

Summary
Toll-like receptors (TLR) are transmembrane pattern recognition receptors that recognize microbial ligands and signal for production of inflammatory cytokines and type I interferon in macrophages and DC. Whereas TLR-induced inflammatory mediators are required for pathogen clearance, many are toxic to the host and can cause pathological inflammation when over-produced. This is demonstrated by the role of TLR-induced cytokines in autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, and systemic lupus erythematosus. Because of the potent effects of TLR-induced cytokines, we have diverse mechanisms to dampen TLR signaling. Here, we highlight three pathways that participate in inhibition of TLR responses in macrophages and DC, and their implications in autoimmunity; A20, encoded by the TNFAIP3 gene, Lyp encoded by the PTPN22 gene, and the BCAP/PI3K pathway. We present new findings that Lyp promotes TLR responses in primary human monocytes and that the autoimmunity risk Lyp620W variant is more effective at promoting TLR-induced IL-6 than the non-risk Lyp620R protein. This suggests that Lyp serves to downregulate a TLR inhibitory pathway in monocytes, and we propose that Lyp inhibits the TREM2/DAP12 inhibitory pathway. Overall, these pathways demonstrate distinct mechanisms of negative regulation of TLR responses, and all impact autoimmune disease pathogenesis and treatment.
Keywords: Toll-like receptors, autoimmune disease, signal transduction, macrophages, dendritic cells
Toll-like receptors
The principal function of the innate immune system is to recognize microbes and induce inflammatory responses that promote immediate pathogen control as well as initiate adaptive immunity. The innate immune system uses a diverse system of pattern recognition receptors (PRR) to survey both extracellular and intracellular niches to identify microbial pathogens and signal for appropriate inflammatory responses. Toll-like receptors (TLR) were the first defined class of PRR, and are found in evolutionarily diverse organisms, such as sponges, flies and mammals. The TLR family contains transmembrane receptors with share a common structure with leucine-rich repeats for ligand binding located extracellularly or in the endosome and a cytosolic signaling domain termed the Toll-IL-1 Receptor (TIR) domain.
TLR4 was the first mammalian TLR to have its specific ligand defined as the prototypical pathogen associated molecular pattern (PAMP) lipopolysaccharide (LPS), found in the cell wall of Gram negative bacteria(1). Since TLR4 was identified as the receptor for LPS, many microbe-specific TLR ligands have been found, including bacterial flagellin for TLR5, acylated lipoproteins for TLR2 heterodimers, and parasite profilin-like molecules for TLR10 and TLR11. As microbe-specific TLR ligands were identified, it was easy to classify TLR as receptors that distinguish between self and non-self. But this distinction was quickly found to be incorrect, as TLR that reside in endocytic compartments recognize nucleic acids, which are common to microbes and the host(2). TLR9 recognizes double stranded DNA, TLR3 recognizes double stranded RNA, and TLR7 and TLR13 recognize single stranded RNA(3). All of these nucleic acid sensing receptors have the potential to recognize both foreign and self nucleic acids. These TLR presumably evolved to sense microbial nucleic acids and are therefore localized to endosomes to help protect against detection of self nucleic acids(4). We now know that some cell-surface localized TLR also have endogenous ligands (discussed below).
Signaling through TLR leads to a well-studied signaling cascade initiated by TIR domain-containing signaling adapters, including MyD88, TRIF, TIRAP and TRAM, which interact with the TIR domain of the TLR(5,6). All TLR except TLR3 signal through MyD88, which leads to activation and nuclear translocation of NF-κB transcription factors, as well as MAPK pathway signaling leading to AP-1 activation, resulting in transcription of hundreds of pro-inflammatory genes (Fig. 1). These TLR-induced inflammatory genes include the prototypical pro-inflammatory cytokines TNF, IL-6 and IL-12 p40, as well as many chemokines that induce leukocyte homing to sites of infection to help begin the inflammatory process. In addition to induction of NF-κB-dependent inflammatory genes, many TLR also induce transcription of type I IFN genes that include the many genes encoding IFNα and the single IFNβ gene. TLR3 and TLR4 use TRIF, whereas TLR7 and TLR9 use MyD88, to induce activation and nuclear translocation of IRF3 and IRF7, resulting in IFNα and IFNβ gene transcription. Additionally, TLR7 and TLR9 use IRF5 to promote transcription of both inflammatory cytokines and type I interferon (IFN) (5). Thus TLR use several mechanisms to induce transcription of pro-inflammatory genes and type I IFN, both of which serve to alert the host to the presence of infection and promote immune activation and pathogen clearance.
