

Abstract
Background
Hypoplastic left heart syndrome (HLHS) is a heterogeneous, lethal combination of congenital malformations characterized by severe underdevelopment of left heart structures, resulting in a univentricular circulation. The genetic determinants of this disorder are largely unknown. Evidence of copy number variants (CNVs) contributing to the genetic etiology of HLHS and other congenital heart defects has been mounting. However, the functional effects of such CNVs have not been examined, particularly in cases where the variant of interest is found in only a single patient.
Methods and Results
Whole‐genome SNP microarrays were employed to detect CNVs in two patient cohorts (N = 70 total) predominantly diagnosed with some form of nonsyndromic HLHS. We discovered 16 rare or private variants adjacent to or overlapping 20 genes associated with cardiovascular or premature lethality phenotypes in mouse knockout models. We evaluated the impact of selected variants on the expression of nine of these genes through quantitative PCR on cDNA derived from patient heart tissue. Four genes displayed significantly altered expression in patients with an overlapping or proximal CNV verses patients without such CNVs.
Conclusion
Rare and private genomic imbalances perturb transcription of genes that potentially affect cardiogenesis in a subset of nonsyndromic HLHS patients.
Keywords: cardiovascular diseases, genetics, heart defects, congenital
Introduction
Congenital heart defects (CHDs) not only comprise the single largest class of birth defects,1 they account for 4.2% of all neonatal (i.e., less than 28 days) deaths and nearly one‐quarter of all neonatal deaths attributable to birth defects in the United States.2 The most commonly specified CHD causal for both neonatal deaths2 and deaths during the first year of life3 is hypoplastic left heart syndrome (HLHS). HLHS encompasses a diverse spectrum of cardiac structural malformations of the left side,4, 5 with left ventricular hypoplasia as the hallmark feature. The resulting undersized chamber is insufficient for sustaining systemic circulation, making this condition universally fatal without surgical intervention. Even with advancements in staged surgical palliation, one year transplant‐free survival remains poor at 68.7%.6
In addition to efforts to improve surgical outcomes, research on determining genetic causes and mechanisms of HLHS has been ongoing. A heritability analysis demonstrated that this disease is determined primarily by genetic factors.7 Genetic linkage studies have identified multiple significant loci in HLHS kindreds, including several that overlap loci found in kindreds with other left ventricular outflow tract malformations, providing evidence of genetic heterogeneity and shared etiology.8, 9, 10 Association with several disorders caused by chromosomal aneuploidy (e.g., Jacobsen syndrome, Turner syndrome, and Down syndrome) is further evidence of the principal role genetics plays in the development of this condition. Although large‐scale chromosomal abnormalities (i.e., greater than 1 Mb in length) are found in a high percentage of patients that also display extracardiac defects, the majority of HLHS cases do not harbor large‐scale changes with additional phenotypes. The genetic alterations involved in the etiology of these nonsyndromic cases are still being sought.
A potential source of causal variants for nonsyndromic HLHS is single gene mutations. Mutations in the cardiac transcription factor NKX2‐5 11 and the signaling receptor NOTCH1 12, 13, 14 have been identified in a handful of these cases. Similarly, a small number of patients have been found to harbor mutations in genes that control left‐right embryonic axis patterning, but these patients also display the typical extracardiac defects observed in heterotaxy. Thus, the variants discovered in single genes so far account for a very small fraction of nonsyndromic HLHS cases, necessitating additional research to find other sources of genetic variation contributing to this ailment.
Recently, genomic structural variation, in the form of copy number variants (CNVs), has been explored for association with both syndromic and nonsyndromic CHDs.13, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 Potentially etiologic CNV loci have been identified using various criteria, including size, rarity, gene content, and de novo status. Yet the phenotypic consequences of altering the dosage of genes within these loci were infrequently addressed. Additionally, transcriptional effects of these variants on overlapping genes of interest were not determined. In this report we detail our own search for CNVs in HLHS cases, as well as our efforts toward verifying their functional impact on transcription in order to demonstrate etiologic potential.
Methods
Patient cohorts
Samples from two independent subject cohorts were obtained for this study. The first cohort consisted of 23 patients with CHDs, primarily HLHS (Table S1). Most of the HLHS cases (14 out of 16) had no family history. Two of these cases displayed dysmorphia, indicative of a potential genetic syndrome, at the time of sample collection. Samples consisted of genomic DNA extracted from peripheral blood.
The second cohort, detailed in Table S2, contained over 50 patients with nonfamilial, nonsyndromic HLHS or a variant thereof, with only two (HLHS30 and HLHS53) having a previously diagnosed genetic syndrome (Turner syndrome). All of these patients underwent heart transplantation, at which time tissue from the explanted heart was collected, snap frozen in liquid nitrogen, and stored at –80°C. DNA was extracted from these tissues with either a commercially available kit (DNeasy Blood & Tissue Kit, Qiagen, Valencia, CA, USA) or by proteinase K digestion in lysis buffer (50 mM Tris, pH 8.0; 100 mM EDTA; 100 mM NaCl; 1% SDS), followed by ethanol precipitation.
