

Abstract
Purpose
Breast cancer is the second leading cause of cancer mortality among women worldwide. The major problem with current treatments is tumor resistance, recurrence, and disease progression. ErbB-2 positive breast tumors are aggressive and frequently become resistant to trastuzumab or lapatinib. We showed previously that Notch-1 is required for trastuzumab resistance in ErbB-2 positive breast cancer.
Experimental Design
Here, we sought to elucidate mechanisms by which ErbB-2 attenuates Notch signaling and how this is reversed by trastuzumab or lapatinib.
Results
The current study elucidates a novel Notch inhibitory mechanism by which PKCα downstream of ErbB-2: 1. Restricts the availability of Jagged-1 at the cell surface to trans activate Notch; 2. Restricts the critical interaction between Jagged-1 and Mindbomb-1, an E3 ligase that is required for Jagged-1 ubiquitinylation and subsequent Notch activation; 3. Reverses trastuzumab resistance in vivo; and 4. Predicts better outcome in women with ErbB-2 positive breast cancer.
Conclusions
The clinical impact of these studies is PKCα is potentially a good prognostic marker for low Notch activity and increased trastuzumab sensitivity in ErbB-2 positive breast cancer. Moreover, women with ErbB-2 positive breast tumors expressing high Notch activation and low PKCα expression could be the best candidates for anti-Notch therapy.
Keywords: Breast Cancer, ErbB-2, Jagged-1, Notch, Trastuzumab, Resistance, PKCα
Introduction
Breast cancer is the second leading cause of cancer-related deaths in women. Main hurdles for women are drug resistance, recurrence, and disease progression (1, 2). Breast cancer is a heterogeneous disease comprising at least four major subtypes, i.e. luminal A, luminal B, HER2, and basal/triple negative (3). The majority of breast cancers express estrogen, progesterone receptors (ER, PR), and/or the human epidermal growth factor receptor-2 (HER2 or ErbB-2). The ErbB-2 proto-oncogene is amplified in 15–25% of breast cancers (4, 5). ER and/or ErbB-2 are targets of therapies which include selective estrogen receptor modifiers (SERMS) such as tamoxifen (6), aromatase inhibitors (7), or selective estrogen receptor disruptor (SERD) fulvestrant (8) for ER/PR-positive breast cancer or trastuzumab (9) for ErbB-2-positive breast cancer.
Despite trastuzumab’s efficacy and dramatic effects on survival, 20–50% of women with ErbB-2–positive, metastatic breast cancer exhibit intrinsic resistance (10). Furthermore, 10–15% of the women treated with trastuzumab plus chemotherapy developed acquired resistance within the first year (11). Thus, understanding resistant mechanisms is critical for identifying novel targets to prevent and/or reverse resistance. Numerous mechanisms are implicated in resistance including loss of PTEN, overexpression of IGF-1R, truncation of ErbB-2, hyperactivation of PI3-K and mTOR signaling, among others (12).
Protein kinase C-α (PKCα) is another mediator of the ErbB-2 pathway. ErbB-2 activates phospholipase C γ (PLCγ) which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), triggering an elevation of intracellular Ca+2 and activation of PKCα (13). PKCα overexpression promotes tamoxifen resistance (14) and invasion (15). Recently, PKCα was shown to mediate breast cancer stem cell survival (16) and is a therapeutic target in triple negative breast cancer (17). However, the role of PKCα in the ErbB-2-positive breast cancer and its significance for anti-ErbB-2-targeted therapy remains unclear.
Notch ligands (Delta-like 1, 3, and 4 and Jagged-1 and -2) and receptors (Notch-1, -2, -3, and -4) are implicated in breast cancer development and drug resistance (18). They require cell-cell contact for engagement and subsequent cleavage of membrane-bound receptors to intracellular transcriptional activators. The ligand-induced Notch activation is regulated by E3 ubiquitin ligases, Mindbomb1 (18) and Neuralized (19). Co-overexpression of Notch-1 and Jagged-1 predicts for the poorest overall survival (20). Notch-1, Notch-3, and Notch-4 are breast oncogenes and potent regulators of cell differentiation, proliferation, and apoptosis (21). High Notch-1 and Notch-4 expression correlates to poor prognosis (22) and Notch-3 promotes ErbB-2 negative breast cancer cell proliferation (23). Notch receptor-ligand interactions are important for mammary stem cell differentiation (24) and tumor-initiating cells (25).
We showed that trastuzumab or a dual EGFR/ErbB-2 inhibitor increased Notch-1 activity, and trastuzumab resistance was reversed by Notch-1 knockdown or a γ-secretase inhibitor (26). Subsequently, we demonstrated that dual targeting of ErbB-2 and Notch prevented recurrence and partially reversed resistance to trastuzumab (27). Here, we demonstrate a novel mechanism of action: PKCα attenuates Mib-1-mediated Jagged-1-Notch activation to predict sensitivity to trastuzumab. Furthermore, breast cancers expressing low PKCα might be more sensitive to anti-ErbB-2 agents. Importantly, anti-Jagged-1 therapy could reverse resistance to trastuzumab by attenuating Jagged-1-mediated Notch activity.
Materials and Methods
Cell Culture and Reagents
MDA-MB-453, BT474, and HCC1954 breast cancer cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) within the last 6 years. BT474 trastuzumab resistant (BT474 Resistant) cells were generated by treating parental BT474 cells with increasing concentrations of trastuzumab for 6 months (26). All cell lines were authenticated using STR allelic profiling (DCC Medical, Fairfield, OH).
Biotinylation Assay
MDA-MB-453 cells were seeded in 10 cm plates at a density of 40 – 50% confluency. After 48 hours, cells were treated with either mouse IgG (20 μg/ml in PBS) or trastuzumab (20 μg/ml in PBS) for 48 hours. The cells were washed 3 times in PBS and a cell impermeable biotinylation reagent EZ-Link Sulfo-NHS-Biotin Reagent (Pierce Chemicals, Rockford, IL, USA) as previously described (28) was added to cells to label cell surface proteins at 4°C under constant shaking. Cells were scraped, centrifuged at 1000 rpm for 1 min, washed twice with PBS, and lysed in RIPA lysis buffer. The biotinylated cell surface proteins were precipitated from the total protein lysate using 30 μl of Immobilized Neutravidin Protein (Cat. 29200, Pierce Chemicals, Rockford, IL, USA). The beads were washed 4 times with PBS and the protein was eluted with 4X SDS Laemmli buffer and then heated at 95°C for denaturation. Western blotting was performed to detect Jagged-1 and ErbB-2 as described in the supplemental section.
Confocal Immunofluorescence microscopy
MDA-MB-453, BT474HS, or BT474HR cells were either transfected with control siRNA, PKCα siRNA or ErbB-2 siRNA, or transfected with LRZS-linker alone or LRZS-PKCα and subsequently plated into chamber slides, or untransfected cells were treated with PBS or 20 μg/ml trastuzamab directly in the well. Cells were then fixed in the chamber with 3.7% paraformaldahyde, permeabilized with 0.1% saponin, and non-specific interactions were blocked with 1% BSA. The staining protocol for specific proteins of interest are provided in the supplemental section.
Co-Immunoprecipitation (Co-IP)
Jagged-1(Jagged 1 2H28 rabbit monoclonal antibody, Cat. 2620, Cell Signaling) was immunoprecipitated under treatment conditions as described in the figure legends. Lysates at a concentration of 3–5mg/mL were incubated with 6μg of antibody for the specific protein of interest or rabbit isotype control IgG (Cat. SC-2027, Santa Cruz, Santa Cruz, CA, USA) overnight with gentle rocking at 4°C. Thirty microliters of protein A-plus beads (sc-2002, Santa Cruz, CA, USA) were added to the immune complexes for two hours, the beads were washed with lysis buffer and the proteins attached to the pelleted beads were eluted with 20μl of 2X Laemmli sample buffer plus β-mercaptoethanol and heated for 10 minutes at 95°C while vigorously shaking. Immunoblots are described in supplemental section.
Co-culture Assay
MDA-MB-453 cells were plated at a density of 3×106 cells in a 10 cm plate. Mouse fibroblast (LTK) cells expressing no ligand or overexpressing Jagged-1 were added in equal parts (1:1) to previously plated MDA-MB-453 cells. The cells were then treated with PBS or 20μg/ml trastuzumab for 18 hours. Cells were then stained with 10μL of PE-conjugated human-ErbB-2 antibody (Cat. 340552, Becton Dickinson, Franklin Lakes, NJ, USA). Stained cells were then sorted for ErbB-2 expression at the cell surface by FACS. Total RNA was extracted from sorted cells, reverse transcribed to total cDNA, and real-time PCR was performed as previously described. The PCR primers that were used for detection of specific transcripts were Hes-1 and Deltex-1 with RPL13a as a loading control.
