218:248–258. doi: 10.1006/dbio.1999.9585. [DOI] [PubMed] [Google Scholar]
- 11.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 12.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 13.Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA, Zwaka TP. Caspase activity mediates the differentiation of embryonic stem cells. Cell stem cell. 2008;2:595–601. doi: 10.1016/j.stem.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX, BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nature genetics. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 16.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 17.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 18.Jakel HP, Wobus AM, Bloch C, Jakel N, Schoneich J. A micromethod for the determination of alkaline phosphatase in mammalian cells. Biomedica biochimica acta. 1983;42:1123–1128. [PubMed] [Google Scholar]
- 19.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 20.Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell death and differentiation. 2012;19:42–50. doi: 10.1038/cdd.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416:545–548. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- 22.Lee DF, Su J, Ang YS, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF, Gingold J, Wang HL, Zhao R, Sevilla A, et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell stem cell. 2012;11:179–194. doi: 10.1016/j.stem.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nature cell biology. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 24.Lutzker SG, Levine AJ. A functionally inactive p53 protein in teratocarcinoma cells is activated by either DNA damage or cellular differentiation. Nature medicine. 1996;2:804–810. doi: 10.1038/nm0796-804. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto N, Imamura R, Suda T. Caspase-8- and JNK-dependent AP-1 activation is required for Fas ligand-induced IL-8 production. The FEBS journal. 2007;274:2376–2384. doi: 10.1111/j.1742-4658.2007.05772.x. [DOI] [PubMed] [Google Scholar]
- 26.Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. The EMBO journal. 2004;23:3175–3185. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 28.Torres J, Watt FM. Nanog maintains pluripotency of mouse embryonic stem cells by inhibiting NFkappaB and cooperating with Stat3. Nature cell biology. 2008;10:194–201. doi: 10.1038/ncb1680. [DOI] [PubMed] [Google Scholar]
- 29.Richards M, Tan SP, Tan JH, Chan WK, Bongso A. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem cells. 2004;22:51–64. doi: 10.1634/stemcells.22-1-51. [DOI] [PubMed] [Google Scholar]
- 30.Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nature cell biology. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barberi T, Willis LM, Socci ND, Studer L. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS medicine. 2005;2:e161. doi: 10.1371/journal.pmed.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li SS, Yu SL, Kao LP, Tsai ZY, Singh S, Chen BZ, Ho BC, Liu YH, Yang PC. Target identification of microRNAs expressed highly in human embryonic stem cells. Journal of cellular biochemistry. 2009;106:1020–1030. doi: 10.1002/jcb.22084. [DOI] [PubMed] [Google Scholar]
- 34.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 35.Gray DH, Kupresanin F, Berzins SP, Herold MJ, O’Reilly LA, Bouillet P, Strasser A. The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity. 2012;37:451–462. doi: 10.1016/j.immuni.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sevenich L, Pennacchio LA, Peters C, Reinheckel T. Human cathepsin L rescues the neurodegeneration and lethality in cathepsin B/L double-deficient mice. Biological chemistry. 2006;387:885–891. doi: 10.1515/BC.2006.112. [DOI] [PubMed] [Google Scholar]
- 37.Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- 38.Janne PA, Suchy SF, Bernard D, MacDonald M, Crawley J, Grinberg A, Wynshaw-Boris A, Westphal H, Nussbaum RL. Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J Clin Invest. 1998;101:2042–2053. doi: 10.1172/JCI2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nichols KE, Malkin D. Genotype Versus Phenotype: The Yin and Yang of Germline TP53 Mutations in Li Fraumeni Syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015 doi: 10.1200/JCO.2015.61.5757. [DOI] [PubMed] [Google Scholar]
- 40.Biswas S, Shi Q, Matise L, Cleveland S, Dave U, Zinkel S. A role for proapoptotic Bax and Bak in T-cell differentiation and transformation. Blood. 2010;116:5237–5246. doi: 10.1182/blood-2010-04-279687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.