Prior to SNP array genotyping, all DNA samples were diluted to 50 ng/μL, as measured either by the NanoDrop ND‐3300 Fluorospectrometer (NanoDrop Technologies, Wilmington, DE, USA) or the Wallac 1420 Multilabel Counter (PerkinElmer, Waltham, MA, USA), with the Quant‐iT PicoGreen dsDNA Assay Kit (Invitrogen/Molecular Probes, Eugene, OR, USA). Samples were also verified to be of high molecular weight by agarose gel before submittal to the University of Colorado Anschutz Medical Campus Genomics and Microarray Core (Aurora, CO, USA), for genotyping with the HumanOmni2.5 BeadChip (Illumina, San Diego, CA, USA) SNP microarray platform. This study was approved by the Institutional Review Boards of the University of Colorado and the University of California San Diego; informed consent was obtained from the parents of all subjects.
CNV detection and selection
All samples from the first patient cohort, along with 52 samples from the second patient cohort, were genotyped by microarrays. Five samples from the second cohort yielded either unacceptably low genotype call rates or excessive segmentation, and were excluded from further analysis. Table S2 displays the patient information for the 47 samples included from that cohort.
Figure 1 shows the analysis steps performed on the data generated by the Illumina arrays. These data were evaluated using two different workflows. Illumina's GenomeStudio (version 2010.3) and the R/Bioconductor package crlmm 28 independently generated sample genotypes, normalized sample/reference probe intensity ratios (i.e., LogR Ratios), and SNP B allele frequencies (BAF) for evaluating zygosity. GenomeStudio utilized an in‐house derived reference file created from the results of 99 control samples, whereas crlmm employed a reference annotation package created by the software developer. The data generated by GenomeStudio have been deposited in NCBI's Gene Expression Omnibus (GEO), and can be accessed through GEO Series accession number GSE66032.
Figure 1.
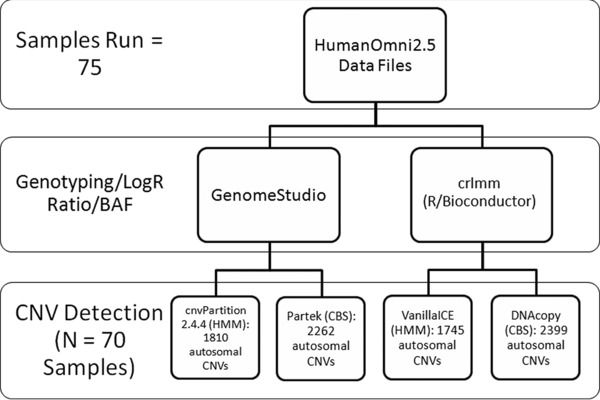
Flow chart of data analysis steps. Raw data were imported into two independent genotyping and sample/reference ratio‐calculating programs. Output from each of these programs was analyzed by software implementing two different classes of CNV detection algorithms, hidden Markov model (HMM) and circular binary segmentation (CBS). These programs generated four lists of putative CNVs. A total of 645 CNV segments, identified in at least three of the four lists, were further examined.
The resulting genotyping and intensity data from each of these software packages were channeled into programs that implement two classes of CNV detection algorithms, the hidden Markov model (HMM) and circular binary segmentation (CBS). We employed the R/Bioconductor packages VanillaICE and DNAcopy to identify CNV segments within crlmm‐derived data via HMM and CBS, respectively. Similarly, the Illumina HMM plug‐in to GenomeStudio (cnvPartition, version 2.4.4) and the genomic segmentation (CBS) tool in Partek Genomics Suite version 6.6 (Partek Incorporated, St. Louis, MO, USA) were executed on GenomeStudio‐derived data. Unless noted, default settings were used for all programs, with requisite parameter specifications for applicable packages detailed in the “Supplemental Methods.”
After CNV detection, concordance between the four methods was evaluated to create a list of putative CNVs that could be prioritized into candidates. This strategy was employed to reduce the number of possible false positive segments. A variant was included if it was identified by at least three of the four detection algorithms. This condition narrowed the list to 645 autosomal CNVs (Table S3).
Prioritization for the variant data was performed at the gene level (Figure 2). Specifically, lists of protein‐coding genes overlapping detected CNV segments or within 50 kb of their breakpoints were compiled. Segment verification was performed by visualization of logR ratios and B allele frequencies in both Partek and GenomeStudio, via its Illumina Genome Viewer. List entries were evaluated for potential cardiovascular development roles by queries in the Mouse Genome Informatics Database (http://www.informatics.jax.org/allele), an integrated laboratory mouse database website. Genes whose knockout or transgenic mouse models showed either a cardiovascular phenotype or premature lethality (a possible sign of cardiovascular malformation) were considered to be prime candidates for further investigation. This process highlighted 59 entries (Table S4). To distinguish verified CNVs that are likely benign polymorphisms from rare variants with etiologic potential, segments were assessed for overlap with entries from the Database of Genomic Variants (DGV) (http://dgv.tcag.ca/dgv/app/home) in March of 2014, using a track on the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/cgi‐bin/hgGateway). Segments completely or partially overlapping fewer than 10 DGV entries or containing gene exons that overlap fewer than 10 DGV entries were considered rare, and their corresponding genes retained. This step generated a list of 20 genes for possible follow‐up (Table 1).
