

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with a life expectancy of less than 5 years post diagnosis for most patients. Poor molecular characterization of IPF has led to insufficient understanding of the pathogenesis of the disease, resulting in lack of effective therapies. In this study, we have integrated a label-free LC-MS based approach with systems biology to identify signaling pathways and regulatory nodes within protein interaction networks that govern phenotypic changes that may lead to IPF. Ingenuity Pathway Analysis of proteins modulated in response to bleomycin treatment identified PI3K/Akt and Wnt signaling as the most significant profibrotic pathways. Similar analysis of proteins modulated in response to vascular endothelial growth factor (VEGF) inhibitor (CBO-P11) treatment identified natural killer cell signaling and PTEN signaling as the most significant antifibrotic pathways. Mechanistic/mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK) were identified to be key mediators of pro- and antifibrotic response, where bleomycin (BLM) treatment resulted in increased expression and VEGF inhibitor treatment attenuated expression of mTOR and ERK. Using a BLM mouse model of pulmonary fibrosis and VEGF inhibitor CBO-P11 as a therapeutic measure, we identified a comprehensive set of signaling pathways and proteins that contribute to the pathogenesis of pulmonary fibrosis that can be targeted for therapy against this fatal disease.
Keywords: BLM mouse model, High-throughput proteomics, Ingenuity Pathway Analysis, Pulmonary fibrosis, Systems biology, VEGF inhibitor
1 Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and invariably fatal disease of unknown etiology that is limited to the lung. The disease carries a very poor prognosis, with most patients dying within 5 years of diagnosis [1–3]. One of the factors contributing to the challenges with treatment of IPF is its poor molecular characterization. Very few molecular targets that directly contribute to the pathogenesis of the disease have been identified, and there are no approved therapies for IPF [4–6]. There is therefore a compelling need to generate novel hypotheses that can be critical to the development of effective diagnostic and therapeutic modalities for treatment of IPF. Delineating IPF-specific signaling pathways using an unbiased global perspective will offer a higher probability of identifying meaningful therapeutic targets, and provide a much deeper understanding of IPF pathogenesis [7].
Although the application of high-throughput comparative proteomics, a robust yet sensitive technique to study disease pathogenesis and biomarker discovery has been successfully applied in cancer research, it has found strikingly limited utility in IPF research [8–13]. Given that all proteins interact with each other to form networks of signal transduction pathways, analyzing high-throughput differential protein expression collectively using systems biology tools is a more robust way of interpreting proteomic data. Such methodology allows not only for the delineation of groups of differentially expressed proteins that direct overall phenotypic change, but also for identification of highly connected central nodes within these protein groups that dictate modulation of the identified groups of key deregulated protein networks [14–16].
The purpose of this study was to use an untargeted protein based approach to gather an unbiased and nonintuitive view of the important regulatory pathways that dictate bleomycin (BLM) induced pulmonary fibrosis. The BLM mouse model for pulmonary fibrosis is well characterized and widely used for understanding the pathogenesis of IPF [17]. Typical features of human IPF including intra-alveolar buds, incorporation of collagen, and invasion of alveolar space are recapitulated in the BLM mouse model [18]. Vascular endothelial growth factor (VEGF), a proinflammatory cytokine and an important regulator of angiogenesis has been shown to play an important role in lung injury and fibrosis [19]. Although inhibition of angiogenesis has been shown to attenuate fibrosis, the key VEGF-related protein networks that mediate pulmonary fibrosis have not yet been elucidated [20]. We therefore used the VEGF inhibitor CBO-P11 as a therapeutic measure within our high-throughput proteomic approach to delineate important angiogenesis-related pathways and proteins in our BLM mouse model.
In this study, we provide evidence for a set of novel signal transduction pathways and proteins that are deregulated in the pulmonary tissue of mice challenged with BLM. From a therapeutic perspective, we report several signaling pathways and molecules altered as a result of intervention using CBO-P11, which may provide attractive targets for the treatment of pulmonary fibrosis.
2 Materials and methods
2.1 Cell culture
The human lung fibroblast CRL-1490 cell line was obtained from ATCC. The cells were maintained in Eagle's minimum essential medium (MEM) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. They were cultured at 37°C in 5% CO2 incubator. The cells were passaged at preconfluent densities using a solution containing 0.05% trypsin and 0.5 mM EDTA.
2.2 Animal maintenance and study design
For the animal studies, 6- to 8-week old C57BL/6 mice were used (Jackson Laboratories, Bar Harbor, ME). Mice were housed in a barrier facility with specific pathogen-free conditions, and all experiments were performed using protocols approved by the Old Dominion University (ODU) animal facility. Briefly, Mice were divided into groups of five animals for each treatment group and anesthetized with isoflurane. BLM sulfate (1 U/kg body weight (BW)) or equal volumes of saline as control was administered intranasally. In a separate set, one group of BLM-treated mice was cotreated every other day by intraperitoneal injection of VEGF inhibitor CBO-P11 (0.3 mg/kg BW), starting at day 0 and continued until the mice were euthanized. All mice were euthanized at day 28, and the lungs were perfused with 5 mL of cold saline through the left ventricle and surgically removed. The left lungs were used to evaluate the fibrotic score by histological examination, and the right lungs were homogenized to analyze protein expression levels.
2.3 Immunohistochemistry
Mice were euthanized and the left lung was fixed in 10% formalin overnight and embedded in paraffin. Paraffin sections (3 μm thick) were stained with H&E. The pathological grade of fibrosis was evaluated under 4× magnification. Histological assessment of the extent and severity of pulmonary fibrosis was independently determined by one pathologist and two researchers in a blind study using the Ashcroft method [21] on sections of the left lung stained with H&E.
2.4 Collagen detection assay
Soluble collagen levels were quantified by Sircol® Assay (Biocolor Ltd., Belfast, UK) as per manufacturer's protocol. Briefly, mice lung homogenates were mixed with 1 mL Sircol dye for 30 min then centrifuged to pellet collagen. The pellet was washed and resuspended in alkyne solution and the absorption was measured at 550 nm (Synergy HI Hybrid Reader, BioTek).
2.5 Protein extraction for label-free protein quantification
For mass spectrometric analysis, the tissue samples were recovered from cryopreservation, weighed and placed in impact-resistant tubes containing 6.5 mm garnet and ceramic sphere matrix (MP Biomedical, Santa Ana, CA) with 1 mL of 8 M urea, 300 mM Tris-HCL, 10 mM DTT (pH8.5) per 100 mg tissue sample. The sample was subjected to mechanical disruption in a FastPrep-24 instrument (MP Biomedical) for 20 s at a speed of 4 m/s twice, the slurry was then centrifuged at 10 000 g for 10 min at 4°C and the supernatant transferred to a new microcentrifuge tube. The protein concentration of the supernatant was determined using a DTT compatible BCA assay (Thermo Fisher Scientific, San Jose, CA) and 100 μg of extracted protein sample was run on a NuPAGE reducing gel (4–12% Bis-Tris Gel; Life Technologies, Carlsbad, CA) with NuPAGE MOPS SDS 1 × buffer run at 200 V for about 10 min. After the protein band had migrated 3–5 mm, the gel was stained with Page Blue (Bio-Rad, Hercules, CA) and the entire protein band cut out into 1–2 mm gel plugs. These gel plugs were destained and washed three times in 50 mM NH4HCO3, 50% acetonitrile, and 80% acetonitrile. The gel-bound proteins were reduced with 1 mL of 40 mM DTT for 25 min at 56°C. The plugs were rinsed with 1 mL of 50 mM NH4HCO3 buffer and the reduced proteins alkylated with 1 mL of 50 mM iodoacetamide for 30 min at 25°C in the dark with constant mixing. Iodoacetamide was discarded and the gel bound proteins were digested with 0.5 mL of trypsin (20 ng/μL; Promega, Madison, WI) in 50 mM NH4HCO3 buffer at 37°C with constant mixing for 12 h. After digestion, the tryptic fraction was collected by washing the gel plugs with 50 mM NH4HCO3. The eluent containing the tryptic peptides was dried using a Speed-Vac apparatus (Thermo Fisher Scientific) and stored at 4°C prior to mass spectrometric analysis.