Trastuzumab Resistant Xenografts
Five million BT474 trastuzumab resistant cells were injected bilaterally into mammary fat pads of ovariectomized, FoxN1nu/nu athymic nude mice (Harlan Sprague-Dawley, Madison, WI, USA) followed by implantation of a 17β-estradiol-containing silastic capsule of 0.3cm in length with a constant release providing 83–100pg/mL as described previously (29). The identity of each mouse and tumor was tracked by an ear tag. Once tumors grew to a mean cross sectional area (CRA) of 0.30–0.50 cm2, mice were randomized to four treatment groups: 1. Vehicle control (PBS), 2. trastuzumab (10mg/Kg in a total volume of 100 μL sterile PBS, i.p. once weekly), 3. CTX-033 (5mg/kg, i.p. once weekly), or 4. trastuzumab plus CTX-033. In case of BT474HR cells retrovirally transduced with the LRZS-linker or LRZS-PKCα, these tumors were randomized to two treatment groups each: 1. PBS or 2. trastuzumab. Tumor area (l x w) was measured weekly using Vernier calipers and cross sectional area [(l x w)Π)/4] was calculated and graphed. All animal study protocols were approved by Loyola University’s Institutional Animal Care and Use Committee.
Flow Cytometry
MDA-MB-453 cells were treated with indicated dose of lapatinib/trastuzumab for 48 hours. DMSO and IgG were used as negative controls. Cells were harvested using Cellstripper™ (Cat. 25-056-CI Corning Cellgro) and were stained with fluorescein isothiocyanate (FITC) conjugated Jagged-1 antibody as per the manufacturer’s protocol. The cells were then resuspended in Flow Cytometry Staining Buffer (Cat. FC001 R&D Systems) and analyzed with BD FACS Canto™ II. Data acquisition was done using BD FACSDiva™ software and data analysis was performed using FlowJo software.
Proliferation Assay
MDA-MB-453 cells were seeded into 10cm dishes at 5×106 cells. HCC-1954 cells were seeded into 10cm dishes at 3.5×106 cells. The cells were transfected as described in the RNA interference section of these methods. After completion of the siRNA transfection, the cells were seeded in triplicate into 6-well plates at a density of 50,000 for MDA-MB-453 cells or HCC-1954 cells. BT474HS Sensitive cells were seeded into 6-well plates at a density of 60,000 cells. BT474HR resistant cells were seeded into 6-well plates at a density of 60,000 cells. After plating into the 6-well plates, cells were treated with PBS or 20μg/ml trastuzamab on a daily basis. Cells were counted at days 2, 4, 6, and 8 post treatment, cells were trypsinized into single cell suspensions and counted using the Countess Automated Cell Counter (Cat. C10310, Life Technololgies, Grand Island, NY, USA). The live cell number was then used to calculate the total number of live cells over the number of live cells plated.
Immunohistochemical staining of Human ErbB-2 positive breast tumors
TMA sections were placed in a 58–60°C oven overnight for tissue to adhere. The sections were deparaffinized in xylene, rehydrated through graded ethanol and washed with PBS before being treated with 1X Reveal in a Decloaking Chamber (Biocare Medical, CA) for antigen retrieval following the manufacturer’s protocol. After rinsed in PBS for 15 min, the sections were soaked in 3% H2O2 in PBS for 20 min. to quench endogenous peroxidase activity. Sections were incubated for 60 min in 3% normal rabbit serum (Vector Laboratories, CA) in PBS at room temperature to block non-specific binding sites and then probed with primary antibodies (PKCα antibody was Santa Cruz C20 at 1μg/ml, with non-immune IgG used as controls. The details of the staining protocol are in the supplemental section.
Statistical Analysis
Most experiments were performed at least three times. Means plus or minus standard deviations were calculated on at least N=3 experiments. A two-tailed Student’s T-test was performed on results with two comparisons. ANOVA was performed on results with multiple comparisons. Linear regression analysis was performed on tumor growth studies as each mouse was tagged with a number and each tumor was measured independently followed by ANOVA for multiple comparisons. A Kaplan-Meier curve for recurrence free survival or overall survival of human patients was generated by the Kaplan-Meier Plotter software (30) or generated by GraphPad Prism software and analyzed with a Log rank (Mantel-Cox) test.
Supplemental Material and Methods: More detailed methods are provided in the supplemental section.
Results
Jagged-1 is required for trastuzumab-induced Notch activation
We demonstrated that inhibiting ErbB-2 increased NICD1 expression, CBF-1-driven reporter activity, DELTEX1 and HEY1 transcripts, and thus most likely canonical Notch activation (26). Jagged-1 predicts poor outcome in women with breast cancer (31). Here, we sought to understand if ErbB-2 attenuates Notch signaling by regulating Jagged-1 expression and/or cellular localization in breast cancer cells. Figure 1A confirms that ErbB-2 inhibits Notch transcriptional activity. Conversely, ErbB-2 inhibition using a kinase-dead mutant, ErbB-2 siRNA, or lapatinib increased RNA transcripts of Notch gene targets, HES1, DELTEX1, and/or HEY1 (Figure 1A). Figure 1B shows that trastuzumab or lapatinib increased Jagged-1 protein expression on the cell surface by flow cytometry. Furthermore, this increase in Jagged-1 protein expression by trastuzumab treatment is restricted to the cell surface as measured by a cell surface-specific biotinylation assay (Figure 1C). These results indicate that ErbB-2 inhibition promoted the accumulation of Jagged-1 at the cell surface.
Figure 1.
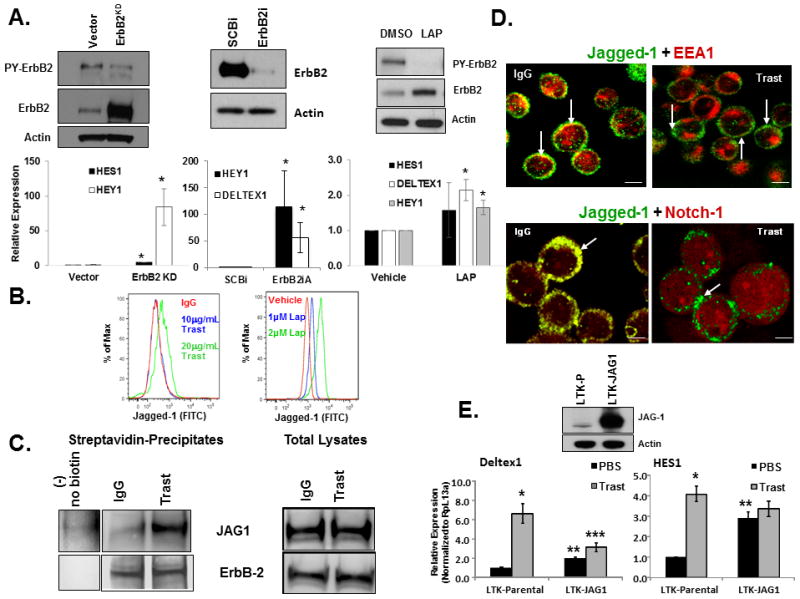
ErbB2 Inhibits Notch Signaling and Jagged-1 Cell Surface Expression. A, ErbB2 expression and activity inhibits Notch activation. Left panel: MDA-MB-453 cells were stably transfected with vector alone or kinase dead ErbB2 (ErbB2KD) in an expression plasmid. Middle panel: MDA-MB-453 cells were transfected with a scrambled control siRNA (SCBi) or an ErbB2 siRNA. Right panel: MDA-MB-453 cells were treated with DMSO or lapatinib (LAP) for 6 days. Total protein from cell lysates were subjected to SDS-PAGE followed by western blotting to detect tyrosine phosphorylated ErbB2 (PY-ErbB2), total ErbB2, and Actin proteins. Lower panels: similar studies were performed as described in A, and real-time PCR was performed to detect RNA transcript levels for HES1, HEY1, and/or DELTEX1. The bar graphs depict means plus or minus standard deviations of three independent experiments. *Denotes statistical significance p<0.05 using a non-paired Student’s T-test. B, Trastuzumab or lapatinib increases cell surface Jagged-1 protein expression. MDA-MB-453 cells were treated with IgG control, DMSO, or increasing concentrations of trastuzumab or lapatinib for 48 hours. Flow cytometry was performed on live cells to detect cell surface expression of Jagged-1 protein. The results are representative of at least three independent studies. C, Inhibition of ErbB-2 increases cell surface Jagged-1 expression. MDA-MB-453 cells were treated with IgG or trastuzumab for 48 hours. Cell surface proteins were biotinylated as described in Experimental Procedures followed by Streptavidin precipitation and western blotting to detect the following proteins: Jagged-1 (JAG1) and total ErbB-2 in both precipitates and total lysates. D, Trastuzumab promotes Jagged-1 surface localization and Notch-1 activation as shown by confocal immunofluorescence. Upper panel scale bars = 20μm; lower panel scale bars = 10μm. E, Jagged-1 mediated trans-activation of Notch. MDA-MB-453 cells were co-cultured with mouse fibroblasts expressing no ligand (LTK-Parental) or overexpressing Jagged-1 (LTK-JAG1) in a 1:1 ratio and then treated with PBS or trastuzumab for 18 hours. Cells were sorted by flow cytometry for ErbB-2 positive cells. RNA was extracted and real-time PCR was performed to detect relative transcript levels: HES1 (right panel) or Deltex1 (left panel). Western blotting was performed on LTK cells to detect levels of Jagged-1 protein (middle panel). *Denotes statistical significance p<0.05 between PBS and Trast, ** denotes statistical significance between LTK-P and LTK-JAG1, and *** denotes statistical significance between LTK-P and LTK-JAG1 in response to Trast using an ANOVA.