Figure 2.
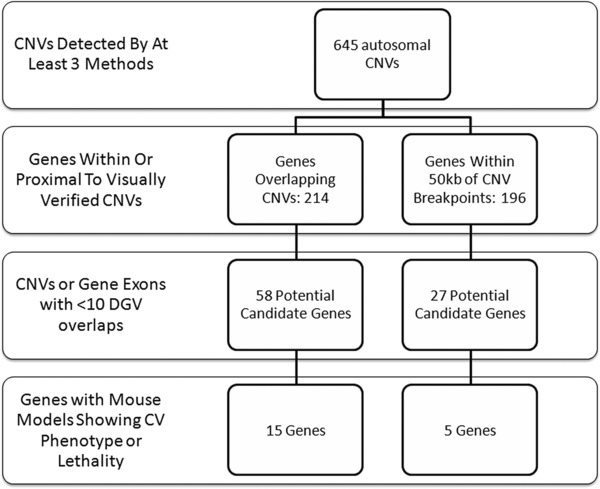
Flow chart of candidate prioritization steps. CNV data were first evaluated for protein‐coding gene overlap/proximity, and then graded on aspects of these genes.
Table 1.
Candidate CNVs discovered in both patient cohorts with Illumina arrays
| Sample | Cytoband | Coordinates (hg19) | Length (bp) | Estimated copy number | Candidate genes within or near CNV | qPCR‐verified CNV? |
|---|---|---|---|---|---|---|
| PG05 | 5q33.3 | 158899789–159627399 | 727,611 | 3 | ADRA1B | Yes |
| HLHS56 | 6p24.1 | 12306232–12320786 | 14,555 | 1 | EDN1 | Yes |
| HLHS24 | 11p15.4 | 10368614–10404266 | 35,653 | 3 | ADM | Yes |
| PG07 | 11q13.2–q13.3 | 68100519–69469069 | 1,368,551 | 3 | CPT1A, IGHMBP2 | Yes |
| HLHS27, HLHS39 | 16q23.3 | 83196349–83209154 | 12,806 | 1 | CDH13 | Yes (39 only) |
| HLHS08 | 1q24.2 | 170560604–170613602 | 52,999 | 1 | PRRX1 | Not tested |
| HLHS57 | 2p21 | 45408893–45974339 | 565,447 | 3 | PRKCE | Not tested |
| HLHS22 | 3p22.3 | 32953920–32985486 | 31,567 | 1 | TRIM71 | Not tested |
| HLHS27 | 7q21.12 | 87524013–87656083 | 132,071 | 3 | ADAM22 | Not tested |
| HLHS44 | 9q21.32–q21.33 | 86881490–87359570 | 478,081 | 3 | NTRK2 | Not tested |
| PG16 | 15q26.1 | 90108613–90171306 | 62,694 | 3 | KIF7 | Not tested |
| HLHS23 | 16q12.2 | 56653745–56694878 | 41,134 | 1 | MT1E, MT1F | Not tested |
| HLHS44 | 17q12 | 32441969–32566206 | 124,238 | 3 | ASIC2, CCL2 | Not tested |
| HLHS23 | 21q21.3 | 27513837–28194874 | 681,038 | 3 | APP | Not tested |
| HLHS23 | 21q21.3 | 28202149–29000785 | 798,637 | 3 | ADAMTS1, ADAMTS5 | Not tested |
| HLHS41 | 21q22.2 | 41034215–41435172 | 400,958 | 3 | DSCAM | Not tested |
The candidate gene(s) may not be the only gene(s) within or near the CNV.
Quantitative PCR (qPCR) validation
Selected copy number variants were validated using predesigned TaqMan Copy Number Assays (Applied Biosystems, Carlsbad, CA, USA). Each sample reaction contained approximately 20 ng of genomic DNA, and four replicates per sample were assayed. All reactions were performed in an Applied Biosystems 7300 Real Time PCR System, with SDS v1.3.1 software applying manufacturer‐recommended parameters. Copy number determinations were provided by Applied Biosystems CopyCaller Software v1.0, using analysis settings of no calibrator sample and a most frequent sample copy number of two.
In addition to patient samples, a portion of the DNA Polymorphism Discovery Resource Collection (Coriell Institute for Medical Research, Camden, NJ, USA) was analyzed with the same assays for copy number. The first 90 samples, a manufacturer‐defined subset of the panel, were tested in the same manner as the patient samples. This subset served as an additional gauge for the rarity of an assayed CNV within the entire human population, since the collection consists of samples representative of ancestries from the major geographic regions of the world.29
Gene expression assays
Table S5 displays the putative candidate genes assessed for expression in cardiac tissue with the listed Applied Biosystems TaqMan Gene Expression Assays. Most of these genes (ADAMTS1, ADM, EDN1, and NTRK2) were selected based on exhibition of a cardiac structural phenotype, in addition to premature lethality, in mouse knockout models.30, 31, 32, 33 The only gene tested lacking a cardiovascular phenotype in a mouse model was DSCAM, although the model does show perinatal lethality,34 and the gene lies in a defined critical region of Down syndrome‐associated congenital heart disease.35, 36 Thus, all of the presumptive candidates chosen for evaluation have plausible links to cardiac physiology or structural development.