2.6 MS analysis
The dried samples were dissolved with 20 μL of 0.1% formic acid/water, 2 μL of each sample was then analyzed by LC/ESI-MS/MS on a Q-Exactive (Thermo Fisher Scientific) mass spectrometer with an Easy NanoLC-1000 system, using data-dependent acquisition with dynamic exclusion (DE = 1) settings. The data-dependent acquisition settings used were a top 12 higher energy collision induced dissociation for the Q-Exactive MS. Resolving power for Q-Exactive was set at 70 000 for the full MS scan, and 17 500 for the MS/MS scan at m/z 200. LC/ESI-MS/MS analyses were conducted using a C18 column (75 μm × 150 mm). The mobile phases for the reverse phase chromatography were (A) 0.1% formic acid/water and (B) 0.1% formic acid in acetonitrile. A four-step, linear gradient was used for the LC separation (column preequilibration with 2% B for 10 min; 2–30% B in the first 47 min, followed by 80% B in the next 1 min and holding at 80% B for 12 min).
2.7 Protein identification and database searching
A label-free precursor ion detection method (Proteome Discoverer, version 1.3, Thermo Fisher Scientific) was used with specific retention times on precursors/fragments within 5 ppm mass accuracy. The Sequest algorithm was used to identify peptides from the resulting MS/MS spectra by searching against the combined mouse protein database (a total of 25 320 sequences) extracted from Swissprot (version 57) using taxonomy “Mus musculus”. Searching parameters for parent and fragment ion tolerances were set as 15 ppm and 60 mmu for the Q-Exactive MS. Other parameters used were a fixed modification of carbamidomethylation–Cys, variable modifications of phosphorylation (S, T, Y), and oxidation (Met). Trypsin was set as the protease with a maximum of two missed cleavages. Only those proteins that had more than two peptides identified (or >50% of protein covered by a single peptide) were included in the comparative quantitative analysis steps, and resulted in a correct protein identification probability of p < 0.05. Peptides that have yet to be definitively linked to a protein were excluded from further analysis.
2.8 Differential protein expression quantification
Three technical replicates for one mouse within each treatment groups (control, BLM, and combination of VEGF inhibitor with BLM) were analyzed. A protein was classified as being expressed if it was identified in all three technical replicates from individual mice. The AUC for the respective deisotoped peptide and charge reduced multiple tryptic peptides was calculated for each identified protein within technical replicates, which was then used to calculate the mean AUC value for an expressed protein for each individual mice. In the event that a protein was not detectable in a particular cohort, an AUC value of 1 was assigned for that protein. Proteins were classified as being up- or downregulated comparing the mean AUC for a protein from one treatment group to the mean AUC for the corresponding protein in other treatment group. The Wilcoxon–Mann–Whitney test was used to identify proteins whose expression differed (by at least 1.5-fold) at the 5% significance level.
2.9 Ingenuity Pathway Analysis
Differentially regulated proteins identified by MS were further analyzed using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Mountain View, CA). IPA was used to interpret the differentially expressed proteins in terms of an interaction network and predominant canonical pathways [22]. The Ingenuity Pathways Knowledge Base (IKB) is a frequently updated curated database that consists of interactions between different proteins culled from scientific literature. IPA uses this database to construct protein interaction clusters that involve direct and indirect interactions. The networks are displayed graphically as nodes (proteins) and edges (the biological relationship between the proteins). A protein interaction network was generated as follows. A dataset containing the up- and downregulated proteins, called the focus proteins, for a particular comparison was uploaded into the IPA. These focus proteins were overlaid onto a global molecular network developed from the information in the IKB. Networks of these focus proteins were then algorithmically generated by including as many focus proteins as possible and other nonfocus proteins from the IKB that are needed to generate the network based on connectivity. Canonical pathways are identified from the IPA library based on their significance to the dataset. The significance of the association between the dataset and canonical pathway is measured in two ways: (i) a ratio of the number of proteins in the dataset that map to the pathway divided by the total number of proteins that exist in the canonical pathway and (ii) a p-value that is obtained by comparing the number of genes/proteins of interest relative (i.e., focus genes) to the total number of genes/proteins in all functional/pathway annotations stored in the IKB (i.e., a right-tailed Fisher's exact test with the Benjamini–Hochberg correction for multiple hypothesis testing).
2.10 Immunoblotting
Mouse lung homogenates were resolved on a 10% SDS-PAGE and transferred onto a nitrocellulose membrane. The protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL), and equal amount of protein was loaded per sample. The membrane was blocked with TBS-T (0.1% Tween-20 in TBS) containing 5% dry milk, and incubated with primary antibody overnight at 4°C. After three washes with TBS-T, the membrane was incubated with HRP-conjugated secondary antibody for 1 h and then washed with TBS-T. Immunoreactive proteins were detected by chemiluminescence (Supersignal® West Femto, Pierce, Rockford, IL). The following antibodies were used: rabbit anti-p44/42 mitogen-activated protein kinase (MAPK) (Erk1/2) antibody, rabbit anti-β-catenin antibody, rabbit anti-mTOR antibody, rabbit antivimentin antibody, HRP-conjugated anti-rabbit IgG antibody, HRP-conjugated anti-mouse IgG antibody (Cell Signaling Technology, Danvers, MA), and mouse anti-β-actin antibody (Sigma Chemical Co., St. Louis, MO). Immunoreactive proteins were detected by chemiluminescence (Supersignal® West Femto, Pierce, Rockford, IL) using MyECL Imager (Thermo Scientific) and quantified by imaging densitometry, using ImageJ digitizing software (NIH, Image analysis using Java). Mean densitometry data from independent experiments were normalized to the control.
3 Results
3.1 Effect of VEGF inhibitor (CBO-P11) on BLM-induced fibrotic response in mice
We characterized BLM-induced fibrotic response in mice and assessed the effect of VEGF inhibitor CBO-P11. Lung histology data showed interstitial fibrosis of the alveolar wall in mice receiving BLM (Fig. 1A). The extent of lung fibrosis was determined by quantitative histology according to Ashcroft's method [21]. A significant reduction in BLM-induced fibrosis was observed in mice cotreated with CBO-P11, bringing it down to control levels (Fig. 1A and B). Analysis of total collagen content by Sircol® assay showed that BLM-induced collagen production was significantly inhibited in mice cotreated with CBO-P11 (Fig. 1C). Figure 1D shows that collagen type III, one of the most abundant collagen proteins of the extracellular matrix (ECM), was induced by BLM treatment and this effect was significantly inhibited in mice cotreated with CBO-P11.
Figure 1.
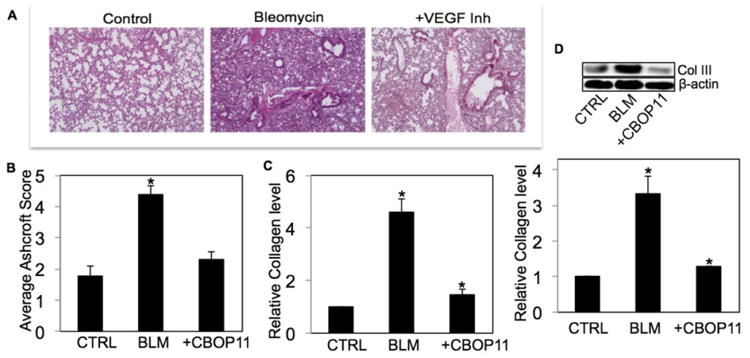
Effect of VEGF inhibitor (CBO-P11) on BLM-induced fibrotic response in mice. Mice treated with BLM (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg) or equal volumes of saline as control were euthanized at 28 days. (A) Representative immunohistochemical micrographs of treated mice lung tissue stained with H&E. (B) Calculated fibrosis score based on the histopathological assessment of H&E-stained slides. (C) Lung homogenate from treated mice was analyzed for soluble collagen levels by Sircol® Assay. (D) Lung homogenate from treated mice was analyzed for collagen III levels by Western blotting. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. Plots are mean ± S.E.M of five animals per group. *p < 0.05 versus nontreated control.