Figure 1. Model of TLR signaling with select negative regulators.
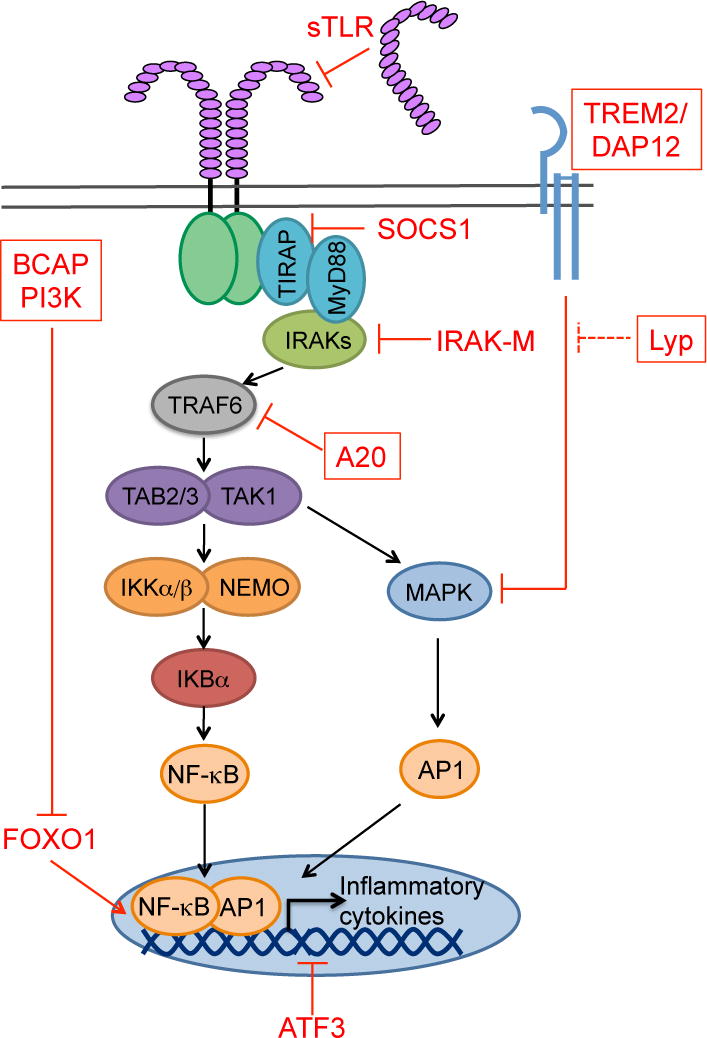
MyD88-dependent TLR signaling can be inhibited at any step along the signaling pathway. We have highlighted in red text select inhibitors that exemplify inhibition at different steps of the pathway. Those in a red box are discussed further in this review. For example, soluble forms of TLR2 and TLR4 inhibit signaling by blocking ligand binding to the TLR. SOCS1, A20 and IRAK-M block key steps in signal transduction downstream of most TLR (A20 and IRAK-M) or TLR4 (SOCS1). ATF3 is a transcription factor that dampens the transcription of inflammatory genes by recruiting histone deacetylases to the promoters of pro-inflammatory genes. BCAP is a signaling adapter that inhibits TLR signaling through PI3-kinase (PI3K) activation. One downstream mediator of PI3K inhibition of TLR responses is the transcription factor FOXO1, whose removal from the nucleus helps terminate transcription of some pro-inflammatory genes. The TREM2/DAP12 receptor signals in response to endogenous glycoproteins and lipids, thereby sensing the extracellular milieu of the macrophage. Signaling through TREM2/DAP12 inhibits TLR responses through reducing MAPK (Erk1/2) activation. Here we propose this pathway is regulated by the tyrosine phosphatase Lyp.