Candidate expression was assessed in total RNA extracted from several sources. One source was pooled whole hearts, dissected from either E9.5 or E10.5 129S6 mouse embryos and placed into RNAlater Stabilization Solution (Ambion, Carlsbad, CA, USA) prior to extraction. The tissue from six E10.5 and seven E9.5 hearts was lysed by vortexing in Buffer RLT (Qiagen), with 2‐mercaptoethanol (Fluka Analytical, St. Louis, MO, USA) added. Lysates were homogenized in a Qiagen QIAshredder spin column prior to RNA cleanup and concentration with the Qiagen RNeasy MinElute Kit, following the manufacturer's protocol. Another source of RNA was explanted human heart ventricle tissue from the HLHS patients evaluated for copy number variation. Approximately 500 ng of extracted RNA was obtained from the seven patients harboring CNVs of interest, in addition to 13 gender‐matched patients without these CNVs. Finally, we purchased pooled Human Heart Total RNA and pooled Human Fetal Heart Total RNA from Clontech (Mountain View, CA, USA) to use as normal controls for comparison.
Prior to expression assessment, RNA samples were converted to cDNA using the Applied Biosystems High‐Capacity cDNA Reverse Transcription Kit, according to the manufacturer's protocol. Volumes were adjusted to create RNA reaction concentrations of approximately 10 ng/μL. One microliter from each of these reverse transcription reactions was used as the template for each gene expression reaction assembled; three replicates per sample were assayed. All expression experiments were performed in an Applied Biosystems 7300 Real Time PCR System, with SDS v1.3.1 software applying manufacturer‐recommended parameters. In addition to the candidate genes, an endogenous reference gene, GAPDH, was evaluated and used to normalize the results.
Mouse assay data were analyzed for a relative change in gene expression between E9.5 and E10.5 by the 2−ΔΔ CT method.37 Alternatively, we compared normalized patient cycle threshold (CT) values with an unpaired, two‐tailed t‐test using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). To normalize, we computed the average CT value of GAPDH for each patient sample and subtracted this value from each corresponding patient test gene CT (ΔCT). The ΔCT values of a patient sample harboring the CNV of interest were compared to the values of the remaining patient samples without the CNV. The pooled adult and fetal heart samples were not included in these comparisons.
Results
Putative candidate CNV identification and validation
We leveraged the availability of software for Illumina array results interpretation to create a consensus‐based analysis strategy. Two genotyping and four segmentation algorithms generated lists of putative CNVs (see Methods and Figure 1). These lists were evaluated for concordant entries; 645 putative CNVs were identified by at least three of the four algorithms executed (Table S3). The concordant entries were ranked by features of the genes contained within or near them. Prioritization of these entries via the scheme detailed in Figure 2 yielded 20 candidate genes potentially impacted by 16 rare or novel variants, 15 of which were found in separate patients (Table 1).
In addition to concordance, validation of aberrant copy number at several candidate loci was performed via qPCR. Having a greater confidence in what was identified due to the array's high marker density and multiple algorithm detection, we only chose a subset of CNVs to verify. The shortest, a 12806 bp deletion covering a single exon of the cadherin 13 gene (CDH13), was detected in two patients, HLHS39 and HLHS27, by one and three packages, respectively. Unfortunately, only patient HLHS39 had a sufficient amount of sample remaining for confirmatory testing. The other four loci chosen for validation were discovered in individual patients.
Copy number variation was verified for all five selected loci in the predicted samples. Surprisingly, the amplification identified in sample HLHS24 near ADM displayed a calculated copy number of around four by qPCR (Figure 3 A), whereas the array estimated a copy number of three. The remaining case cohort samples yielded concordant qPCR and array copy number results. Another unexpected finding was discovering a single PDR sample with a deletion at the candidate locus near EDN1 (Figure 3 B). All of the other PDR samples displayed a normal copy number at each tested locus. These results corroborate the apparent private nature of most of the CNVs chosen for investigation.
Figure 3.
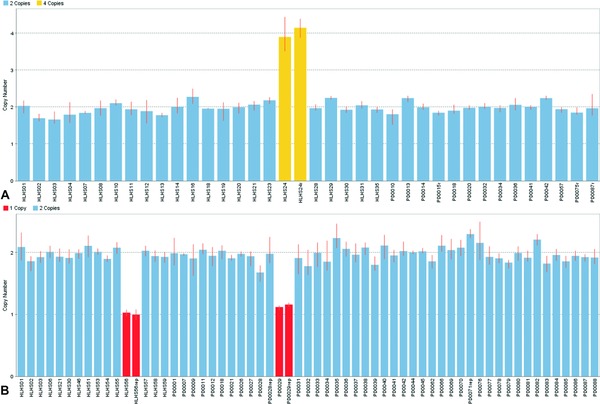
Bar graphs of calculated copy number for two candidate loci. (A) Results for locus near ADM. Sample HLHS24 yielded a calculated copy number near four in two separate runs. (B) Results for locus near EDN1. One case sample (HLHS56) and one control sample (PD0029, from PDR Collection) indicated a single‐copy deletion; each result was confirmed by an additional run.