3.2 Identification and assessment of differentially expressed proteins across treatment groups
Differentially expressed proteins between the treatment groups (control, BLM, and BLM+CBO-P11) were identified by performing chromatographic alignment of the peptides separated on the 50 cm long column and parsing the respective peptide signals using SIEVE 2.0 software. For data analysis, a combination of several search engines including SEQUEST and MASCOT, were employed for protein/peptide identification. Using a fold change cut-off of 1.5 for differential expression of proteins, a total of 1729 common proteins were identified that were deregulated between the control (PBS) and BLM-treated group, of which 666 proteins were upregulated in the BLM-treated group and 616 were downregulated in the BLM-treated group by at least 1.5-fold (Supporting Information Table 1). Likewise, comparison of the BLM alone group with the CBO-P11+BLM group yielded a total of 1241 differentially regulated proteins, of which 192 were upregulated in the CBO-P11+BLM group and 666 were downregulated in the CBO-P11+BLM group (Supporting Information Table 2).
The identified proteins represented broad functional and cellular compartmental classes upon categorization using GO. Figure 2 shows proteins grouped according to their subcellular localization (Fig. 2A), fold change (Fig. 2B), and biological function (Fig. 2C) across the treatment groups. Functional enzymes represented the largest class of proteins identified. Some enzymes such as exonuclease-1 and DNA polymerase, which are involved in increased cell turnover, were found to be upregulated with BLM treatment. Transcription regulators (eukaryotic translation initiation factor 2A) and transport proteins (stabilin-1) were the other major group of proteins identified. Glycogen synthase kinase-3 (GSK-3β), a kinase involved in positive regulation of wound healing was downregulated with BLM treatment, and constituted a group of kinases that were identified to be differentially regulated. Thus, our proteomic approach presented an unbiased view of dysregulated proteins that were a part of a cross-section of various biological processes and subcellular compartments. The differentially regulated proteins thus identified were subjected to IPA in order to identify important protein-interaction networks and pathways.
Figure 2.
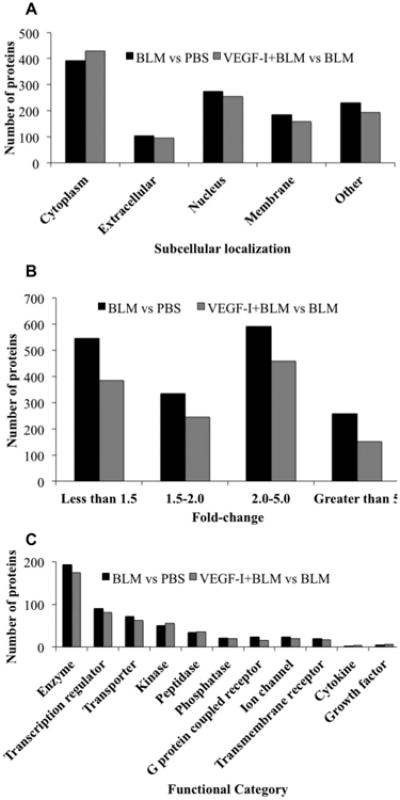
Distribution of identified proteins according to (A) subcellular location, (B) Fold change, and (C) biological process according to GO annotation terms.
3.3 Pathway analysis and network analysis using IPA
The upregulated and downregulated proteins in each comparison group were uploaded into the IPA database to classify proteins according to the associated canonical pathways, and the pathways that had maximum significance values were identified. The significance was determined using Benjamini–Hochberg corrected Fisher's exact test to calculate the p-values for protein datasets within the predetermined canonical pathways, and the ratio of the number of molecules in the dataset associated with a particular pathway to the total number of molecules in that pathway.
Pathways upregulated with BLM treatment included PI3K/Akt signaling (p < 0.0004; ratio = 0.099), epithelial adherens junction signaling, nitric oxide signaling, IL-4 signaling, and endothelin-1 signaling (Fig. 3A). We have successfully validated the involvement of nitric oxide signaling and PI3K/Akt pathways in BLM-induced pulmonary fibrosis, which recapitulates the robustness of our experimental approach in identifying pathways important for the regulation of pulmonary fibrosis [23,24]. The BLM-induced upregulated proteins associated with each of these pathways is shown in Table 1. The pathways associated with proteins downregulated with BLM treatment included Wnt/Ca+ pathway (p < 0.0001; ratio = 0.145), coagulation system, G2/M DNA damage checkpoint regulation, actin cytoskeleton signaling, and integrin signaling (Fig. 3B). The BLM- induced downregulated proteins associated with each of these pathways involved in exacerbating fibrosis are shown in Table 2.
Figure 3.
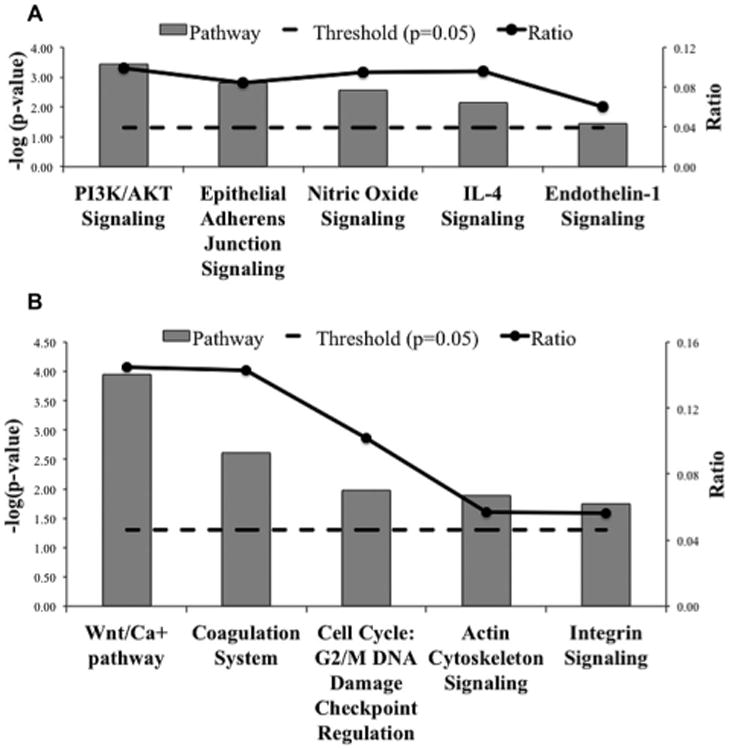
Significant canonical pathways (p < 0.05) for proteins differentially expressed in control versus BLM-treated mice. The negative of the log10(p-value; dashed line) and ratio (number of focus molecules involved in the pathway/total number of molecules in the pathway; solid line joined by circles) are plotted on the primary and secondary Y-axes, respectively. (A) Canonical pathways resulting from proteins upregulated in BLM-treated mice as compared to controls. (B) Canonical pathways resulting from proteins downregulated in BLM-treated mice as compared to controls.