Pathological consequences of TLR signaling
Whereas TLR-induced inflammatory mediators are required for pathogen clearance, many of these molecules are toxic to the host and can cause pathological inflammation. This is most apparent with TNF, which has pleiotropic effects on many cell types and participates in the severe systemic inflammation seen in bacterially-induced sepsis, mainly through its effects on endothelial cells. Chronic TNF production is also found in target tissues of several autoimmune diseases where it promotes local inflammation(7). In Rheumatoid Arthritis (RA), TNF levels are elevated in affected joints and targeting this cytokine therapeutically is effective in many RA patients for alleviating joint inflammation. Elevated TNF is also seen in the intestinal tract of patients with inflammatory bowel disease (IBD) and TNF blockade is also effective in some IBD patients. IL-6, another prototypical TLR-induced inflammatory cytokine is also found in the joints of RA patients and monoclonal antibodies blocking IL-6 are also effective in treatment of this disease(8). The effectiveness of IL-6 blockade in other autoimmune diseases is under investigation. Not only are TLR-induced NF-κB-dependent pro-inflammatory cytokines, such as TNF and IL-6, implicated in autoimmune disease, but the type I IFN cytokine family, induced by TLR signaling through IRF3 and IRF7 is also implicated in autoimmune diseases(9,10). In systemic lupus erythematosus (SLE), elevated type I IFN production and evidence for IFN signaling via the presence of a type I IFN signature is found in the blood and tissues of many patients. The amount of type I IFN or IFN signature in the blood of SLE patients correlates with disease activity, autoantibodies and clinical manifestations of disease(9,10). To date, therapies targeting type I IFN signaling have not proved effective for SLE, but there is some evidence that patients with a low IFN signature may benefit from these therapeutics(11).
It is clear that cytokines associated with TLR-induced responses to pathogens are also implicated in autoimmune diseases—they are abundant in target organs and cytokine or cytokine receptor blockade are some of the most effective therapeutics that have been developed for these diseases. Many of these cytokines can be induced by signaling pathways in addition to TLR. TNF and IL-6 are produced downstream of signaling from the IL-1R, which also signals via MyD88, as well as by TNF signaling itself, causing amplification of an initial TNF signal. Type I IFN are induced not only by TLR signaling, but also by signaling via cytoplasmic PRR, such as the RIG-I-like receptors, RIG-I and MDA5, and cGAS, after RNA and DNA recognition, respectively(4,12). This leads us to ask “what is the evidence for TLR involvement in autoimmune disease?”
The most direct link between TLR signaling and autoimmunity is seen in SLE, where pathogenic autoantibodies recognize autoantigens that contain TLR agonists, in this case ssRNA, the TLR7 ligand, or dsDNA, the TLR9 ligand(13). Autoantibodies in SLE may directly recognize TLR ligands or may recognize cellular proteins that bind nucleic acids, such as chromatin or snRNPs. In mouse models of SLE, genetic loss of TLR7 protects mice from disease, whereas the role of TLR9 is more complex and can either promote or exacerbate disease (reviewed in (14)). Conversely, overexpression of TLR7 in mice via a BAC transgene can recapitulate several aspects of SLE in the non-autoimmune prone B6 mice and TLR7 overexpression due to a gene duplication of TLR7 causes disease in the BxSB SLE model(15,16). Genetic polymorphisms in the TLR7 locus have also been implicated in SLE in people. The rs3853839 single nucleotide polymorphism (SNP) is located in the 3′UTR of the TLR7 gene and is associated with increased risk of SLE (G>C) and increased expression of TLR7 mRNA and protein(17–21). Additionally, inhibitors of TLR7 and TLR9 ameliorated lupus-like disease in a mouse models(22,23), and are being developed for use in SLE patients. Therefore, there is strong evidence that TLR, particularly TLR7, is involved in SLE pathogenesis, though in which cells, plasmacytoid DC, conventional DC or B cells, is less clear.