To confirm the supporting cell surface biochemical assays from Figure 1B and 1C that ErbB-2 overexpression may restrict cell surface expression of Jagged-1. We performed confocal fluorescence microscopy to visualize Jagged-1 cellular localization. The IgG control cells showed that a portion of Jagged-1 co-localized with early endosomal antigen-1 (EEA-1) (Figure 1D). In contrast, trastuzumab treated cells showed that Jagged-1 was no longer retained in EEA-1 positive endosomes and primarily accumulated at the cell surface (Figure 1D). Furthermore, Notch-1 co-localized with Jagged-1 in control cells (Figure 1D). However, trastuzumab treatment induced accumulation of Notch-1 throughout the cell and Jagged-1 at sites of cell-cell contacts (Figure 1D). These results indicate that ErbB-2 overexpression possibly traps both Jagged-1 and Notch-1 in sub-plasma membrane compartments including early endosomes. However, anti-ErbB-2 treatment with trastuzumab disrupts the Jagged-1/Notch-1 co-localization.
If Jagged-1-mediated trans-activation of Notch is attenuated by ErbB-2, we predicted that supplying the Notch-expressing breast cancer cells with stromal cells expressing abundant surface Jagged-1 would restore Notch activation. We co-cultured ErbB-2 expressing MDA-MB-453 breast cancer cells with mouse fibroblasts expressing no ligand (LTK-P) or overexpressing Jagged-1 (LTK-JAG-1) and then treated with vehicle or trastuzumab. Trastuzumab increased both HES1 (4-fold; Figure 1E right panel) and DELTEX1 mRNAs (7-fold; Figure 1E left panel) in ErbB-2+ cells when co-cultured with LTK-P cells. Providing MDA-MB-453 cells with abundant Jagged-1 in trans increased HES1 mRNA by 3-fold (Figure 1E right panel) and DELTEX1 mRNA by more than 2 fold (Figure 1E left panel) in the presence or absence of trastuzumab. Interestingly, trastuzumab treatment in the presence of LTK-JAG1 did not increase DELTEX-1 levels compared to LTK-P cells. It is possible that there are cis-inhibitory mechanisms within the breast cancer cells that limited the activation of the Notch gene target. These results indicate that ErbB-2 most likely restricts Jagged-1 cell surface localization and limits trans-activation of Notch.
To determine whether Jagged-1 is necessary for trastuzumab-induced Notch activation, we used a genetic approach to downregulate Jagged-1 in two ErbB-2-positive breast cancer cell lines. Different Jagged-1 siRNAs (JAG1iA or JAG1iNew) decreased Jagged-1 protein expression (Figures 2A, 2B, and Supplemental Figure 1A) and significantly inhibited the trastuzumab-induced increase of Notch gene targets, HES1, DELTEX1, HEY1, and/or NOTCH4 transcripts (Figures 2A, 2B, and Supplemental Figure 1A). These results show that Notch activation mediated by ErbB-2 inhibition is most likely Jagged-1 dependent.
Figure 2.
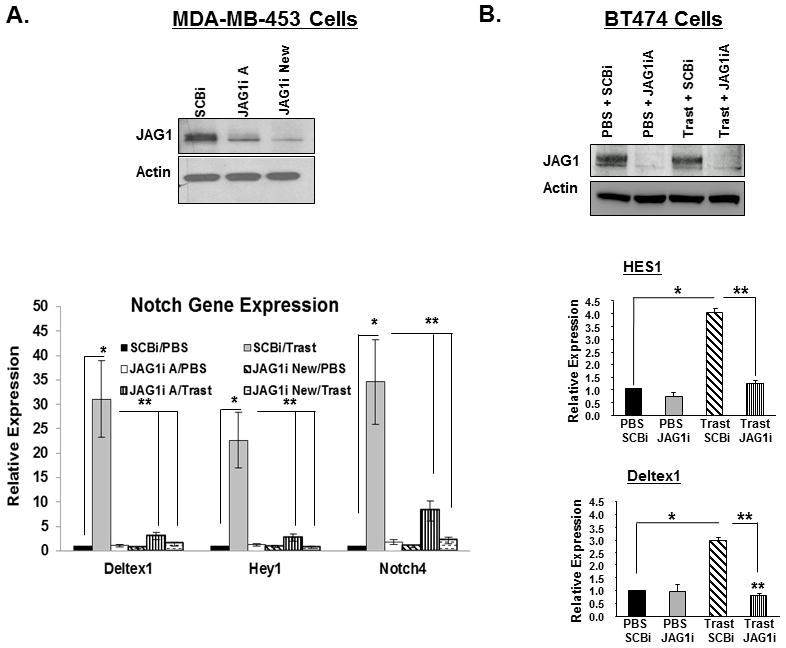
Jagged-1 is required to promote trastuzumab-induced Notch activation. A, Jagged-1 is required for trastuzumab-induced increase of Notch gene targets. MDA-MB-453 were transfected with control siRNA (SCBi) or two distinct Jagged-1 siRNAs (JAG1iA or JAG1i New) and then treated with PBS or trastuzumab. Protein expression of Jagged-1 or Actin was determined by Western blotting (upper panel). Transcript levels of Notch gene targets, Deltex1, Hey1, and Notch4 were measured by real-time PCR (lower two panels). The results are means plus or minus standard deviations of three independent experiments. B, BT474 cells were transfected with SCBi or JAG1iA siRNA and then treated with PBS or trastuzumab for 48 hours. Western blotting was performed to detect efficiency of Jagged-1 knock down (upper panel) and real-time PCR was performed to detect transcripts of Notch gene targets: HES1 and Deltex1 (lower panels). The results are means plus or minus standard deviations of three independent experiments. * and **Denotes Statistical significance of p<0.05 as determined by an ANOVA.
Dual blockade of ErbB-2 and Jagged-1 inhibits proliferation in-vitro and reverses resistance in-vivo
We tested whether decreasing Jagged-1 by siRNA would enhance trastuzumab’s effectiveness in a panel of ErbB-2-positive breast cancer cells that are sensitive (MDA-MB-453 and BT474-Sensitive) or resistant (BT474-Resistant and HCC1954) to trastuzumab. Figure 3A shows that proliferation of MDA-MB-453 cells was decreased by 50% upon trastuzumab treatment. Two distinct Jagged-1 siRNAs (JAG1iA or JAG1iNew) decreased the proliferation similarly to trastuzumab, by almost 50% (Figure 3A). However, the proliferation of MDA-MB-453 cells was decreased by almost 75% when cells were treated with both trastuzumab and a Jagged-1 siRNA (Figure 3A). Proliferation of BT474 (Sensitive) was slightly decreased upon Jagged-1 knockdown (Figures 3B). Proliferation of trastuzumab resistant BT474 cells was significantly decreased upon Jagged-1 knockdown alone. In addition, resistance was reversed upon Jagged-1 knockdown (Figure 3B). Additionally, HCC1954 cells that are inherently resistant to trastuzumab were partially inhibited by Jagged-1 knockdown and trastuzumab treatment (Supplemental Figure 1B). These results indicate that Jagged-1 is required for trastuzumab resistance.
Figure 3.