Table 1. Proteins upregulated with BLM treatment and associated with each of the enriched profibrotic pathways.
| Profibrotic enriched pathways | Proteins upregulated with BLM treatment |
|---|---|
| PI3K/AKT signaling | MTOR, HSP90B1, ITGA3, RRAS2, HSP90AB1, INPP5B, SOS2, TSC2, HSP90AA1, INPPL1, NOS3, OCRL |
| Epithelial adherens junction signaling | MYH4, MYH6, RRAS2, MYH13, SORBS1, MYO7A, MYH3, MYL4, IQGAP1, PTPRM, NOTCH1, APC |
| Nitric oxide signaling | HSP90B1, HSP90AB1, RYR2, GUCY2F, CACNA1C, HSP90AA1, NOS3, ATP2A2, ATP2A1 |
| IL-4 signaling | MTOR, RRAS2, INPP5B, SOS2, INPPL1, NFATC4, OCRL |
| Endothelin-1 signaling | RRAS2, GNA15, PLB1, GPLD1, PLA2R1, GNA11, GUCY2F, PLCL2, NOS3, ADCY10 |
Table 2. Proteins downregulated with BLM treatment and associated with each of the enriched profibrotic pathways.
| Profibrotic enriched pathways | Proteins downregulated with BLM treatment |
|---|---|
| Wnt/Ca+ pathway | DVL2, PLCE1, CAMK2A, PDIA3, PDE6C, GSK-3β, PLCD4, EP300 |
| Coagulation system | SERPINC1, F9, F8, F5, THBD |
| Cell cycle: G2/M DNA damage checkpoint regulation | YWHAQ, YWHAZ, TOP2A, ATR, EP300 |
| Actin cytoskeleton signaling | ITGB1, DOCK1, ACTR3, MPRIP, ARHGEF7, PAK7, TLN1, MYLK, ARHGAP24, TTN, SSH1, FGF7 |
| Integrin signaling | RAP1B, ITGB1, DOCK1, ACTR3, MPRIP, ARHGEF7, PAK7, TLN1, MYLK, GSK-3β, TTN |
To evaluate the mechanistic pathways by which VEGFinhibitor may attenuate fibrosis progression, we compared the treatment groups CBO-P11+BLM versus BLM alone. Pathways enriched with proteins upregulated by VEGF inhibitor treatment included natural killer (NK) cell signaling (p < 0.015; ratio = 0.04), PTEN signaling, intrinsic prothrombin activation, gap junction signaling, and SAPK/JNK signaling (Fig. 4A). Proteins upregulated in response to VEGF inhibitor are shown in Table 3. Similarly, therapeutic pathways enriched by the downregulation of proteins with VEGF inhibitor treatment included epithelial adherens junction signaling (p < 8 × 10−11; ratio = 0.16), tight junction signaling, actin cytoskeleton signaling, integrin-linked kinase (ILK) signaling, and focal adhesion kinase (FAK) signaling (Fig. 4B and Table 4).
Figure 4.
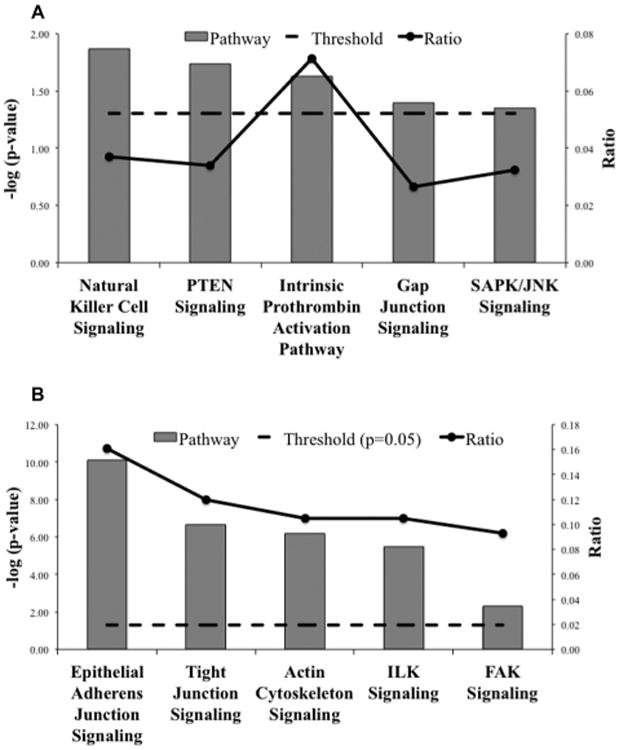
Significant canonical pathways (p < 0.05) for proteins differentially expressed in BLM and VEGF inhibitor cotreated mice as compared to BLM-treated mice. The negative of the log10 (p-value; dashed line) and ratio (number of focus molecules involved in the pathway/total number of molecules in the pathway; solid line joined by circles) are plotted on the primary and secondary Y-axes, respectively. (A) Canonical pathways resulting from proteins upregulated with VEGF inhibitor treatment as compared to BLM. (B) Canonical pathways resulting from proteins downregulated with VEGF inhibitor treatment as compared to BLM.
Table 3. Proteins upregulated with VEGF inhibitor treatment and associated with each of the enriched antifibrotic pathways.
| Antifibrotic enriched pathways | Proteins upregulated with VEGF inhibitor treatment |
|---|---|
| Natural killer cell signaling | PAK6, SOS1, SH3BP2, INPP5D |
| PTEN signaling | FLT1, SOS1, KDR, INPP5D |
| Intrinsic prothrombin activation pathway | FGG, COL3A1 |
| Gap junction signaling | SOS1, PLCD4, HTR2A, MAP3K2 |
| Endothelin-1 signaling | RIPK1, SOS1, MAP3K2 |
Table 4. Proteins downregulated with VEGF inhibitor treatment and associated with each of the enriched antifibrotic pathways.
| Antifibrotic enriched pathways | Proteins downregulated with VEGF inhibitor treatment |
|---|---|
| Epithelial adherens junction signaling | TUBA1B, MYH4, MYH10, MYH6, MYH9, MYH13, MYH8, TUBB4B, MYH14, MLLT4, MYH7, TUBB, IQGAP1, PTPRM, ACTR3, RRAS2, TUBA1A, SORBS1, AKT3, TUBB4A, JUP, VCL, MYH1 |
| Tight junction signaling | MYH4, MYH10, MYH6, MYH9, MYH13, MYH8, TJP1, MYH14, PRKAR2A, MLLT4, MYH7, CASK, NFKB2, CGN, PRKAG2, AKT3, GOSR1, VCL, INADL, MYH1 |
| Actin cytoskeleton signaling | MYH10, MYH4, MYH6, MYH9, MPRIP, MYH13, MYH8, MYH14, MYH7, TLN1, IQGAP1, LIMK1, DOCK1, PAK1, ACTR3, RRAS2, APC2, FLNA, VCL, MYH1, ITGA4, MSN |
| ILK signaling | MYH4, FLNB, MYH10, MYH6, MYH9, MYH13, MYH8, MYH14, FERMT2, VIM, MYH7, NFKB2, DOCK1, MTOR, FLNC, FLNA, AKT3, DSP, MYH1 |
| FAK signaling | DOCK1, PAK1, RRAS2, AKT3, TLN1, VCL, TNS1, ITGA4 |
The network that was generated using proteins upregulated with BLM treatment had functions associated with cellular movement and dermatological diseases and disorder with a p-value of 10−36, with 28 molecules in the dataset being mapped onto the master gene network (Fig. 5A). Similarly, the network generated using proteins downregulated with VEGF inhibitor treatment had functions associated with cancer and organismal injury and abnormalities, with a p-value of 10−35 and 27 molecules of the dataset were mapped onto the network (Fig. 5B). Analysis of the networks demonstrated the highly interconnected central node was occupied by ERK, an integral component of the MAPK/ERK signaling cascade that regulates cellular processes associated with fibrosis including growth, proliferation and survival.
Figure 5.