Expression of putative candidate genes in mouse heart
To substantiate the potential of selected candidate genes to impact cardiac structural development, we verified their expression in embryonic mouse heart using TaqMan Assays (Table S5). All of the putative candidate genes tested were expressed in the heart at the two time points studied, E9.5 and E10.5. Since two time points were examined, the relative change in expression of these genes from E9.5 to E10.5 could be determined. Figure 4 graphically displays the results of this comparison. Transcript levels of Dscam were virtually unchanged during the period. Only the transcript for Adm showed a slight, yet significant, decrease in expression, whereas the other genes studied showed significant increases. Two of them, Cdh13 and Ntrk2, were upregulated approximately fourfold, a substantial amount that is suggestive of involvement with an emerging process during that time. Several steps of cardiogenesis commence during this time frame, including formation of endocardial cushions, the precursors to heart valves. It has been suggested that severe valve malformation underlies the development of HLHS.7 Thus, we contemplate that alterations in the dosage of these presumptive candidates may disrupt the normal formation of valves and other cardiac structures that are beginning to take shape during this period.
Figure 4.
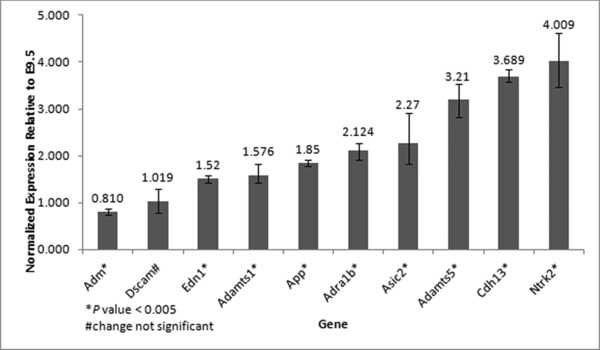
Relative change in expression of putative candidate genes from E9.5 to E10.5. The change was significant for every gene except Dscam (based on unpaired, two‐tailed t‐test, assuming equal variances). Error bars denote maximum and minimum fold change values based on the highest and lowest ΔCT values for each gene tested.
Expression of putative candidate genes in patient hearts
Having established the expression of our candidate genes in the heart during a relevant period of structural development, we next wanted to determine if this expression was significantly altered in specific patients. The majority of the patient samples tested were derived from explanted cardiac ventricle tissue removed at the time of transplantation. This afforded the opportunity to directly measure candidate gene dosage effects on ventricular expression. Of the ten patient CNV‐associated candidates chosen for expression studies, nine had corresponding patient RNA available to test, with ADRA1B being the lone exception. One of the candidates, CDH13, appeared to be disrupted by an identical deletion in two patients, although the CNV could only be verified in one patient due to the total consumption of the DNA sample from the other patient. Fortunately, RNA extracts from both patients were on hand. Two other candidates, ADAMTS1 and ADAMTS5, are adjacent to each other on chromosome 21 and overlapped by the same amplification. This circumstance can serve as a test of whether the expression of all genes within a structural variant are affected similarly (i.e., a strict dosage effect).
To explicitly determine possible effects of dosage on cardiac transcription, comparisons of candidate gene expression were made between samples from patients harboring CNVs that overlap or are proximal (i.e., within 50 kb) to the candidates, and samples from patients without these same CNVs. Using gender‐matched patients from the same cohort should help control for possible effects from disease state and patient age, although both of these factors are fairly heterogeneous in our sample set. To see if age and disease state had a noticeable effect on expression of these candidates, pooled normal adult and fetal heart RNA samples were obtained and assayed along with the cohort samples. A qualitative inspection of the data from both of the pooled samples revealed similar values, and these values were well within the range of those exhibited by the patient samples (Figure S1).
Having ruled out disease status and age as confounders, we proceeded with the comparisons. Four of the nine candidate genes demonstrated a significant difference in expression between patients with and without overlapping or proximal CNVs, at a level of significance of p < 0.05 (Figure 5). ADAMTS1 was completely covered by a genomic amplification, whereas only the first 9–12 exons (out of 13–21, depending on transcript variant) of NTRK2 were overlapped by a different amplification. Both genes displayed an increase in expression, consistent with a copy number increase. Likewise, the qPCR‐verified patient sample displaying a heterozygous deletion of a single exon in CDH13 showed decreased expression of the transcript specific to this exon. Another patient sample with a heterozygous deletion 8,806 bp from the 3′ end of EDN1 demonstrated reduced expression of this proximal gene, implying the disruption of a cis‐regulatory element by the CNV. Therefore, in some cases alterations of gene or regulatory element dosage by overlapping CNVs appear to have a significant impact on cardiac gene expression. Interestingly, a strict dosage effect does not seem to exist, since expression of ADAMTS5, a gene adjacent to ADAMTS1 and completely overlapped by the same CNV in a patient, did not display the same significant increase (Figure S2). Alternatively, other regulatory mechanisms may compensate for the altered dosage of particular genes.