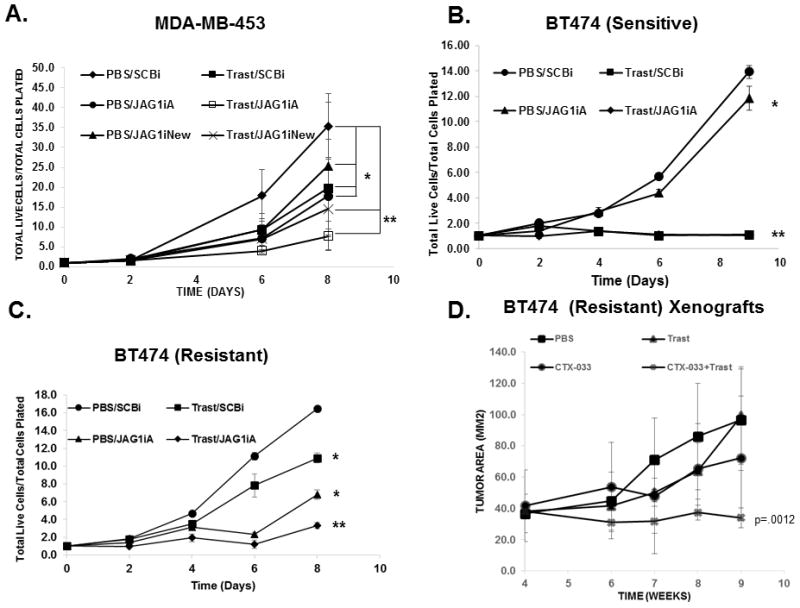
Jagged-1 is necessary for Trastuzumab Resistance. A, MDA-MB-453 cells were transfected with SCBi or JAG1iA or JAG1iNew siRNAs and then treated with PBS or trastuzumab once daily for up 8 days. Proliferation was measured and calculated as total live cells/total live cell plated at the designated days. B and C, BT464 parental cells (B) and BT474 trastuzumab resistant cells (C) were transfected with SCBi or JAG1iA and treated as described in A. * Denotes statistical significance p<0.05 and ** p<0.01 were calculated based on means plus or minus standard deviations of three replicates using an ANOVA D, Trastuzumab resistant breast tumors are inhibited by Jagged ProbodyTM CTX-033 plus trastuzumab. BT474 trastuzumab resistant cells were injected into mammary fat pads of 40, 5-6 week old female nude, athymic mice. Tumors were allowed to grow to a mean tumor cross sectional area of 40mm2 and then randomized to four treatment groups: 1. PBS, 2. Trastuzumab (Trast), 3. CTX-033, or 4. CTX-033 plus Trast for up to 9 weeks. Tumor area (LXW) was measured weekly using Vernier calipers. The results are means plus or minus standard deviation of 5-10 tumors per group. Statistical significance was calculated using an ANOVA.
Previously, Pandya et al demonstrated partial restoration of trastuzumab sensitivity and prevention of ErbB-2 positive breast tumor recurrence using a combination of trastuzumab plus a γ-secretase inhibitor (GSI) (27). GSIs are pan-Notch inhibitors and might not specifically target Notch-1. To determine if Jagged-1 is required for trastuzumab resistance in vivo, we chose a novel format of an anti-Jagged antibody, termed a Probody therapeutic (CTX-033) (32, 33). The anti-Jagged Probody, CTX-033 has demonstrated anti-tumor activity in a mouse xenograft model of pancreatic cancer (34). Figure 3D showed that xenograft breast tumors generated from trastuzumab resistant cells were resistant to trastuzumab following 4 weeks of treatment. Treatment with the CTX-033 inhibited tumor growth when used alone. However, trastuzumab plus CTX-033 significantly decreased tumor growth compared to CTX-033 or trastuzumab alone (Figure 3D). These data suggest that using a combination of trastuzumab plus CTX-033 can partially reverse trastuzumab resistance in this model.
ErbB-2 limits Mib-1-mediated ubiquitinylation of Jagged-1 and subsequent trans-activation of Notch
Since ubiquitinylation of Jagged-1 by Mib-1 is required to activate Notch (35, 36), we hypothesized that ErbB-2 attenuates expression of Mib-1 and/or its association with Jagged-1 to inhibit Notch activity. To test this hypothesis, Jagged-1 was immunoprecipitated from MDA-MB-453 cells-treated with trastuzumab or lapatinib and Western blotting was performed to detect Jagged-1 and Mib-1 proteins. Co-immunoprecipitation demonstrated that trastuzumab or lapatinib increased the amount of Mib-1 in a complex with Jagged-1 (Figure 4A). From these results, we conclude that ErbB-2 possibly limits the interaction between Jagged-1 and Mib-1.
Figure 4.
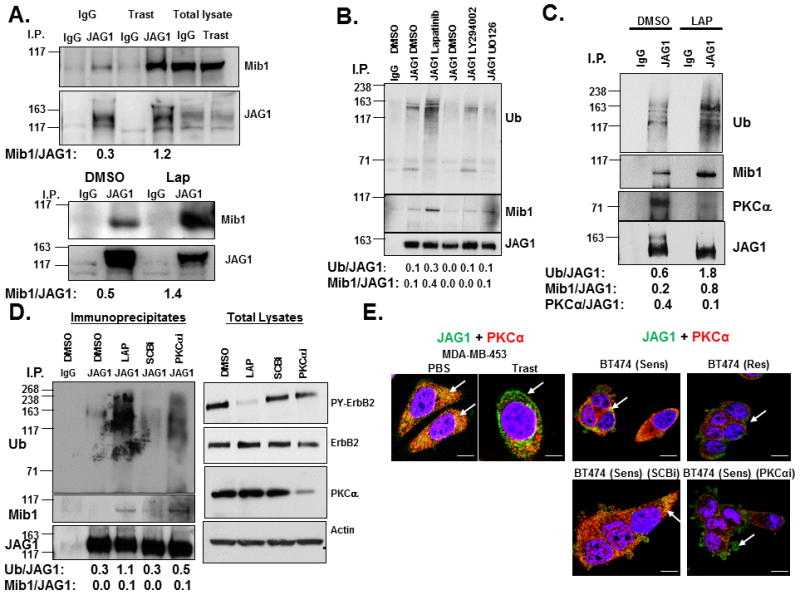
ErbB-2 or PKCα Attenuates Mib-1-mediated Ubiquitinylation of Jagged-1. A, Trastuzumab or lapatinib increases the interaction between Mib-1 and Jagged-1. MDA-MB-453 cells were treated with controls (IgG or DMSO) or trastuzumab or lapatinib, respectively for 30 minutes. A Jagged-1 antibody was used to immunoprecipitate Jagged-1 from treated cell lysates. An IgG antibody was used as a control for nonspecific immunopreciptation. Western blotting was performed to detect co-immunoprecipitated proteins such as Mib-1 (Mib1). Jagged-1 (JAG1) protein was detected to determine specificity of the immunoprecipitation. Western blotting was also performed on total lysates to detect levels of total Mib1 and JAG1. B, Lapatinib-induced Jagged-1 ubiquitinylation is independent of PI3-K and MAPK signaling. Cells were treated with DMSO, lapatinib, LY294002, or U0126 and then lysed. Jagged-1 was immunoprecipitated and Western blotting was performed to detect ubiquitinylation (Ub), Mib-1, and JAG1. C, The recruitment of PKCα to Jagged-1 is inhibited by lapatinib. MDA-MB-453 cells were transfected with the FLAG-tagged Ub for 48 hours and treated with DMSO or lapatinib for 30 minutes. JAG1 was immunoprecipitated from cell lysates followed by western blotting to detect Ub, Mib1, PKCα, and JAG1. D, PKCα inhibits recruitment of Mib-1 and Jagged-1 ubiquitinylation. MDA-MB-453 cells were transfecteda a SCBi or PKCαi siRNA for 72 hours. Cells were then treated with DMSO or lapatinib for 30 minutes followed by Jagged-1 immunoprecipitation and western blotting to detect Ub, Mib1, and JAG1. Western blotting as was also performed on total lysates to detect PY-ErbB2, ErbB2, PKCα, and Actin. E, Jagged-1 and PKCα co-localize in ErbB-2 positive breast cancer cells. MDA-MB-453 cells were treated with PBS or trastuzumab for 48 hours. BT474 trastuzumab sensitive (Sens) and resistant (Res) cells were transfected with SCBi or PKCαi siRNA for 72 hours. Cells were fixed and stained for JAG1 (Green) and PKCα (Red) and visualized using Confocal immunofluorescence microscopy. The results are representative of three independent experiments. Left two panels scale bare = 10μm; right panels scale bars = 20μm. Image J software was used to measure densitometry of protein bands on Western blots. Ratios of proteins are presented below images.