There is also evidence for a role of TLR in autoimmune diseases other than SLE. Many reports document increased TLR expression and increased TLR responses in peripheral blood mononuclear cells (PBMC) and/or monocytes from patients with a variety of autoimmune diseases, including type 1 diabetes, RA, and SLE. The increased TLR responses may be secondary to inflammation due to ongoing disease in these patients or be directly caused by genetic polymorphisms or epigenetic changes impacting TLR signaling. In RA, there is evidence for TLR signaling in amplifying inflammation in the joint both in synovial fibroblasts and macrophages (reviewed in(24,25)), and blocking or inhibiting TLR4, TLR8, TLR2, MyD88 or TIRAP in cultures of synovial membranes from RA patients reduces inflammatory cytokine production (reviewed in (26)). Assessing the role of TLR in mouse models of autoimmune disease requires using spontaneously occurring models as many induced diseases use adjuvants that require TLR signaling for their function. The common RA model collagen-induced arthritis (CIA) and common multiple sclerosis (MS) model experimental autoimmune encephalomyelitis (EAE) both use complete freund’s adjuvant to induce disease. In a spontaneous model of RA driven by IL-1R antagonist deficiency, TLR4-deficiency or blockade ameliorated disease(27–29). Surprisingly, TLR2 deficiency exacerbated disease due to effects on regulatory T cells in this model(27). Thus in RA, data from both human and mouse disease implicate TLR, but the identity and source of the TLR agonists is not clear. Some infections have been associated with RA and may be a source of TLR agonists, but endogenous TLR agonists from intestinal microbiota or from damaged tissues (discussed below) have also been implicated(24–26). Associations of TLR with RA, SLE and other autoimmune diseases suggest targeting these pathways may be efficacious, and inhibitors of TLR4 and nucleic acid sensing TLR are under clinical development.
Negative regulation of TLR signaling
Because of the potent effects of TLR-induced cytokines, we have evolved a variety of mechanisms to dampen the magnitude and duration of TLR signaling. These negative regulators of TLR signaling also help protect against aberrant activation of this signaling cascade by endogenous TLR ligands. In the case of nucleic acid sensing TLR, while localization to the endosomal system protects the innate immune system from responding to extracellular RNA and DNA released from necrotic cells, this does not protect from sensing nucleic acids from apoptotic cells that are phagocytosed into this very compartment. TLR4 and TLR2/1 or TLR2/6 heterodimers signal not only in response to microbe-derived PAMPs, but also in response to endogenous ligands termed damage associated molecular patterns (DAMPs), some of which are released from dead and dying cells. These include HMGB1, a variety of heat shock proteins, fragments of extracellular matrix components such as hyaluronic acid, oxidized phospholipids and oxidized LDL, among others. Microbiota found in the intestines, lung, skin and other epithelial surfaces are also a rich source of TLR agonists. Therefore, specialized mechanisms are also required to ensure inflammation is limited in response to low levels of DAMPs, to nucleic acids in apoptotic cells, and to TLR agonists derived from microbiota. A diverse array of proteins serve as negative regulators of TLR signaling to perform this critical function.
Negative regulators of TLR signaling have been found which intersect the TLR signaling pathway at almost every step. These negative regulators show diversity not only in how they inhibit TLR signaling, but also in expression. Some negative regulators are expressed ubiquitously, whereas several are specific to macrophages or epithelial cells. There is also diversity in the timing of expression. Expression of some negative regulators is induced by TLR signaling itself, and these inhibitors appear important in controlling the magnitude of the peak response as well as turning off the response. Other negative regulators are constitutively expressed and as such can influence the sensitivity of the initial response as well as the peak and duration of TLR signaling. Mice with deficiency in some constitutive inhibitors display spontaneous inflammatory or autoimmune diseases, suggesting they are important for controlling the basal inflammatory response to endogenous TLR ligands, whether host or microbiota-derived. The large number and diverse actions of TLR negative regulators underscores the toxicity of pro-inflammatory cytokines and type I IFNs, which when overproduced can lead to pathological consequences. The abundance and function of these inhibitors must be finely tuned to allow for a productive response to pathogens, while limiting responses to self.
When thinking about the many ways that TLR signaling can be inhibited, it is useful to break down the TLR recognition and signaling pathways into discrete steps that can be interfered with by host negative regulators (Fig. 1). Ligand binding—molecules that bind to PAMPs thereby blocking their binding to TLR or that degrade TLR ligands. Cytosolic signal transduction—proteins that interfere with any stage of signal transduction from TLR proximal TIR-domain adapters to activation of cytosolic transcription factors for translocation to the nucleus. This is by far the largest class of inhibitors that have been defined. Transcription of inflammatory mediators—interference with NF-κB, IRF3/7 or AP1 DNA binding and/or activity in the nucleus. Sensing of the extracellular milieu—this class of inhibitors are cells surface receptors with endogenous ligands whose signaling can inhibit TLR responses. TLR co-engagement—receptors that are co-engaged with TLR or that sense the TLR ligand delivery method, such as in apoptotic cells, which inhibit or modify the TLR signal. As several reviews have covered TLR negative regulators in a broad manner (30,31), here, we will focus on three specific negative regulatory pathways that fall in several of these classes, and their implications for autoimmune disease.