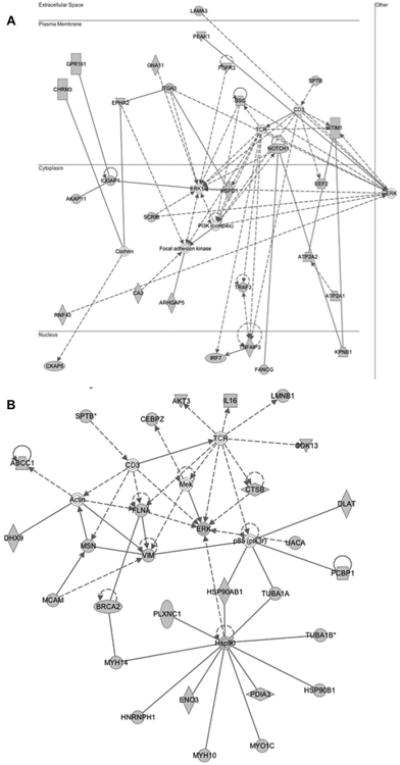
Proteins differentially expressed were overlaid onto a global molecular network developed from information contained in the IKB. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). Solid lines indicate a direct relationship and dashed lines indicate an indirect relationship between nodes. The gray nodes represent the proteins differentially expressed by greater than 1.5-fold identified using proteomics profiling. White nodes represent the IKB molecules associated with focus genes. Networks reflect (A) interaction network for proteins upregulated with BLM treatment (function: cellular movement, dermatological diseases, and disorder; p-value = 10−36) (B) interaction network for proteins downregulated with VEGF inhibitor treatment (function: cancer, organismal injury, and abnormalities; p-value = 10−35).
3.4 Target validation using Western blotting
Immunoblotting was used to validate the high-throughput differential protein expression data and the IPA-inferred pathway and network analysis (Fig. 6). Involvement of mTOR, vimentin, ERK, and β-catenin in BLM-induced pulmonary fibrosis and their regulation by VEGFinhibitor CBO-P11 was validated in vitro using human lung fibroblast cell line and in vivo using mouse model. mTOR, a putative fibrotic marker, was identified to be upregulated 3.9-fold with BLM treatment and downregulated 2.7-fold with VEGF inhibitor treatment using the proteomics approach. Western blotting results confirmed mTOR upregulation in response to BLM treatment and downregulation in response to VEGF inhibitor pretreatment both in in vitro and in vivo samples (Fig. 6). Protein profiling also identified vimentin, an important canonical marker of epithelial-mesenchymal transition (EMT) and hitherto not widely associated with fibrosis, to be downregulated 2.6-fold in response to VEGF inhibitor treatment, which was confirmed in vitro as well as in vivo using Western blotting (Fig. 6). Additionally, upregulation of vimentin was also observed with BLM treatment, although protein profiling did not identify vimentin in this comparison group. Probing for β-catenin, the driver of the canonical Wnt cascade, showed upregulation in response to BLM treatment and downregulation with VEGF inhibitor treatment in human lung fibroblast and mouse lung homogenate samples, validating the involvement of Wnt/Ca+ pathway (Fig. 6). Likewise, immunoblotting for ERK validated the protein interaction networks where ERK featured as a central node. BLM upregulated ERK expression and pretreatment with VEGF inhibitor reversed the effect in in vitro and in vivo samples (Fig. 6).
Figure 6.
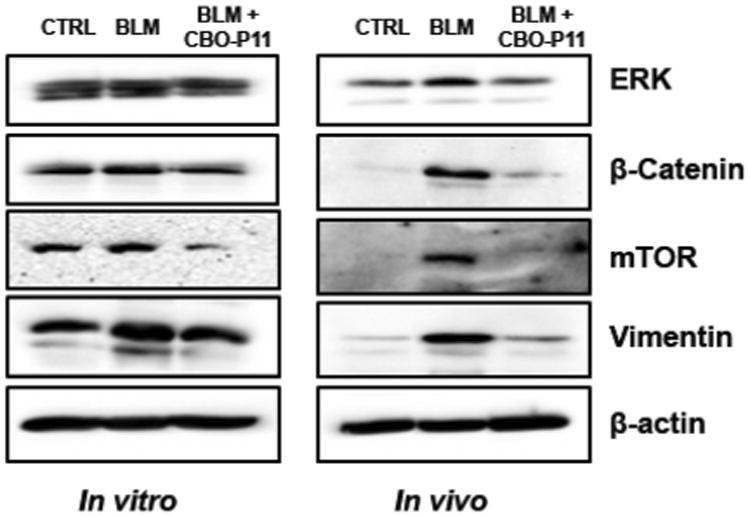
Validation of proteomics and bioinformatics data. Human lung fibroblasts CRL 1490 were treated with BLM (25 mU/mL) and BLM in combination with CBO-P11 (10 μM) for 24 h following which cell lysate was collected. Mice treated with BLM (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg every other day) or equal volumes of saline as control were euthanized at 28 days. Lung homogenate from treated mice and cell lysates was analyzed for mTOR, vimentin, ERK, and β-catenin levels by Western blotting. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. Representative Western blots for mTOR, vimentin, ERK, and β-catenin levels with BLM and VEGF inhibitor treatment with β-actin as a loading control are shown.
4 Discussion
In this study, we have identified a cohort of deregulated proteins and pathways that are involved in the BLM mouse model of pulmonary fibrosis using a combination of high-throughput proteomics and systems biology. More importantly, using the VEGF inhibitor CBO-P11 as a potential therapeutic agent, we have identified a set of important proteins and pathways that can serve as possible therapeutic targets against this chronic and progressive disease. Using our proteomics and systems biology findings, we validated the involvement of mTOR and ERK in pulmonary fibrosis. Some of the key pathways and dysregulated proteins within those modulated pathways that were found to be involved in the progression and attenuation of fibrosis in our study have been discussed below.
4.1 Profibrotic pathways involved in BLM-induced pulmonary fibrosis
4.1.1 PI3K/Akt signaling
The role of PI3K/Akt signaling pathway (Fig. 3A), associated with proteins upregulated with BLM treatment in this study, has been earlier reported by our group in BLM-induced fibrogenesis of human lung fibroblasts in vitro [23]. Furthermore, we have confirmed the involvement of PI3K/Akt pathway in BLM-induced pulmonary fibrosis in vivo [24]. NO was observed to play a key role in BLM-induced pulmonary fibrosis through regulation of VEGF via the PI3K/Akt pathway. The PI3K/Akt pathway has recently been shown to be a therapeutic target for systemic sclerosis, another fibrotic disorder of dermal tissue, and in hepatic fibrosis [25,26]. mTOR, identified to be upregulated with BLM treatment and associated with PI3K/Akt pathway, has been shown to positively regulate the translational efficiency of epithelial cells, sustaining their proliferation and thereby leading to pulmonary fibrosis [27]. Another study reported elevated mTOR levels in lung tissues from patients with IPF exhibiting decreased autophagy. Induction of autophagy using rapamycin, resulted in decreased mTOR levels, increased autophagic flux and led to the abatement of IPF [28]. We demonstrate that cotreatment with VEGF inhibitor leads to downregulation of BLM-induced mTOR expression indicating that mTOR may play a key role in the inhibition of BLM-induced fibrosis in mice treated with the VEGF inhibitor.
4.1.2 Epithelial adherens junction signaling
Epithelial injury is postulated to be one of the primary causative factors in the pathogenesis of IPF. One study investigating the role of adherens junctions in IPF identified that expression levels of both tight and adherens junction proteins were elevated in the alveolar epithelium in IPF as compared to the normal alveolar epithelium [29]. We identified increased flux of epithelial adherens junction signaling (Fig. 3A) in BLM-induced lung injury with increased expression of Ras GTPase-activating-like protein (IQGAP1) and Notch1. IQGAP1, a scaffold protein that regulates cell migration and polarization, has been reported to be elevated in interstitial lung disease [30]. We observed that BLM caused upregulation of Notch1, a Notch family protein involved in the regulation of several developmental processes including proliferation, differentiation, and apoptosis [31]. Activation of Notch pathway has been reported to positively regulate Hes-1, a downstream transcription factor, in patients with IPF [32]. Moreover, Notch-regulated overexpression of Hes-1 has been shown to be involved in interstitial renal fibrosis, and inactivation of Notch1 signaling leads to inhibition of Hes-1, thereby ameliorating renal fibrosis [33]. The identification of the epithelial adherens junction signaling as an enriched pathway highlights its value for therapeutic targeting based on elevated levels of the aforementioned proteins that are directly involved in pulmonary fibrosis.