Figure 5.
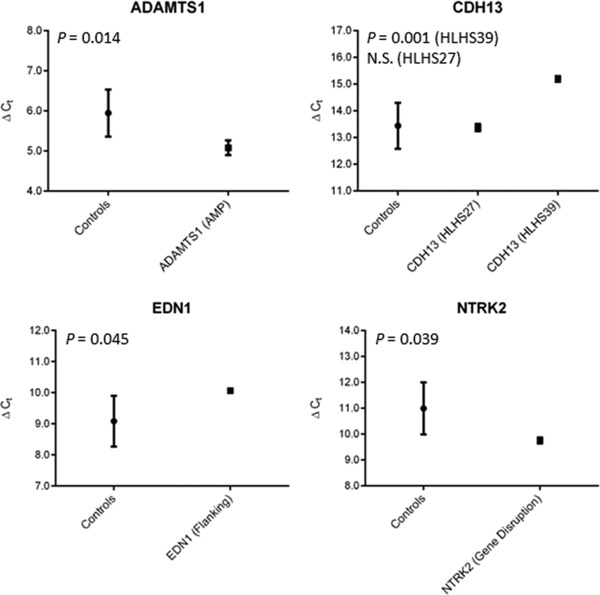
Plots of the ΔCT mean and standard deviation for genes found to differ significantly between patients with and without overlapping or proximal CNVs. N.S. indicates not significant.
Discussion
In this report, we document our search for genomic imbalances that could play a role in the etiology of congenital heart defects, particularly nonsyndromic HLHS. Our strategy for identifying potentially deleterious CNVs was primarily based on gene content, similar to other reports.15, 20, 26 Unlike other studies, we also considered genes that were near a CNV breakpoint (i.e., within 50 kb) to account for possible cis‐regulatory element dosage effects. Our approach highlighted 20 putative candidate genes potentially altered by 16 variants, four of which had no gene overlap. Only one of the 16 CNVs was identified in more than a single patient, although this result could only be verified in one of the patients due to a lack of sample.
Hence, nearly all of our highlighted CNVs appear to be private; comparable observations have been noted elsewhere.20, 23, 27 A private mutation model of disease would befit a heterogeneous, mostly sporadic condition such as HLHS. Yet previous studies, including our own, generally lacked the power to detect recurrent, yet extremely rare, causative CNVs due to insufficient sample sizes. Therefore, we adopted an alternative strategy: assessing the effects of some of our noted CNVs on the expression of putative candidate genes overlapped by or proximal to these variants.
First, we verified candidate gene expression in embryonic mouse heart during a period that corresponds to the start of valvulogenesis, a process that is often disrupted in HLHS patients. In addition to finding that every candidate was expressed, we discovered that several of these genes became substantially upregulated during the time frame. This result hints at the participation of these candidates in the formation of valves or other cardiac structures arising at that time.
Second, we measured nine of these genes for differential expression in human ventricular tissue between a patient harboring the gene‐overlapping or proximal CNV and patients without the same CNV. Four of the nine, including one (EDN1) that was not overlapped by but proximal to a CNV, demonstrated significantly altered expression in the same direction as the copy number change, loss or gain (Figure 5). These findings underscore possible dosage sensitivity and involvement of several novel genes in the etiology of HLHS.
One of these genes, ADAMTS1, has also been identified by other groups within genomic imbalances of CHD patients that are absent in controls.21, 23, 25 The gene encodes a metalloproteinase that regulates, through its proteolysis of the extracellular matrix proteoglycan versican, several vital cardiac developmental processes in mice, including remodeling of endocardial cushions38 and the outflow tract,39 as well as myocardial trabeculation.32 It is also located on human chromosome 21, whose trisomy results in Down syndrome, a condition associated with a high frequency of CHDs, though not typically HLHS. Thus, ADAMTS1 has multiple lines of evidence linking its aberrant dosage with cardiac structural defects, making it a strong candidate for involvement in the development of HLHS and other CHDs.
Unlike ADAMTS1, the other candidates we discovered, to our knowledge, have not been previously reported in human CHD studies. However, several of them have been implicated in normal mouse heart development, including EDN1 (endothelin 1)31, 40 and NTRK2 (neurotrophic tyrosine kinase, receptor, type 2; also known as TrkB).33, 41 Whether or not these candidates influence valvulogenesis or the formation of septal structures remains to be elucidated. Likewise, in some cases the determination of the precise mechanism of how specific patient CNVs are altering gene expression will require additional experiments. Yet the demonstration of their aberrant expression in human samples, combined with previous mouse data, supports their inclusion as valid candidate genes worthy of further investigation. We anticipate that as more candidate loci are identified, their functions and the cell types that they act upon will start to converge upon a limited number of cardiogenic networks or pathways. This could permit the development of biomarker screening panels to determine which processes are perturbed in a fetus, and perhaps direct appropriate intervention measures if available in the future.
Conclusion
We have identified rare and seemingly private copy number variants in HLHS patients that display a significant impact on the transcription of neighboring or overlapping cardiovascular genes. Our approach permits the discovery of credible candidates for involvement in the etiology of diseases with high heterogeneity and relatively low frequency.