A20—a deubiquitinase that inhibits TLR signaling and protects from autoimmunity
Proteins that directly inhibit the TLR signaling cascade are the largest group of identified TLR negative regulators(31). A20 is an example of a TLR negative regulator that fits within this class. A20 is a deubiquitinase with specificity for lysine 63 (K63)-linked ubiquitin chains, a protein modification associated with signal transduction via recruitment of ubiquitin binding proteins, rather than proteasome-mediated degradation as is associated K48-linked ubiquitin chains (reviewed in(32,33)). After TLR crosslinking and subsequent recruitment and/or activation of MyD88, IRAK4 and then IRAK1/2 are recruited to the forming signaling node or “myddosome”. IRAK activation recruits TRAF6, a ubiquitin E3 ligase, which mediates the addition of K63-linked ubiquitin chains to itself along with the E2 complex formed with Ubc13 and Uev1a proteins. This K63-linked polyubiquitylated TRAF6 can then bind TAB2 and TAB3, thereby recruiting TAK1 and mediating activation of the IKK complex and downstream NF-κB activation(5). A20 inhibits this process by cleaving K63-kinked ubiquitin chains from TRAF6, thereby impeding activation of this key point in TLR signal transduction(32,33). Interestingly, a large number of TLR negative regulators in addition to A20 also inhibit TRAF6 function, showing this is a critical node in TLR signal transduction(30,31).
The inhibitory function of A20 is supported by both human and mouse studies. Human genetic studies have identified many SNPs in and around the TNFAIP3 gene encoding A20 associated with increased risk of autoimmune and inflammatory diseases. This broad list of diseases includes RA, SLE, psoriasis, Crohn’s disease, T1D, Sjogren’s syndrome, Systemic sclerosis and Juvenile idiopathic arthritis(32). Several TNFAIP3 polymorphisms associated with risk of autoimmunity that have been studied in detail cause reduced A20 function or expression, including the non-synonymous SNP rs2230926 (34) and a polymorphic dinucleotide in a putative 3′ enhancer (35), consistent with idea that A20 is an inhibitor of immune responses. Interestingly, reduced A20 expression has been documented in cells from patients with several autoimmune diseases, and reduced A20 expression and polymorphisms in TNFAIP3 have been associated with response to TNF blocking therapies in RA, psoriasis and Crohn’s disease(32,36).
What is the evidence that A20 inhibition of TLR signaling in myeloid cells is important in autoimmune disease? This is an important question because A20 is broadly expressed and can inhibit multiple signaling pathways. Though A20 has been documented to inhibit TLR signaling in both macrophages and DC, A20 also targets other pro-inflammatory signaling pathways in these cells, including Nod2 signaling by targeting RIP2 and TNFR1 signaling by targeting RIP1(32,33). A20 also inhibits IL-1R signaling as this receptor also signals through TRAF6. Additionally, A20 is broadly expressed in the immune system and in non-hematopoietic cells, where it may promote autoimmune disease through targeting non-myeloid cells. Evidence for the importance of A20 in inhibiting TLR responses in myeloid cells for prevention of autoimmunity comes from mouse models. A20 deficiency in all cells leads to a fatal inflammatory disease causing perinatal lethality that does not depend upon the presence of B or T cells, highlighting the potency of A20 as an inhibitor of inflammation in the non-lymphoid compartment(37). Conditional deletion using a floxed allele of Tnfaip3 in macrophages and neutrophils with the Lysm-Cre strain caused the development of a spontaneous polyarticular arthritis similar to RA(38). This disease depended upon both MyD88 and TLR4 and could be ameliorated by treatment with blocking antibodies to IL-6, but not TNF. Thus, inhibition of TLR signaling in macrophages and neutrophils by A20 is important in protection from RA.