4.1.3 Wnt/Ca+ pathway
The canonical Wnt/β-catenin pathway (Fig. 3B) is a key regulator of cellular differentiation, proliferation, and polarity. The Wnt/β-catenin pathway has been shown to be activated in BLM-induced lung fibrosis through upregulation of β-catenin and TGF-β, and inhibition of this pathway by using siRNA for β-catenin downregulated TGF-β, resulting in inhibition of pulmonary fibrosis [34,35]. A recent report indicates that BLM treatment causes downregulation of glycogen synthase kinase-3 beta (GSK-3β), a protein that positively regulates wound healing. GSK-3 β knockout mice has been shown to exhibit increased collagen production, increased fibrogenesis, decreased apoptosis and increased myofibroblast formation [36]. Downregulation of GSK-3β has been associated with increased Wnt signaling, resulting in accumulation of β-catenin that binds TCF/LEF transcription factors to induce target gene expression [37]. β-Catenin upregulation observed in BLM-treated mouse tissue was completely ablated in the presence of VEGF Inhibitor (Fig. 6) in our study, indicating the involvement of Wnt/Ca+ pathway in BLM-induced pulmonary fibrosis.
4.1.4 G2/M DNA damage checkpoint regulation
G2/M DNA damage checkpoint was identified as a dysregulated mechanism in response to BLM treatment (Fig. 3B). Cell-cycle deregulation in pathogenesis of fibrosis is poorly characterized, and the only study to associate cell-cycle arrest in the G2/M phase with fibrogenesis was reported in acute kidney injury [38]. The study found that G2/M arrest in renal cells activated JNK signaling, leading to higher production of profibrotic cytokines. Bypassing G2/M arrest with a p53 inhibitor rescued fibrosis in ischemic injured kidney [38]. To our knowledge, ours is the first report positing a role for G2/M DNA damage checkpoint in the pathogenesis of pulmonary fibrosis.
4.2 Antifibrotic pathways regulated in response to VEGF inhibitor treatment
4.2.1 NK cell signaling pathway
NK cell signaling, a pathway associated with increased protein expression with VEGF inhibitor treatment (Fig. 4A), is an early stage innate immune response involved in host defenses against pathogen invasion, tumorigenesis, and fibrogenesis [39]. NK cell stimulated cytotoxicity mediated through STAT1 has been shown to have an inhibitory effect on hepatic fibrosis [40]. On the other hand, antifibrotic effects of NK cell signaling were suppressed during advanced liver fibrosis mediated through increased expression of TGF-β and SOCS1 [41]. We observed increased expression of INPP5D (SH2 containing inositol-5′-phosphatase-1 or SHIP-1) inlung homogenates of mice treated with VEGF inhibitor. SHIP-1 is a negative regulator of cell proliferation and survival, and SHIP-1-deficient mice have been reported to have decreased number of NK cells [42]. The potential of SHIP-1 as a therapeutic target in BLM-induced pulmonary fibrosis was recently demonstrated where AQX-1125, a SHIP-1 activator, inhibited pulmonary fibrosis in a murine model [43]. We observed from proteomic analysis that VEGF inhibitor treatment induced upregulation of SHIP-1, indicating a potential antifibrotic response through increased flux of NK cell signaling pathway (Table 3).
4.2.2 PTEN signaling
PTEN, a negative regulator of angiogenesis and tumorigenesis, is also known to play major roles in embryonic development, cell migration, and apoptosis [44,45]. Loss of PTEN in acute renal injury is reported to cause a loss of differentiation, growth arrest, vimentin upregulation, and profibrotic JNK activation causing renal fibrosis [46]. PTEN deficiency has also been reported to be a contributing factor in dermal and pulmonary fibrosis [47,48]. It is reported that loss of PTEN and SHIP-1 interferes with the levels of phosphatidylinositol (3,4,5)P3, resulting in neutrophils losing their polarity upon adhesion and impairing chemotaxis [49]. PTEN overexpression has been shown to inhibit LPS-induced pulmonary fibrosis through inactivation of PI3K/Akt/GSK-3β, an effect that was observed to be reversible with a selective PTEN inhibitor [50]. Proteomics findings indicate VEGF inhibitor induced SHIP-1 overexpression, resulting in increased PTEN signaling flux (Fig. 4A) thereby mediating an antifibrotic effect in response to BLM.
4.2.3 Epithelial adherens junction signaling
It was interesting to note that epithelial adherens junction signaling was associated with the proteins upregulated with BLM treatment as well as proteins that were downregulated with VEGF inhibitor treatment (Fig. 4B). IQGAP1, a protein that regulates cell migration and invasion was downregulated with VEGF inhibitor treatment in our dataset. Targeted knockdown of IQGAP1 has also been shown to inhibit the progression of esophageal carcinogenesis, thereby demonstrating its important role in regulating cell proliferation and migration [51]. We demonstrate CBO-P11 treatment downregulated Akt3, a protein found to be overexpressed in breast, prostate, and ovarian cancers, and hypothesized to promote cell proliferation by accelerating G2/M transition [52]. Akt3 is a prime therapeutic target in cancer therapy and Akt3 knockdown or inhibition has been shown to induce apoptosis in tumor cells [53, 54]. The therapeutic potential of Akt3 in fibrosis has been recently unraveled where miRNA29b was used to knockdown Akt3, resulting in reduced collagen production and induced apoptosis in hepatic fibrosis [55]. Ours is the first report where profibrotic and antifibrotic involvement of epithelial adherens junction signaling involving its several mediators such as IQGAP1, Notch1, and Akt3 has been reported in pulmonary fibrosis.
4.2.4 ILK signaling
ILK is a serine threonine protein kinase that is involved in regulating integrin-mediated processes such as cell adhesion, proliferation, and extracellular matrix deposition and is a mediator of EMT [56,57]. The only study exploring the role of ILK in BLM-induced pulmonary fibrosis identified that although ILK expression was not altered and did not directly regulate pulmonary fibrosis, it mediated its profibrotic effect through the upregulation of vimentin [58]. Pharmacological inhibition of ILK signaling attenuated renal fibrosis, which was mediated via inhibition of TGF-β-induced phosphorylation of Akt and GSK-3β [59]. Vimentin, a protein downregulated with VEGF inhibitor treatment in our study, has been shown to positively regulate fibrosis through stabilization of collagen mRNAs [60].
4.3 Validation of proteomics and systems biology data
Several proteins were identified to be upregulated with BLM and downregulated with VEGF inhibitor treatment including mTOR and IQGAP1 that could serve as putative diagnostic and therapeutic markers of pulmonary fibrosis. Additionally, we identified oncogenes such as RRAS2, AKT3, and vinculin that induced and abrogated fibrosis with BLM and CBO-P11 treatment, respectively. IPF is a lethal lung disorder that is analogous to cancer in that it results from genetic alterations, is a result of uncontrolled and aberrant cellular proliferation, altered cell-cell communication, and tissue invasion, with the only exception that IPF is not known to metastasize [61]. These oncogenes, hitherto uncharacterized in IPF, are a novel finding that could serve as additional targets for treating IPF.
A disruption of balance between the angiogenic and angiostatic factors regulating blood vessel homeostasis is a factor attributed to this poorly characterized disease [62,63]. Vascular remodeling using an angiogenesis inhibitor has been successfully demonstrated to attenuate fibrosis in vivo [64,65]. We hypothesized that since VEGF inhibitor as a therapeutic agent would modulate the angiogenic pathway, it would be one of the enriched pathways. The robustness and specificity of our approach was validated by the identification of “inhibition of angiogenesis by thrombospondin-1” (p = 0.25; Supporting Information Table 3), identified as enriched pathway associated with the proteins downregulated with VEGF inhibitor treatment. Thrombospondin is reported to play a role in fibrosis through activation of TGF-β [66, 67]. The higher p-value associated with the pathway is a result of only two focus genes along the pathway, HSPG2 and Akt3, exceeding the 1.5-fold change threshold.