In order to investigate the role of Mib-1 on Notch-l activity, the effect of a Mib-1 siRNA on the expression of HES1 protein levels was measured. As measured by Western blot analysis, Mib-1 protein levels were decreased upon Mib-1 knocked down (Supplemental Figure 2). Either trastuzumab or lapatinib increased HES1 protein (Supplemental Figure 2). Importantly, Mib-1 siRNA decreased the induction of HES1 (Supplemental Figure 2), indicating that Mib-1 is necessary for Notch activation by ErbB-2 inhibitors.
In order to determine whether ErbB-2 or downstream mediators such as PI3-K or MEK attenuate Jagged-1 ubiquitinylation, we performed an immunoprecipitation of Jagged-1 similar to Figure 4A except we used the PI3-K inhibitor, LY294002 or the Mek1/2 inhibitor, U0126 in addition to lapatinib. Lapatinib increased Jagged-1 ubiquitinylation at 150 kilodaltons and enhanced the recruitment of Mib-1 to a Jagged1 complex (Figure 4B). Neither LY294002 nor U0126 induced recruitment of Mib-1 to Jagged-1 or Jagged-1 ubiquitinylation compared to lapatinib (Figure 4B). Western blots confirmed that the kinase inhibitors inhibited their targets (Supplemental Figure 3A). These results suggest that ErbB-2 restricts the interaction of Mib-1 to Jagged-1 and its ubiquitinylation possibly through a different kinase downstream of ErbB-2.
ErbB-2 promotes an association between Jagged-1 and PKCα
To determine the mechanism by which ErbB-2 attenuates the association between Jagged-1 and Mib-1, the protein sequences of Jagged-1 and Mib-1 were scanned using Prosite Expasy and several high scoring, putative PKC phosphorylation sites were identified. ErbB-2 activates PKCα in breast cancer cells via PLCγ (13) or by up-regulating c-Src (37). Co-immunoprecipitation demonstrated that Jagged-1 was associated with PKCα when ErbB-2 was active but failed to bind PKCα when cells were treated with lapatinib (Figure 4C). Downregulation of PKCα using a siRNA facilitated the association of Mib-1 with Jagged-1 and subsequent ubiquitinylation of Jagged-1 similarly to lapatinib treatment (Figure 4D). Control Western blots were performed to detect protein levels of active ErbB-2 (PY), total ErbB-2, and PKCα to confirm the efficiency of the PKCα siRNA and that it had little effect on the expression and activity of ErbB-2 (Figure 4D).
Jagged-1 and PKCα co-localize in ErbB-2-positive breast cancer cells and dissociate upon trastuzumab treatment
We performed confocal fluorescence microscopy to explore whether Jagged-1 and PKCα co-localize in ErbB-2 overexpressing breast cancer cells. Jagged-1 co-localizes with PKCα (Figure 4E, left panel) in PBS-treated cells. In contrast, Jagged-1 was no longer colocalized with PKCα (Figure 4E, left panel) in response to trastuzumab. Similar, results were observed in BT474 trastuzumab sensitive cells (Figure 4E, right panels). Interestingly, trastuzumab resistant BT474 cells express less PKCα and Jagged-1 is primarily near the cell surface (Figure 4E, right, upper panels). PKCα knockdown in BT474 sensitive cells disrupts the Jagged-1-PKCα colocalization similar to what we observed in resistant cells (Figure 4E, right, lower panels).
PKCα inhibits Jagged-1-mediated Notch activation and restores trastuzumab sensitivity in vivo
To determine if PKCα is sufficient to restrict Notch activation, we tested whether a constitutive active PKCα (Δ22–28) would prevent the lapatinib-induced Mib-1 and Jagged-1 interaction, Jagged-1 ubiquitinylation, and increase in Notch gene targets. Figure 5A showed lapatinib increased recruitment of Mib-1 to a Jagged-1 complex and increased Jagged-1 ubiquitinylation in cells transduced with control LRZS-linker. In contrast, lapatinib treatment of cells-transduced with LRZS-PKCα 22–28 which increased PKC activation as shown in Figure 5A, right panel, failed to increase Mib-1 recruitment to Jagged-1 and ubiquitinylation (Figure 5A). Furthermore, Figure 5C showed that lapatinb significantly increased 3 out of 5 Notch gene targets (DELTEX1, HEY1, and NOTCH-4) in cells transduced with LRZS-linker but not in cells-expressing PKCα 22–28. In contrast, knockdown of PKCα increased expression of HES1 and HEY1 proteins (Figure 5B, left panel) and DELTEX1 transcripts (Figure 5B, right panel), respectively. Taken together, these results indicate that ErbB-2 facilitates a PKCα-Jagged-1 association to limit Mib-1-mediated ubiquitinylation of Jagged-1 and activation of Notch.
Figure 5.
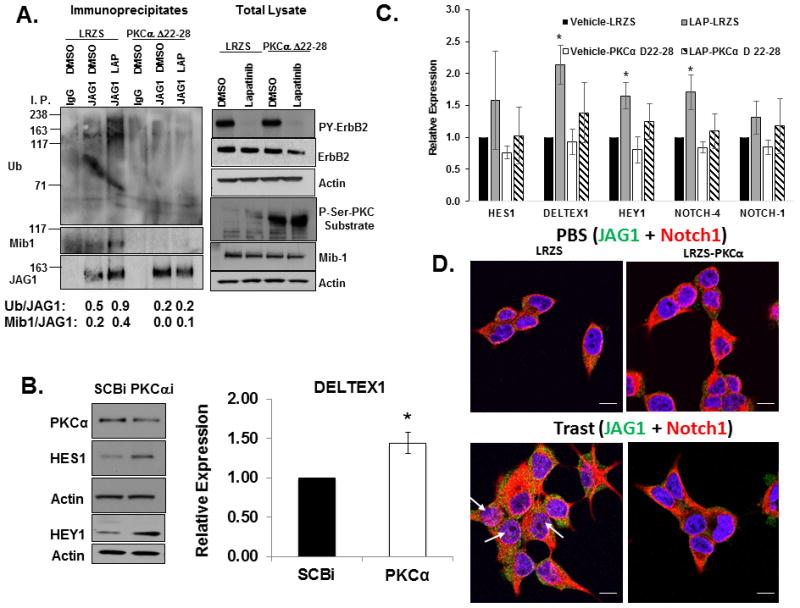
PKCα is necessary and sufficient to inhibit Notch activation. A, Active PKCα prevents Mib1-mediated Jagged-1 ubiquitinylation. MDA-MB-453 cells were transduced with a retrovirus LRZS-linker or LRZS-PKCα 22–28 for 48 hours and then treated with DMSO or lapatinib for 30 minutes. Jagged-1 was immunoprecipitated from cell lysates followed by western blotting to detect Ub, Mib1, and JAG1 proteins. Total lysates were subjected with western blotting to detect total PY-ErbB-2, ErbB-2, P-Ser-PKC substrate, Mib1, and Actin. B, PKCα is necessary to inhibit Notch target gene expression. MDA-MB-453 cells were transfected with SCBi or PKCαi siRNA for 72 hours. Cell lysates were prepared and western blotting performed to detect PKCα, HES1, HEY1, and Actin proteins. Additionally, RNA was extracted from cells and real-time PCR was performed to detect relative expression of DELTEX1 transcripts normalized to HPRT. The results from the PCR represent means plus or minus standard deviation of three independent experiments. C, Active PKCα attenuates lapatinib-induced Notch target gene expression. MDA-MB-453 cells were transduced with a retrovirus LRZS-linker or LRZS-PKCα 22–28 for 48 hours and then treated with DMSO or lapatinib for 24 hours. RNA was extracted from cells and real-time PCR was performed to detect relative expression of HES1, DELTEX1, HEY1, NOTCH-1, and NOTCH-4 transcripts. The results are shown as means plus or minus standard deviations of three independent experiments. *Denotes statistical significance p<0.05 was calculated using an ANOVA. D, PKCα is sufficient to prevent trastuzumab-induced Notch-1 nuclear localization. BT474 trastuzumab sensitive cells were transduced with a retrovirus LRZS-linker or LRZS-PKCα for 48 hours and then treated with PBS or trastuzumab for 18 hours. Cells were fixed and stained for JAG1 (Green) and Notch-1 (Red) followed by visualization using Confocal immunofluorescence microscopy. Scale bars = 20μm.
To determine whether PKCα expression limits Notch-1 nuclear localization, we performed confocal immunofluorescence. The results showed that cellular localization of Notch-1 and Jagged-1 was relatively unchanged in cells expressing LRZS-linker alone or LRZS-PKCα upon vehicle treatment (Figure 5D, upper panels). In contrast, trastuzumab treatment of cells-transduced with the LRZS-linker induced nuclear localization of a proportion of the Notch-1 protein while Jagged-1 accumulated in the cytoplasm or near the cell surface (Figure 5D, lower left panel). PKCα overexpression almost completely prevented Notch-1 nuclear localization (Figure 5D, lower right panel). These results indicate that PKCα is sufficient to prevent Notch-1 nuclear localization and activation in ErbB-2 positive breast cancer cells.