Conditional deletion of A20 in DC with the CD11c-Cre strain also caused spontaneous autoimmune disease, with one group finding lupus-like disease and arthritis and another group finding IBD-like colitis with associated arthritis(39,40). Both of these strains showed highly activated DC that spontaneously produced inflammatory cytokines and which were hyper-responsive to TLR agonists. By crossing these mice with Myd88-deficient mice, one group found that the increased TLR-induced IL-6 secretion in A20-deficient DC was MyD88 dependent, but that the increased DC activation was MyD88-independent(38). Thus, the increased DC activation may be not be due to augmented TLR signaling or may be TLR-dependent, but depend upon TRIF rather than MyD88. Together, these three mouse models with macrophage/neutrophil-specific A20 deletion and DC-specific A20 deletion clearly show that the ability of A20 to inhibit TLR responses in these myeloid cell populations is important in protection from autoimmune disease.
A20 is the prototype for a TLR negative regulator with strong associations with autoimmune disease. Not only are SNPs in A20 associated with a variety of autoimmune diseases, but mouse models of A20 deficiency have been informative in teasing apart the relative contributions of A20 in different hematopoietic lineages. A20 also highlights the common promiscuity among TLR negative regulators—A20 inhibits not only TLR signaling, but also several other signaling pathways that promote inflammatory responses. Therefore, dissecting the relative contribution of each pathway in mouse models is important. Several important questions have yet to be addressed about the role of A20 in autoimmunity. While A20 deficiency has been a useful tool in the mouse, mouse strains that model more precisely the major autoimmunity risk variants of TNFAIP3 in a ubiquitous and cell type specific manner will allow for a better understanding of how these risk variants promote disease. Future studies will also more precisely define the function of myeloid cells from subjects carrying the autoimmunity risk alleles of TNFAIP3.
TLR regulation by Lyp and the autoimmunity associated Lyp620W variant
One of the strongest genetic associations for increased risk of autoimmunity after the HLA region is a SNP in the PTPN22 gene(41). The PTPN22-1858T allele is associated with increased risk of developing many autoimmune diseases, particularly those associated with pathogenic autoantibodies, including type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus, and Graves disease, among others(41). The association of the PTPN22-1858T allele with so many diverse autoimmune diseases suggests that it may have a broad role in promoting immune responses(42), similar to variants in TNFAIP3 discussed above. This idea is supported by several studies showing that subjects with the PTPN22-1858T allele are protected from pulmonary tuberculosis(43–45) and from severe bacterial infections post allogeneic hematopoietic stem cell transplantation(46), though this association with increased bacterial clearance was not seen in all studies of tuberculosis (47) or other infections (48–50). Thus, the PTPN22-1858T allele is associated not only with many autoimmune diseases, but may also be associated with increased protection from bacterial infection, particularly infection with Mycobacterium tuberculosis.
PTPN22 encodes lymphocyte tyrosine phosphatase (Lyp). As its name suggests, Lyp is a protein tyrosine phosphatase expressed in B and T cells and has been primarily studied in these cell types. The normal function of Lyp has been most extensively studied in T cells, through knockdown, overexpression, and knockout models. Together, data from these systems show the normal function of Lyp and its mouse ortholog PEP in T cells is to negatively regulate T cell receptor (TCR) signaling through its interactions with kinases that participate in transducing TCR signals, including Lck, Fyn, and Zap-70 (reviewed in (51,52)). Lyp has also been reported to interact with a variety of important signaling molecules that may regulate its phosphatase function, such as Csk, and, conversely, Lyp may impact the function of its binding partners. In contrast, there is little evidence from PEP-deficient mice for a B cell-intrinsic role of Pep in regulating B cell receptor (BCR) signaling in an analogous manner(53). Though PEP-deficient mice have increased numbers of spontaneous germinal centers, this is thought to be because of T cell hyper-responsiveness, not intrinsic defects in the B cells(51,53). However, in humans Lyp is thought to influence B cell development and survival. Increased Lyp expression is present in chronic lymphoid leukemia (CLL) and is thought to contribute to the escape of these cells from BCR-mediated death. In human cell lines Lyp inhibits BCR signaling via Syk and PLCγ, but also enhances AKT activity by preventing the activation of the CD22/SHIP regulatory circuit(54). Therefore, from mouse models of PEP deficiency and knockdown studies, Lyp/Pep primarily functions as a negative regulator of TCR signaling, however, human overexpression studies indicate that Lyp also impacts BCR signaling(54).