To further consolidate our work, we chose to validate our experimental and analytical findings on three levels, which includes protein, network, and pathway level using Western blotting. At the protein level, vimentin was identified to be downregulated with VEGF inhibitor treatment, however it was not identified in the BLM-treated group. Vimentin, an oncogene with limited evidence of its role in wound healing and remodeling, has not been widely associated with fibrosis. Given vimentin's downregulation with VEGF inhibitor, we hypothesized that vimentin is overexpressed with BLM treatment, suggesting its involvement in fibrogenesis. Figure 6 validated this hypothesis, where vimentin overexpression was observed with BLM treatment and its downregulation with VEGF inhibitor treatment confirmed the proteomics findings. On a network level, ERK was identified as the central node in the protein interaction networks, associated with the networks showing both induction (Fig. 5A) as well as inhibition of fibrogenesis (Fig. 5B). Though ERK downregulation was observed with VEGF inhibitor treatment and it was a central focus protein in the antifibrotic network, its involvement as a nonfocus protein in the BLM-induced profibrotic network remained unclear. Consistent with the recent evidence that showed ERK activation in induction and its inhibition in amelioration of fibrosis, we observed ERK upregulation with BLM treatment (Fig. 6) [68,69]. ERK upregulation validated the profibrotic network generated using proteins upregulated with BLM treatment. Lastly, as a means of pathway validation, we chose the Wnt pathway that showed an increased flux with BLM. Sustained activation of Wnt/β-catenin pathway is required for the pathogenesis of fibrotic disorders [70]. Though we identified an increased flux of Wnt pathway on account of GSK-3β downregulation, β-catenin, the key driver of Wnt pathway was not identified as a focus protein. Therefore, we probed for β-catenin levels in BLM and VEGF inhibitor treated mouse lung homogenates. We observed a significant increase in β-catenin levels (Fig. 6) following BLM treatment, which returned to basal levels following VEGF inhibitor treatment, validating the involvement of Wnt pathway in driving fibrosis and its potential as a possible therapeutic target for the treatment of IPF.
4.4 Conclusion
Understanding the mechanisms that regulate transition from pulmonary injury to fibrosis is critical, because it is difficult to switch off or reverse fibrosis once initiated. The novel protein targets and pathways reported in this study with a pulmonary fibrosis model will not only help delineate the etiology of IPF but also will help broaden the focus of therapeutic targets. Further targeted studies designed to test the hypotheses involving the novel proteins and pathways identified in this study will aid in better understanding of the pathogenesis of pulmonary fibrosis and will pave way for the development of novel therapeutic strategies against pulmonary fibrosis.
Supplementary Material
Significance of the study.
Idiopathic pulmonary fibrosis, an invariably fatal disease of an unknown etiology, has no effective therapies due to the poor characterization of molecular targets that contribute to the pathogenesis of the disease. Proteomics, an effective biomarker discovery tool successfully applied in cancer research, has been strikingly underutilized in pulmonary fibrosis. In this study, we have used an untargeted protein based approach togather anunbiased and nonintuitive view of the important regulatory pathways that dictate bleomycin (BLM) induced pulmonary fibrosis. The significance of this study not only lies in the increased understanding of the modalities that dictate pathogenesis of pulmonary fibrosis but also in the development of potential therapeutic and preventive strategies for this fatal disease. In the longer term, the novel proteins and pathways identified in this study may pave way for the development of novel therapeutic strategies against pulmonary fibrosis.
Acknowledgments
We thank the Animal facility staff at ODU for maintaining the animals. We thank Dr. Carlos Castro at Magee Women's Research Institute (MWRI), University of Pittsburgh for assistance with the IHC studies. None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. This work was supported by National Institutes of Health grants HL112630 and CA173069.
Abbreviations
- BLM
bleomycin
- IKB
Ingenuity Pathways Knowledge Base
- ILK
integrin-linked kinase
- IPA
Ingenuity Pathway Analysis
- IPF
idiopathic pulmonary fibrosis
- NK
natural killer
- VEGF
vascular endothelial growth factor
Footnotes
The authors have declared no conflict of interest.
Colour Online: See the article online to view Fig. 1 in colour.
Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- 1.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 2.Renzoni E, Srihari V, Sestini P. Pathogenesis of idiopathic pulmonary fibrosis: review of recent findings. F1000Prime Rep. 2014;6:69. doi: 10.12703/P6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azad N, Iyer AK, Wang L, Liu Y, et al. Reactive oxygen species-mediated p38 MAPK regulates carbon nanotube-induced fibrogenic and angiogenic responses. Nanotoxicology. 2013;7:157–168. doi: 10.3109/17435390.2011.647929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem. 2009;16:1400–1417. doi: 10.2174/092986709787846497. [DOI] [PubMed] [Google Scholar]
- 5.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 6.Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012;5:11. doi: 10.1186/1755-1536-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaminski N, Rosas IO. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: can we identify the right target genes? Proc Am Thorac Soc. 2006;3:339–344. doi: 10.1513/pats.200601-011TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korfei M, von der Beck D, Henneke I, Markart P, et al. Comparative proteome analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF), non-specific interstitial pneumonia (NSIP) and organ donors. J Proteomics. 2013;85:109–128. doi: 10.1016/j.jprot.2013.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Landi C, Bargagli E, Bianchi L, Gagliardi A, et al. Towards a functional proteomics approach to the comprehension of idiopathic pulmonary fibrosis, sarcoidosis, systemic sclerosis and pulmonary Langerhans cell histiocytosis. J Proteomics. 2013;83:60–75. doi: 10.1016/j.jprot.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Foster MW, Morrison LD, Todd JL, Snyder LD, et al. Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res. 2015;14:1238–1249. doi: 10.1021/pr501149m. [DOI] [PubMed] [Google Scholar]
- 11.Landi C, Bargagli E, Carleo A, Bianchi L, et al. A system biology study of BALF from patients affected by idiopathic pulmonary fibrosis (IPF) and healthy controls. Proteomics Clin Appl. 2014;8:932–950. doi: 10.1002/prca.201400001. [DOI] [PubMed] [Google Scholar]
- 12.Gucek M. Proteomics approaches to fibrotic disorders. Fibrogenesis Tissue Repair. 2012;5:S10. doi: 10.1186/1755-1536-5-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decaris ML, Gatmaitan M, FlorCruz S, Luo F, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol Cell Proteomics. 2014;13:1741–1752. doi: 10.1074/mcp.M113.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ideker T, Sharan R. Protein networks in disease. Genome Res. 2008;18:644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Zhang Y, Marian C, Ressom HW. Identification of aberrant pathways and network activities from high-throughput data. Brie Bioinform. 2012;13:406–419. doi: 10.1093/bib/bbs001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirouac DC, Saez-Rodriguez J, Swantek J, Burke JM, et al. Creating and analyzing pathway and protein interaction compendia for modelling signal transduction networks. BMC Syst Biol. 2012;6:29. doi: 10.1186/1752-0509-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Usuki J, Fukuda Y. Evolution of three patterns of intra-alveolar fibrosis produced by bleomycin in rats. Pathol Int. 1995;45:552–564. doi: 10.1111/j.1440-1827.1995.tb03503.x. [DOI] [PubMed] [Google Scholar]
- 19.Simler NR, Brenchley PE, Horrocks AW, Greaves SM, et al. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax. 2004;59:581–585. doi: 10.1136/thx.2003.009860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi: 10.1183/09031936.00152106. [DOI] [PubMed] [Google Scholar]