Source of Funding
This work was supported by NIH grants HL089508‐04, HL014985‐39, and UL1 TR000154‐05.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Table S1. Details of first subject cohort.
Table S2. Details of second subject cohort (all HLHS + patient ID number) with array results.
Table S3. Autosomal CNVs identified by at least three detection algorithms.
Table S4. Genes with a mouse model phenotype that overlap or are near a verified CNV.
Table S5. Putative candidate genes evaluated for gene expression in cardiac tissue.
Figure S1. Box plots of the ΔCT values for the patient heart samples analyzed for gene expression, compared to the pooled normal fetal and adult heart samples. The gene results displayed correspond to the genes in Figure 5.
Figure S2. Plot of the ΔCT mean and standard deviation for ADAMTS5. The expression of this gene did not differ significantly between a patient with an overlapping amplification (HLHS23) and other patients without such a CNV.
Acknowledgments
We thank Karin Nunley for her preparation of the human ventricular RNA samples analyzed, and Andre Tavares for technical assistance with the mouse experiments.
References
- 1. Nemer M. Genetic insights into normal and abnormal heart development. Cardiovasc Pathol. 2008; 17(1): 48–54. [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention . Racial differences by gestational age in neonatal deaths attributable to congenital heart defects—United States, 2003–2006. MMWR Morb Mortal Wkly Rep. 2010; 59(37): 1208–1211. [PubMed] [Google Scholar]
- 3. Gordon BM, Rodriguez S, Lee M, Chang RK. Decreasing number of deaths of infants with hypoplastic left heart syndrome. J Pediatr. 2008; 153(3): 354–358. [DOI] [PubMed] [Google Scholar]
- 4. Sedmera D, Cook AC, Shirali G, McQuinn TC. Current issues and perspectives in hypoplasia of the left heart. Cardiol Young. 2005; 15(1): 56–72. [DOI] [PubMed] [Google Scholar]
- 5. Tchervenkov CI, Jacobs JP, Weinberg PM, Aiello VD, Beland MJ, Colan SD, Elliott MJ, Franklin RC, Gaynor JW, Krogmann ON, et al. The nomenclature, definition and classification of hypoplastic left heart syndrome. Cardiol Young. 2006; 16(4): 339–368. [DOI] [PubMed] [Google Scholar]
- 6. Ohye RG, Sleeper LA, Mahony L, Newburger JW, Pearson GD, Lu M, Goldberg CS, Tabbutt S, Frommelt PC, Ghanayem NS, et al. Comparison of shunt types in the Norwood procedure for single‐ventricle lesions. N Engl J Med. 2010; 362(21): 1980–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hinton RB, Jr. , Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson DW. Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol. 2007; 50(16): 1590–1595. [DOI] [PubMed] [Google Scholar]
- 8. Hinton RB, Martin LJ, Rame‐Gowda S, Tabangin ME, Cripe LH, Benson DW. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J Am Coll Cardiol. 2009; 53(12): 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin LJ, Ramachandran V, Cripe LH, Hinton RB, Andelfinger G, Tabangin M, Shooner K, Keddache M, Benson DW. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet. 2007; 121(2): 275–284. [DOI] [PubMed] [Google Scholar]
- 10. McBride KL, Zender GA, Fitzgerald‐Butt SM, Koehler D, Menesses‐Diaz A, Fernbach S, Lee K, Towbin JA, Leal S, Belmont JW. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur J Hum Genet. 2009; 17(6): 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, et al. Cardiac homeobox gene NKX2‐5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. 2003; 41(11): 2072–2076. [DOI] [PubMed] [Google Scholar]
- 12. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005; 437(7056): 270–274. [DOI] [PubMed] [Google Scholar]
- 13. Iascone M, Ciccone R, Galletti L, Marchetti D, Seddio F, Lincesso AR, Pezzoli L, Vetro A, Barachetti D, Boni L, et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin Genet. 2012; 81(6): 542–554. [DOI] [PubMed] [Google Scholar]
- 14. McBride KL, Riley MF, Zender GA, Fitzgerald‐Butt SM, Towbin JA, Belmont JW, Cole SE. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand‐induced signaling. Hum Mol Genet. 2008; 17(18): 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Breckpot J, Thienpont B, Arens Y, Tranchevent LC, Vermeesch JR, Moreau Y, Gewillig M, Devriendt K. Challenges of interpreting copy number variation in syndromic and non‐syndromic congenital heart defects. Cytogenet Genome Res. 2011; 135(3–4): 251–259. [DOI] [PubMed] [Google Scholar]
- 16. Erdogan F, Larsen LA, Zhang L, Tumer Z, Tommerup N, Chen W, Jacobsen JR, Schubert M, Jurkatis J, Tzschach A, et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet. 2008; 45(11): 704–709. [DOI] [PubMed] [Google Scholar]
- 17. Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, Khokha MK, Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left‐right patterning. Proc Natl Acad Sci USA. 2011; 108(7): 2915–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glessner JT, Bick AG, Ito K, Homsy JG, Rodriguez‐Murillo L, Fromer M, Mazaika E, Vardarajan B, Italia M, Leipzig J, et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res. 2014; 115(10): 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009; 41(8): 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hitz MP, Lemieux‐Perreault LP, Marshall C, Feroz‐Zada Y, Davies R, Yang SW, Lionel AC, D'Amours G, Lemyre E, Cullum R, et al. Rare copy number variants contribute to congenital left‐sided heart disease. PLoS Genet. 2012; 8(9): e1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Payne AR, Chang SW, Koenig SN, Zinn AR, Garg V. Submicroscopic chromosomal copy number variations identified in children with hypoplastic left heart syndrome. Pediatr Cardiol. 2012; 33(5): 757–763. [DOI] [PubMed] [Google Scholar]