The autoimmunity risk allele PTPN22-1858T encodes a tryptophan at amino acid 620 (Lyp620W), whereas the non-risk 1858C allele encodes an arginine at this position (Lyp620R)(41). Since the first association of the PTPN22-1858T allele with autoimmunity, the function of the Lyp620W variant has been under intensive investigation in both mouse and human systems, including in mouse strains engineered to express either the human Lyp-620W variant or the analogous mouse PEP-619W variant. While there have been some discordant findings in the field, studies performed in mice expressing the PEP619W demonstrate TCR and BCR hyper-responsiveness and subsequent alterations in T and B cell repertoire and selection consistent with a loss of function(55,56), and this has also been supported by studies in human cell lines(57). In human primary T and B cells the autoimmunity risk Lyp-620W variant demonstrates altered function with blunted TCR and BCR responses compared to the Lyp-620R protein, and additional alterations in signaling downstream of the antigen receptor that impact cell fate and survival ((58,59), reviewed in (51)). Interestingly, both human and mouse studies show the function of the Lyp-620W variant in promoting autoimmunity is not only important in T cells, but also in B cells(55,58). Thus, the role of Lyp and the Lyp620W variant in T and B cells is complex, and Lyp620W likely has unique functions compared to Lyp620R.
Lyp is also expressed in myeloid cells, such as monocytes, macrophages, DC, and neutrophils (60), and therefore the ability of the Lyp620W variant to promote autoimmunity may also have a contribution from these cell types that can both initiate and amplify immune responses. In parallel to the function of A20, possibly Lyp is an inhibitor of TLR responses due to dephosphorylation of a key tyrosine required for TLR signaling, this function is reduced by the Lyp620R variant, resulting in increased TLR responses. Alternatively, Lyp may serve to dephosphorylate an inhibitor of TLR signaling, thereby promoting TLR responses, and Lyp620W may be more effective at turning off this inhibitory pathway. Lastly, the non-risk variant of Lyp, Lyp620R, may have no role in regulating TLR responses, but the Lyp620W variant may have a novel target that regulates TLR signaling. Several recent studies, including our own described below, help shed light on the function of Lyp and Lyp620W on TLR signaling in myeloid cells.
Several groups have recently compared the function of the Lyp620R and Lyp620W proteins in regulating TLR responses in myeloid cells, predominantly in mouse models. Zhang et al. generated knockin mice that expressed the mouse ortholog of the Lyp620W variant, Lyp619W, and found that bone marrow-derived DC from these mice displayed increased LPS-induced activation as measured by CD40 upregulation, IL-12 secretion and ability to induce antigen-specific T cell proliferation, compared to wild-type mice expressing the Lyp619R protein(56). In the spleen, Lyp619W mice had increased DC numbers and increased CD40 expression constitutively. Though these authors assessed the responses of human primary B and T cells from subjects with the PTPN22-1858C and PTPN22-1858T alleles, they did not assess whether DC from these subjects also showed DC hyper-responsiveness(56). Additionally, Zhang et al. propose that the Lyp620W variant is a hypomorph due to increased calpain-mediated degradation, suggesting that Lyp is a negative regulator of DC TLR responses in the mouse. Subsequent work has challenged the notion that the Lyp620W protein has reduced stability and expression compared to the Lyp620R protein(55,61), implying that the increased DC TLR responses in the Lyp619W mice is due to a lack of a specific TLR inhibitory function of Lyp in these cells rather than a lack of expression of Lyp.
Subsequently, Wang et al. examined macrophage and DC TLR responses in both Ptpn22−/− mice and in a model of expression of Lyp620R or Lyp620W under the control of the endogenous promoter elements from a human BAC transgene expressed in Ptpn22−/− mice(61). They found that macrophages and DC from Ptpn22−/− mice produced less TLR-induced type I IFN than from WT mice. This was true downstream for TLR3 and TLR4 signaling in bone marrow-derived macrophages and DC, and for TLR7 and TLR9 signaling in bone marrow-derived plasmacytoid DC (pDC), and this defect in type I IFN production was shown both in vitro and in vivo. In contrast to the defect in TLR-induced type I IFN production, Wang et al. found no change in TLR-induced inflammatory cytokine production, including TNF, IL-6 and IL-12 p40 production(61). From these and other experiments using Ptpn22−/− mice, the authors concluded that Lyp is a positive regulator of TLR-induced type I IFN production and has no effect on TLR-induced inflammatory cytokine production.