- 21.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni YM, Suarez V, Klinke DJ. 2nd, Inferring predominant pathways in cellular models of breast cancer using limited sample proteomic profiling. BMC Cancer. 2010;10:291. doi: 10.1186/1471-2407-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Y, Azad N, Wang L, Iyer AK, et al. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. 2010;42:432–441. doi: 10.1165/rcmb.2009-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer AK, Ramesh V, Castro CA, Kaushik V, et al. Nitric Oxide Mediates Bleomycin-Induced Angiogenesis and Pulmonary Fibrosis via Regulation of VEGF. J Cell Biochem. 2015;116:2484–2493. doi: 10.1002/jcb.25192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang M, Lv J, Chu H, Wang J, et al. Vertical inhibition of PI3K/Akt/mTOR signaling demonstrates in vitro and in vivo anti-fibrotic activity. J Dermatol Sci. 2014;76:104–111. doi: 10.1016/j.jdermsci.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Son MK, Ryu YL, Jung KH, Lee H, et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci Rep. 2013;3:3470. doi: 10.1038/srep03470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stella GM, Inghilleri S, Pignochino Y, Zorzetto M, et al. Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl Oncol. 2014;7:650–655. doi: 10.1016/j.tranon.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel AS, Lin L, Geyer A, Haspel JA, et al. Autophagy in idiopathic pulmonary fibrosis. PloS One. 2012;7:e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lappi-Blanco E, Lehtonen ST, Sormunen R, Merikallio HM, et al. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum Pathol. 2013;44:895–907. doi: 10.1016/j.humpath.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Bogatkevich GS, Ludwicka-Bradley A, Singleton CB, Bethard JR, Silver RM. Proteomic analysis of CTGF-activated lung fibroblasts: identification of IQGAP1 as a key player in lung fibroblast migration. Am J Physiol. 2008;295:L603–611. doi: 10.1152/ajplung.00530.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 32.Plantier L, Crestani B, Wert SE, Dehoux M, et al. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax. 2011;66:651–657. doi: 10.1136/thx.2010.151555. [DOI] [PubMed] [Google Scholar]
- 33.Bielesz B, Sirin Y, Si H, Niranjan T, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040–4054. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Carron B, Yee HT, Yie TA, et al. Wnt pathway in pulmonary fibrosis in the bleomycin mouse model. J Environ Pathol Toxicol Oncol. 2009;28:99–108. doi: 10.1615/jenvironpatholtoxicoloncol.v28.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim TH, Kim SH, Seo JY, Chung H, et al. Blockade of the Wnt/beta-catenin pathway attenuates bleomycin-induced pulmonary fibrosis. Tohoku J Exp Med. 2011;223:45–54. doi: 10.1620/tjem.223.45. [DOI] [PubMed] [Google Scholar]
- 36.Kapoor M, Liu S, Shi-wen X, Huh K, et al. GSK-3beta in mouse fibroblasts controls wound healing and fibrosis through an endothelin-1-dependent mechanism. J Clin Invest. 2008;118:3279–3290. doi: 10.1172/JCI35381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 40.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 41.Jeong WI, Park O, Suh YG, Byun JS, et al. Suppression of innate immunity (natural killer cell/interferon-gamma) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53:1342–1351. doi: 10.1002/hep.24190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang JW, Howson JM, Ghansah T, Desponts C, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295:2094–2097. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- 43.Grant RS, Lloyd FM, Emanuale E, Salvatore C, et al. Prophylactic or therapeutic AQX-1125, a small molecule SHIP1 activator, inhibits bleomycin-induced pulmonary fibrosis in mice, May. 2013:A5660. [Google Scholar]
- 44.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 45.Jiang BH, Liu LZ. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res. 2009;102:19–65. doi: 10.1016/S0065-230X(09)02002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lan R, Geng H, Polichnowski AJ, Singha PK, et al. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol. 2012;302:F1210–F1223. doi: 10.1152/ajprenal.00660.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parapuram SK, Thompson K, Tsang M, Hutchenreuther J, et al. Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2-dependent mechanism. Matrix Biol. 2015;43:35–41. doi: 10.1016/j.matbio.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 48.Liu S, Parapuram SK, Leask A. Fibrosis caused by loss of PTEN expression in mouse fibroblasts is crucially dependent on CCN2. Arthritis Rheum. 2013;65:2940–2944. doi: 10.1002/art.38121. [DOI] [PubMed] [Google Scholar]
- 49.Mondal S, Subramanian KK, Sakai J, Bajrami B, Luo HR. Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol Biol Cell. 2012;23:1219–1230. doi: 10.1091/mbc.E11-10-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He Z, Deng Y, Li W, Chen Y, et al. Overexpression of PTEN suppresses lipopolysaccharide-induced lung fibroblast proliferation, differentiation and collagen secretion through inhibition of the PI3-K-Akt-GSK3beta pathway. Cell Biosci. 2014;4:2. doi: 10.1186/2045-3701-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang XX, Wang K, Li XZ, Zhai LQ, et al. Targeted knockdown of IQGAP1 inhibits the progression of esophageal squamous cell carcinoma in vitro and in vivo. PloS One. 2014;9:e96501. doi: 10.1371/journal.pone.0096501. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Cristiano BE, Chan JC, Hannan KM, Lundie NA, et al. A specific role for AKT3 in the genesis of ovarian cancer through modulation of G(2)-M phase transition. Cancer Res. 2006;66:11718–11725. doi: 10.1158/0008-5472.CAN-06-1968. [DOI] [PubMed] [Google Scholar]
- 53.Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2009;22:400–419. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stahl JM, Sharma A, Cheung M, Zimmerman M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Chu ES, Chen HY, Man K, et al. microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget. 2014;6:7325–7338. doi: 10.18632/oncotarget.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 57.Shafieian M, Chen S, Wu S. Integrin-linked kinase mediates CTGF-induced epithelial to mesenchymal transition in alveolar type II epithelial cells. Pediatr Res. 2015;77:520–527. doi: 10.1038/pr.2015.8. [DOI] [PubMed] [Google Scholar]
- 58.Kavvadas P, Kypreou KP, Protopapadakis E, Prodromidi E, et al. Integrin-linked kinase (ILK) in pulmonary fibrosis. Virchows Arch. 2010;457:563–575. doi: 10.1007/s00428-010-0976-7. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Tan X, Dai C, Stolz DB, et al. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:1907–1918. doi: 10.1681/ASN.2008090930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Challa AA, Stefanovic B. A novel role of vimentin filaments: binding and stabilization of collagen mRNAs. Mol Cell Biol. 2011;31:3773–3789. doi: 10.1128/MCB.05263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- 62.Kalluri R, Sukhatme VP. Fibrosis and angiogenesis. Curr Opin Nephrol Hypertens. 2000;9:413–418. doi: 10.1097/00041552-200007000-00013. [DOI] [PubMed] [Google Scholar]
- 63.Hanumegowda C, Farkas L, Kolb M. Angiogenesis in pulmonary fibrosis: too much or not enough? Chest. 2012;142:200–207. doi: 10.1378/chest.11-1962. [DOI] [PubMed] [Google Scholar]
- 64.Wang X, Zhu H, Yang X, Bi Y, Cui S. Vasohibin attenuates bleomycin induced pulmonary fibrosis via inhibition of angiogenesis in mice. Pathology. 2010;42:457–462. doi: 10.3109/00313025.2010.493864. [DOI] [PubMed] [Google Scholar]
- 65.Tabata C, Tabata R, Kadokawa Y, Hisamori S, et al. Thalidomide prevents bleomycin-induced pulmonary fibrosis in mice. J Immunol. 2007;179:708–714. doi: 10.4049/jimmunol.179.1.708. [DOI] [PubMed] [Google Scholar]
- 66.Hugo C. The thrombospondin 1-TGF-beta axis in fibrotic renal disease. Nephrol Dial Transplant. 2003;18:1241–1245. doi: 10.1093/ndt/gfg159. [DOI] [PubMed] [Google Scholar]
- 67.Frolova EG, Sopko N, Blech L, Popovic ZB, et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012;26:2363–2373. doi: 10.1096/fj.11-190728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Madala SK, Schmidt S, Davidson C, Ikegami M, et al. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol. 2012;46:380–388. doi: 10.1165/rcmb.2011-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leask A. MEK/ERK inhibitors: proof-of-concept studies in lung fibrosis. J Cell Commun Signal. 2012;6:59–60. doi: 10.1007/s12079-011-0156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akhmetshina A, Palumbo K, Dees C, Bergmann C, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.