Based on results shown so far, we hypothesized that trastuzumab resistant cells which have high Notch-1 activation and require Notch-1 for their survival (26) should have lower PKCα protein levels or activity than their sensitive parental cells. The results show that BT474 trastuzumab sensitive cells express higher PKCα protein than BT474 trastuzumab resistant cells and that trastuzumab treatment of both sensitive and resistant cells decreased levels of PKCα protein and kinase activity as measured by detection of phosphorylated-Serine specific PKC substrates (Supplemental Figure 3B). We hypothesized that overexpression of PKCα might restore trastuzumab sensitivity. To address this, we transduced the BT474 trastuzumab resistant breast cancer cell line with LRZS-linker or LRZS-PKCα and injected these cells (Supplemental Figure 3B) into female, nude mice to determine their sensitivity to trastzuumab treatment in vivo. The results demonstrated that BT474 trastuzumab resistant cells-expressing LRZS-linker are resistant to trastuzumab (Figure 6A). However, PKCα overexpression significantly decreased BT474 breast tumor growth compared to LRZS-linker. This result is not surprising as we have shown previously that Notch-1 knockdown alone in trastuzumab resistant BT474 cells was necessary to inhibit cell proliferation in vitro in the absence or presence of trastuzumab (26). These current results would suggest that PKCα overexpression could also be necessary to inhibit ErbB-2 positive breast tumor growth after acquired resistance to trastuzumab possibly by suppressing Notch signaling. PKCα overexpression partially restored trastuzumab sensitivity as the tumors almost completely regressed upon trastuzumab treatment (Figure 6A). These results demonstrate that PKCα could limit Notch-1 activation and thus possibly trastuzumab resistance in ErbB-2-positive breast cancer cells.
Figure 6.
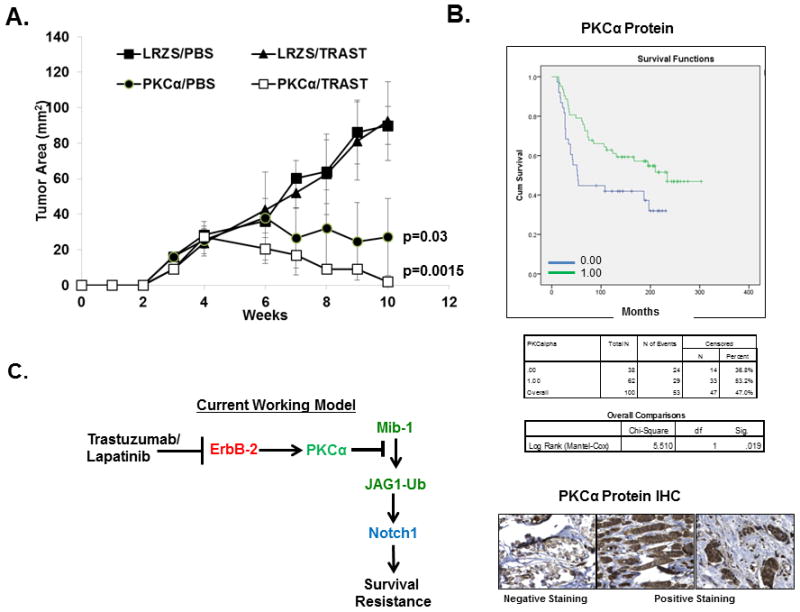
PKCα overexpression reverses trastuzumab resistance and predicts better survival in women with ErbB-2+ human breast cancer. A, Overexpression of PKCα inhibits growth of BT474 trastuzumab resistant tumor xenografts. Five million BT474 trastuzumab resistant cells retrovirally transduced with LRZS-linker or LRZS-PKCα were injected into mammary fat pads of 20 female, nude athymic mice, respectively. Tumors were allowed to grow to a mean cross sectional area of approximately 10–20mm2 and then mice were randomized to PBS or trastuzumab, injected i.p. once weekly for up to 10 weeks. Statistical significance was calculated by a two-way ANOVA. B, Low PKCα protein predicts poorer overall survival in women with ErbB-2 positive breast cancer. The Nottingham cohort of 100 ErbB-2 positive breast tumor tissues were stained for PKCα protein and scored 0.00 for negative staining or 1.00 for positive staining. Overall survival follow up results were collected prospectively up to 400 months. Kaplan-Meier analysis for overall survival was performed on negative and positive expression of PKCα. Statistical significance was calculated using Log Rank (Mantel-Cox) test. Immunohistochemistry for PKCα protein in ErbB-2 positive breast cancer tissue. The three panels are representative of negative expression (top), high expression (middle), and moderate expression (lower). C. Model summarizes conclusion of current study. ErbB-2 through PKCα blocks the interaction of Mib-1 with Jagged-1, subsequently limiting ubiquitinylation of Jagged-1 to restrict Notch activation. Jagged-1-mediated Notch activation by trastuzumab or lapatinib is required for ErbB-2 positive breast cancer survival and resistance.
PKCα and HEY1 inversely predict ErbB-2 positive breast cancer recurrence and survival
These results suggest that ErbB-2 positive breast cancer patients with high PKCα expression should have better outcomes since PKCα can attenuate Notch signaling to promote sensitivity to anti-ErbB-2 therapy. To address this, we used survival analysis via the Kaplan-Meier Plotter software, version 2014 (30) and searched for expression of PRKCA, and HEY1 transcripts in a cohort of 208 ErbB-2 positive breast cancer patients to predict recurrence free survival (RFS) outcome. Results showed that high PRKCA transcript expression based on an average of four probes predicted better recurrence free survival outcome compared to low with a hazard ratio (HR) of 0.64 (Logrank P = 0.037). In contrast, high expression of a Notch gene target, HEY1 predicted poor RFS outcome (HR=1.72, Logrank P=0.011) using an average of two HEY1 probes (Supplemental Figure 3). Interestingly, when we used the multigene analyzer and asked whether the same breast cancer samples that express high HEY1 and low PRKCA predict worse RFS outcomes compared to the individual genes, the results showed a slightly worse outcome with a HR=1.77 and Logrank P=0.0074 (Supplemental Figure 3).
We examined another cohort, the Nottingham primary breast cancer series (the Tenovus cohort) prepared as tissue microarrays (TMA). Outcome data were collected on a prospective basis. These include breast cancer-specific survival (BCSS) defined as the time in months from the date of surgery to the BC-related death (38, 39). 109 ErbB-2 positive breast cancer tissues were stained for PKCα protein by immunohistochemistry, blindly scored for staining intensity (0–3). Samples were classified into two groups: Group 1.0 with high PKCα (staining intensity 2–3) and group 0.0 with low or negative PKCα (staining intensity 0–1) and labeled as “0.00”. The results demonstrated that high PKCα protein expression predicted better BCSS compared to low expressing patients (Logrank P =0.019) (Figure 6B). Figure 6B showed a sample of the immunohistochemistry for PKCα protein stained as negative (top panel), positive sample #1 (middle panel), and positive sample #2 (lower panel). Therefore, based on the results of our current study, we propose a model where PKCα attenuates the association between Jagged-1 and Mib-1 to restrict Jagged-1 ubiquitinylation and subsequent trans-activation of Notch-1 to prevent Notch-1-driven resistance to ErbB-2-targeted therapies (summarized in Figure 6C).
Discussion
ErbB-2 positive breast cancer is currently treated with trastuzumab, lapatinib, pertuzumab, or trastuzumab emtansine. Many patients will not initially respond to these drugs and, among responders, over 25% develop resistance within the first year of therapy (40). Thus, resistance remains a clinical problem which requires identification of new targets for future therapeutic strategies and novel prognostic biomarkers. Our results reveal that PKCα downstream of ErbB-2 limits an association between Mib-1 and Jagged-1 to attenuate Notch-1 activation. The PKCα-mediated attenuation of Notch-1 signaling is necessary and sufficient to enhance sensitivity to trastuzumab and possibly prevent resistance to potentially improve survival for women with ErbB-2 positive breast cancer.