- 22. Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, Yuen T, Rickaby J, Thiruvahindrapuram B, Marshall CR, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012; 8(8): e1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados‐Riveron J, et al. Contribution of global rare copy‐number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012; 91(3): 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns JP, Gewillig M, Vermeesch JR, Devriendt K. Submicroscopic chromosomal imbalances detected by array‐CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J. 2007; 28(22): 2778–2784. [DOI] [PubMed] [Google Scholar]
- 25. Thorsson T, Russell WW, El‐Kashlan N, Soemedi R, Levine J, Geisler SB, Ackley T, Tomita‐Mitchell A, Rosenfeld JA, Topf A, et al. Chromosomal imbalances in patients with congenital cardiac defects: a meta‐analysis reveals novel potential critical regions involved in heart development. Congenit Heart Dis. 2015; 10(3): 193–208. [DOI] [PubMed] [Google Scholar]
- 26. Tomita‐Mitchell A, Mahnke DK, Struble CA, Tuffnell ME, Stamm KD, Hidestrand M, Harris SE, Goetsch MA, Simpson PM, Bick DP, et al. Human gene copy number spectra analysis in congenital heart malformations. Physiol Genom. 2012; 44(9): 518–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane‐Yeboa K, Chung W, Yu L, Wong N, Awad D, et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet. 2014; 133(1): 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ritchie ME, Carvalho BS, Hetrick KN, Tavare S, Irizarry RA. R/Bioconductor software for Illumina's Infinium whole‐genome genotyping BeadChips. Bioinformatics 2009; 25(19): 2621–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998; 8(12): 1229–1231. [DOI] [PubMed] [Google Scholar]
- 30. Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci USA. 2001; 98(2): 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kurihara Y, Kurihara H, Oda H, Maemura K, Nagai R, Ishikawa T, Yazaki Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin‐1. J Clin Invest. 1995; 96(1): 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela‐Arispe ML, et al. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell 2008; 14(2): 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner N, Wagner KD, Theres H, Englert C, Schedl A, Scholz H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005; 19(21): 2631–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amano K, Fujii M, Arata S, Tojima T, Ogawa M, Morita N, Shimohata A, Furuichi T, Itohara S, Kamiguchi H, et al. DSCAM deficiency causes loss of pre‐inspiratory neuron synchroneity and perinatal death. J Neurosci. 2009; 29(9): 2984–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korbel JO, Tirosh‐Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, et al. The genetic architecture of Down syndrome phenotypes revealed by high‐resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA. 2009; 106(29): 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kosaki R, Kosaki K, Matsushima K, Mitsui N, Matsumoto N, Ohashi H. Refining chromosomal region critical for Down syndrome‐related heart defects with a case of cryptic 21q22.2 duplication. Congenit Anom (Kyoto) 2005; 45(2): 62–64. [DOI] [PubMed] [Google Scholar]
- 37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 2001; 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- 38. Kern CB, Twal WO, Mjaatvedt CH, Fairey SE, Toole BP, Iruela‐Arispe ML, Argraves WS. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006; 235(8): 2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kern CB, Norris RA, Thompson RP, Argraves WS, Fairey SE, Reyes L, Hoffman S, Markwald RR, Mjaatvedt CH. Versican proteolysis mediates myocardial regression during outflow tract development. Dev Dyn. 2007; 236(3): 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yanagisawa H, Hammer RE, Richardson JA, Williams SC, Clouthier DE, Yanagisawa M. Role of Endothelin‐1/Endothelin‐A receptor‐mediated signaling pathway in the aortic arch patterning in mice. J Clin Invest. 1998; 102(1): 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, Wang S, Ibanez CF, Rafii S, Hempstead BL. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development 2000; 127(21): 4531–4540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Table S1. Details of first subject cohort.
Table S2. Details of second subject cohort (all HLHS + patient ID number) with array results.
Table S3. Autosomal CNVs identified by at least three detection algorithms.
Table S4. Genes with a mouse model phenotype that overlap or are near a verified CNV.
Table S5. Putative candidate genes evaluated for gene expression in cardiac tissue.
Figure S1. Box plots of the ΔCT values for the patient heart samples analyzed for gene expression, compared to the pooled normal fetal and adult heart samples. The gene results displayed correspond to the genes in Figure 5.
Figure S2. Plot of the ΔCT mean and standard deviation for ADAMTS5. The expression of this gene did not differ significantly between a patient with an overlapping amplification (HLHS23) and other patients without such a CNV.