Notch signaling promotes breast cancer progression (41, 42) and is critical for survival and self-renewal of breast cancer stem cells (24, 25). These cells are hypothesized to be the dormant or slowly self-replicating cell population resistant to standard therapies. We showed that Notch-1 is required for trastuzumab resistance (26) and ErbB-2 breast tumor recurrence (27). Others have shown that Notch cooperates with ErbB-2 to promote a more invasive phenotype in ductal carcinoma in situ (DCIS) (43). Furthermore, Notch expression and activity have been implicated in ErbB-2-driven cancer stem cells (44). Recently, Abravanel et al showed that Notch promotes recurrence of breast tumor cells post anti-ErbB-2 therapy (45). Thus, Notch could promote breast tumor development during early stages of breast tumorigenesis and at later a stage, its activity and function is attenuated as other pathways become dominant. From our studies, we conclude that ErbB-2 attenuates Notch signaling via PKCα under conditions whereby ErbB-2 is the primary growth driver. However, when ErbB-2 is inhibited by trastuzumab or lapatinib, PKCα is inhibited and Jagged-1 is made competent to trans-activate Notch to promote a compensatory survival signal.
The role of PKCα in breast cancer remains controversial as it is not clear whether PKCα is a tumor promoter or a suppressor. PKCα is implicated in human breast cancer progression (46). However, there is conflicting data as some have shown PKCα is decreased in human breast tissue (47). Ectopic expression of PKCα in cell lines promotes a more aggressive phenotype. For example, PKCα overexpression induces tamoxifen resistance in ER-positive cell lines (14). Furthermore, PKCα is required for invasion of breast cancer cells (48). In addition, PKCα activates Notch-4 expression in ER-positive breast cancer cells to promote tamoxifen resistance (49). PKCα is also a mediator of breast cancer stem cell expansion (16), regulates recycling of ErbB2 in breast cancer cells (50), and a therapeutic target in triple negative breast cancer (17). One possible explanation for the conflicting results could be that the activation status of PKCα, its subcellular localization and its immediate substrates in breast cancer remain unclear. As breast cancer is a heterogeneous disease, it would not be surprising that PKCα could have pleotropic functions in different subtypes of breast cancer. For example, patients with ErbB-2 positive tumors expressing low PKCα have poorer RFS than high expressors. These data included both ER-positive and negative tumors. Ideally, if we could stratify and separate these data based on ER expression, results could determine if ER status changes the prediction. However, due to the low number of ErbB-2+ patients from the Kaplan-Meier Plotter dataset and the Nottingham cohort, this could not be done. Increasing the number of retrospective samples and/or a prospective clinical trial will help to increase power and to answer some of these critical questions. Results from clinical trials using PKC inhibitors have been disappointing. The ISIS 3521 (aprinocarsen or LY900003) trial failed to show a response and this could be due to multifactorial roles of PKCs. Over 20 trials from phase I to III have been conducted using various PKC inhibitors in many solid tumors (51). However, results of trials have been disappointing in women with metastatic breast cancer (52) suggesting that targeting PKCα might depend on the cell context.
The mechanism by which PKCα attenuates Jagged-1 vesicular trafficking and recruitment of Mib-1 to Jagged-1 is under investigation. Both Jagged-1 and Mib-1 contain putative PKC phosphorylation sites. Therefore, Jagged-1 and/or Mib-1 could be direct substrates of PKCα. Phosphorylation events regulate recruitment of E3 ligases and thus PKCα could directly phosphorylate Jagged-1 and/or Mib-1 to prevent their interaction. Alternatively, PKCα could regulate vesicular trafficking of Jagged-1 possibly sequestering Jagged-1 in endosomes targeted for recycling. Classical PKCs such as α and βII regulate proteins that are normally recycled by sequestering them in the pericentrion (53). Our results are consistent with this as we showed that Jagged-1 is localized to endosomes when ErbB-2 was hyperactive but accumulated at the cell surface upon ErbB-2 blockade. Thus, PKCα might limit the cell surface availability of Jagged-1 by facilitating sequestration of Jagged-1 in recycling endosomes.
We describe a critical function for PKCα downstream of ErbB-2, which is to attenuate the competency of Jagged-1 to activate Notch-1. Notch ligands are made competent by Mib-1 and subsequent ubiquitinylation which drives endocytosis. We show that Jagged-1 is required for trastuzumab resistance in breast cancer (Figure 3) providing the first pre-clinical proof-of-concept for the use of an anti-Jagged-1, such as the Probody CTX-033, targeted therapy for prevention or reversal of resistance possibly in breast cancers expressing low PKCα levels (Figure 6) and/or high HEY1 levels (Supplemental Figure 3). An anti-Jagged Probody therapeutic may be well positioned for such a strategy given that it demonstrates less systemic toxicity compared to other Notch targeting approaches because it is only activated within the tumor microenvironment (34). The human data from two cohorts of ErbB-2 positive breast tumors predict that women with breast tumors expressing low PKCα mRNA (Supplemental Figure 4) or protein (Figure 6B) have poorer RFS or BCSS, respectively. Conversely, high HEY1 expression predicts poor RFS outcome in women with ErbB-2 positive breast cancer (Supplemental Figure 4). In order to determine whether the same tumors that express low PKCα also express high HEY1 would be better targets for anti-Notch therapy, a prospective clinical trial should be conducted using the Jagged-1 Probody or a Notch specific inhibitors in combination with trastuzumab. These data are consistent with the overall model where the ErbB-2- PKCα axis attenuates Notch activation and this subset of tumors could predict better sensitivity to anti-ErbB-2 targeted agents. Conversely, low PKCα tumors could require concomitant Notch inhibition to prevent or reverse trastuzumab resistance. These results could have clinical impact for the treatment of women with ErbB-2 positive breast cancer. Future clinical trials need to be designed to test whether PKCα protein levels will predict for trastuzumab sensitivity.
Supplementary Material
Translational Relevance.
Breast cancer is the second leading cause of cancer mortality among women worldwide. The major problem with current treatments is tumor resistance, recurrence, and disease progression. The current study elucidates a novel mechanism by which ErbB-2 via PKCα attenuates Jagged-1-mediated Notch signaling in ErbB-2 positive breast cancer. The clinical impact of these studies is PKCα is potentially a good prognostic marker for low Notch activity and increased trastuzumab sensitivity in ErbB-2 positive breast cancer. Moreover, women with ErbB-2 positive breast tumors expressing high Notch activation and low PKCα expression could be the best candidates for anti-Notch therapy. These studies provide a pre-clinical proof of concept for future clinical trials using combinations of trastuzumab/lapatinib plus an anti-Jagged-1 targeted therapy for trastuzumab resistant, ErbB-2 positive breast cancer expressing low levels of PKCα.
Acknowledgments
Funding: This work was supported in part by an NIH grant R01 (CA160378) to C.O.
We thank Dr. Adriano Marchese for generously providing the FLAG-tagged Ub construct and the ubiquitinylation protocols. We also thank Dr. Gerry Weinmaster for generously providing the LTK-parental and LTK-JAG1 mouse fibroblasts. We are grateful to Dr. Maurizio Bocchetta for scientific discussions and advice on this project.
Footnotes
Conflict of Interest Disclosure Statement: Jason Sagert is an employee of CytomX Therapeutics, Inc. with financial interest. Lucio Miele is a consultant for CytomX Therapeutics with financial interest. There are no other conflicts of interests.
Authors’ Contributions:
K.P. wrote the manuscript with D.W. K.P. and D.W. contributed equally to experimental studies. B.G. performed the proliferation assays. D.S. performed flow cytometry. A.B. performed Western blotting for Notch1, Jagged1, and PKCα in BT474 cell lysates. A.Z. performed the statistical analysis and Kaplan-Meier Log rank analysis. J.B. performed the ErbB-2 knockdown studies and Western blots and PCR. A.P., A.G., I.E., A. F., and L.M. provided the PKCα protein IHC and overall survival data in ErbB-2 positive patient tissues. A.R. provided breast cancer reagents. K.S.A. provided clinical breast cancer expertise and assisted in the final conclusions of clinical impact. J.S. provided the Probody™ CTX-033 from CytomX. M.D. provided PKCα reagents and assisted in PKCα-experimental studies. C.O. designed experimental studies, revised the manuscript, and is the corresponding author.
References
- 1.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annual review of medicine. 2011;62:233–47. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta K, Osipo C. Trastuzumab resistance: role for Notch signaling. TheScientificWorldJournal. 2009;9:1438–48. doi: 10.1100/tsw.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer discovery. 2013;3:27–34. doi: 10.1158/2159-8290.CD-12-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 5.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 6.Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocrine-related cancer. 2014;21:R235–46. doi: 10.1530/ERC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masamura S, Adlercreutz H, Harvey H, Lipton A, Demers LM, Santen RJ, et al. Aromatase inhibitor development for treatment of breast cancer. Breast cancer research and treatment. 1995;33:19–26. doi: 10.1007/BF00666067. [DOI] [PubMed] [Google Scholar]
- 8.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. British journal of cancer. 2004;90 (Suppl 1):S2–6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, et al. Adjuvant trastuzumab in HER2-positive breast cancer. The New England journal of medicine. 2011;365:1273–83. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2007 doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 11.Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. The New England journal of medicine. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 12.Nahta R. Molecular Mechanisms of Trastuzumab-Based Treatment in HER2-Overexpressing Breast Cancer. ISRN oncology. 2012;2012:428062. doi: 10.5402/2012/428062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Disatnik MH, Winnier AR, Mochly-Rosen D, Arteaga CL. Distinct responses of protein kinase C isozymes to c-erbB-2 activation in SKBR-3 human breast carcinoma cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1994;5:873–80. [PubMed] [Google Scholar]
- 14.Tonetti DA, Morrow M, Kidwai N, Gupta A, Badve S. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. British journal of cancer. 2003;88:1400–2. doi: 10.1038/sj.bjc.6600923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu TT, Tsai JH, Tsai JH, Kuo SJ, Yu SY, Huang CY, et al. Augmented release of matrix metalloproteinase-9 by PKC activation in organotypic cultures of human breast cancer and adjacent normal breast tissue and fibroadenoma. The Chinese journal of physiology. 2004;47:73–8. [PubMed] [Google Scholar]
- 16.Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer cell. 2013;24:347–64. doi: 10.1016/j.ccr.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu YH, Yao J, Chan LC, Wu TJ, Hsu JL, Fang YF, et al. Definition of PKC-alpha, CDK6, and MET as Therapeutic Targets in Triple-Negative Breast Cancer. Cancer research. 2014;74:4822–35. doi: 10.1158/0008-5472.CAN-14-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koo BK, Lim HS, Song R, Yoon MJ, Yoon KJ, Moon JS, et al. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development. 2005;132:3459–70. doi: 10.1242/dev.01922. [DOI] [PubMed] [Google Scholar]
- 19.Koutelou E, Sato S, Tomomori-Sato C, Florens L, Swanson SK, Washburn MP, et al. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. The Journal of biological chemistry. 2008;283:3846–53. doi: 10.1074/jbc.M706974200. [DOI] [PubMed] [Google Scholar]
- 20.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer research. 2005;65:8530–7. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 21.Reedijk M. Notch signaling and breast cancer. Advances in experimental medicine and biology. 2012;727:241–57. doi: 10.1007/978-1-4614-0899-4_18. [DOI] [PubMed] [Google Scholar]
- 22.Yao K, Rizzo P, Rajan P, Albain K, Rychlik K, Shah S, et al. Notch-1 and notch-4 receptors as prognostic markers in breast cancer. International journal of surgical pathology. 2011;19:607–13. doi: 10.1177/1066896910362080. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi N, Oyama T, Ito E, Satoh H, Azuma S, Hayashi M, et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer research. 2008;68:1881–8. doi: 10.1158/0008-5472.CAN-07-1597. [DOI] [PubMed] [Google Scholar]
- 24.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast cancer research : BCR. 2004;6:R605–15. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010;70:709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene. 2008;27:5019–32. doi: 10.1038/onc.2008.149. [DOI] [PubMed] [Google Scholar]
- 27.Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. British journal of cancer. 2011;105:796–806. doi: 10.1038/bjc.2011.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rizzo P, Miao H, D’Souza G, Osipo C, Song LL, Yun J, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer research. 2008;68:5226–35. doi: 10.1158/0008-5472.CAN-07-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, et al. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. Journal of the National Cancer Institute. 1998;90:1552–8. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- 30.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocrine-related cancer. 2012;19:197–208. doi: 10.1530/ERC-11-0329. [DOI] [PubMed] [Google Scholar]
- 31.Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, et al. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast cancer research and treatment. 2007 doi: 10.1007/s10549-007-9805-3. [DOI] [PubMed] [Google Scholar]
- 32.Polu KR, Lowman HB. Probody therapeutics for targeting antibodies to diseased tissue. Expert opinion on biological therapy. 2014;14:1049–53. doi: 10.1517/14712598.2014.920814. [DOI] [PubMed] [Google Scholar]
- 33.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Science translational medicine. 2013;5:207ra144. doi: 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 34.Sagert JWJ, Vasiljeva O, Richardson J, Desnoyers L, Liu S, Yang A, Wong C, Menendez E, Polu K, Lowman H. Tumor-Specific Inhibition of Jagged-Dependent Notch Signaling Using a Probody Therapeutic. 2013 [Google Scholar]
- 35.Daskalaki A, Shalaby NA, Kux K, Tsoumpekos G, Tsibidis GD, Muskavitch MA, et al. Distinct intracellular motifs of Delta mediate its ubiquitylation and activation by Mindbomb1 and Neuralized. The Journal of cell biology. 2011;195:1017–31. doi: 10.1083/jcb.201105166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah DK, Mohtashami M, Zuniga-Pflucker JC. Role of recycling, Mindbomb1 association, and exclusion from lipid rafts of delta-like 4 for effective Notch signaling to drive T cell development. Journal of immunology. 2012;189:5797–808. doi: 10.4049/jimmunol.1202469. [DOI] [PubMed] [Google Scholar]
- 37.Tan M, Li P, Sun M, Yin G, Yu D. Upregulation and activation of PKC alpha by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors. Oncogene. 2006;25:3286–95. doi: 10.1038/sj.onc.1209361. [DOI] [PubMed] [Google Scholar]
- 38.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, et al. REporting recommendations for tumour MARKer prognostic studies (REMARK) European journal of cancer. 2005;41:1690–6. doi: 10.1016/j.ejca.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 39.McShane MP, Longnecker R. Analysis of fusion using a virus-free cell fusion assay. Methods in molecular biology. 2005;292:187–96. doi: 10.1385/1-59259-848-x:187. [DOI] [PubMed] [Google Scholar]
- 40.Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. British journal of cancer. 2014 doi: 10.1038/bjc.2014.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. Journal of mammary gland biology and neoplasia. 2004;9:145–63. doi: 10.1023/B:JOMG.0000037159.63644.81. [DOI] [PubMed] [Google Scholar]
- 42.Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Seminars in cancer biology. 2004;14:341–7. doi: 10.1016/j.semcancer.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 43.Pradeep CR, Zeisel A, Kostler WJ, Lauriola M, Jacob-Hirsch J, Haibe-Kains B, et al. Modeling invasive breast cancer: growth factors propel progression of HER2-positive premalignant lesions. Oncogene. 2012;31:3569–83. doi: 10.1038/onc.2011.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:2010–21. doi: 10.1158/1078-0432.CCR-08-1327. [DOI] [PubMed] [Google Scholar]
- 45.Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, Sterner CJ, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. The Journal of clinical investigation. 2015;125:2484–96. doi: 10.1172/JCI74883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lahn M, Kohler G, Sundell K, Su C, Li S, Paterson BM, et al. Protein kinase C alpha expression in breast and ovarian cancer. Oncology. 2004;67:1–10. doi: 10.1159/000080279. [DOI] [PubMed] [Google Scholar]
- 47.Kerfoot C, Huang W, Rotenberg SA. Immunohistochemical analysis of advanced human breast carcinomas reveals downregulation of protein kinase C alpha. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2004;52:419–22. doi: 10.1177/002215540405200314. [DOI] [PubMed] [Google Scholar]
- 48.Blobe GC, Obeid LM, Hannun YA. Regulation of protein kinase C and role in cancer biology. Cancer metastasis reviews. 1994;13:411–31. doi: 10.1007/BF00666107. [DOI] [PubMed] [Google Scholar]
- 49.Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, et al. Crosstalk between PKCalpha and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. 2013;2:e60. doi: 10.1038/oncsis.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey TA, Luan H, Tom E, Bielecki TA, Mohapatra B, Ahmad G, et al. A Kinase Inhibitor Screen Reveals PKC-dependent Endocytic Recycling of ErbB2 in Breast Cancer Cells. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.608992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li K, Zhang J. ISIS-3521. Isis Pharmaceuticals. Current opinion in investigational drugs. 2001;2:1454–61. [PubMed] [Google Scholar]
- 52.Mackay HJ, Twelves CJ. Protein kinase C: a target for anticancer drugs? Endocrine-related cancer. 2003;10:389–96. doi: 10.1677/erc.0.0100389. [DOI] [PubMed] [Google Scholar]
- 53.Idkowiak-Baldys J, Becker KP, Kitatani K, Hannun YA. Dynamic sequestration of the recycling compartment by classical protein kinase C. The Journal of biological chemistry. 2006;281:22321–31. doi: 10.1074/jbc.M512540200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.