Wang et al. next investigated the ability of the human Lyp620R and Lyp620W transgenes to rescue type I IFN production in Ptpn22−/− bone marrow-derived DC and macrophages and found that whereas the expression of the non-risk Lyp620R protein greatly increased LPS-induced IFNβ production, expression of the risk variant Lyp620W had little effect(61). Lastly the authors examined PBMC and PBMC-derived DC from healthy PTPN22-1858C/C homozygotes and PTPN22-1858C/T heterozygotes. The authors found lower IFN-stimulated gene (ISG) induction, but no change in TNF, IL6 or IL8 induction, in PTPN22-1858C/T heterozygotes compared to 1858C/C homozygotes, consistent with their findings in mice. Mechanistically, Wang et al. showed that Lyp promotes TLR-induced type I IFN through interactions with TRAF3 resulting in increased TRAF3 K63-ubiquitinylation and that Lyp620W does not have this function (61). More recently, this group followed up their findings in primary human plasmacytoid DC from patients with SLE comparing those homozygous for the PTPN22-1858C allele to those carrying one or two copies of the PTPN22-1858T allele(62). In this study, the authors found that pDCs carrying the PTPN22-1858T allele produced less TLR7-induced IFNα than PTPN22-1858C/C homozygotes. Together, these studies led the authors to conclude that Lyp620R is a positive regulator of TLR-induced type I IFN production and that Lyp620W is defective in this function. This work revealed an interesting new function for Lyp and showed that Lyp620W is a loss-of-function variant in TLR-induced type I IFN production in macrophages and DC. However, it is difficult to reconcile this lack of TLR-induced type I IFN production, particularly from pDC, with the increased susceptibility of carriers of the PTPN22-1858C allele to SLE where TLR-induced type I IFN is thought to promote disease.
We also investigated the role of Lyp in TLR signaling. Our interest stemmed from our previous work showing that signaling from the immunoreceptor TREM2 and its signaling chain DAP12 inhibit TLR responses in mouse macrophages and DC in an immunoreceptor tyrosine-based activation motif (ITAM)-dependent manner requiring the Syk and Btk/Tec tyrosine kinases(63–66). As Lyp inhibits TCR signaling and the Lyp620W protein affects both BCR and TCR signaling, which occur via ITAM-mediated signals similar to those transduced by TREM2/DAP12, we posited that Lyp might also inhibit TREM2/DAP12 signaling. As TREM2/DAP12 signaling inhibits TLR responses, this inhibition of an inhibitory pathway would allow Lyp to serve as a positive regulator of TLR responses.
First, we examined whether inhibition of Lyp phosphatase function in monocytes affected TLR responses. For these experiments, we purified CD14+ monocytes from PBMC of healthy subjects using magnetic bead enrichment, rested the cells for 2 hours, treated with either of two Lyp inhibitors (I-C11 or 8b) or vehicle control, and then activated with LPS and measured TNF and IL-6 secretion after overnight culture(67,68). We performed a careful dose response of LPS on monocytes from several donors to ensure we were using a non-saturating dose, because, from our experience, high doses of TLR agonists can mask regulatory pathways. Both Lyp inhibitors significantly reduced LPS-induced secretion of IL-6 and TNF in monocytes (Fig. 2A and B). Interestingly, Lyp inhibition had a greater effect on IL-6 secretion than on TNF secretion with the I-C11 inhibitor (IL-6, 36.5 +/− 3.4% inhibition; TNF, 15.9 +/− 4.2% inhibition), though we did not see this same effect with the 8b inhibitor (IL-6, 12.6 +/− 2.8% inhibition; TNF, 11.6 +/− 2.4% inhibition) that may be explained by the 5-fold lower concentration of 8b used compared to I-C11. These data show that in primary human monocytes, Lyp phosphatase activity promotes TLR-induced inflammatory cytokines. These data lend support to a model where Lyp turns off an inhibitory pathway, thereby serving as a positive regulator.
Figure 2. Inhibition of Lyp decreases TLR responses in monocytes.
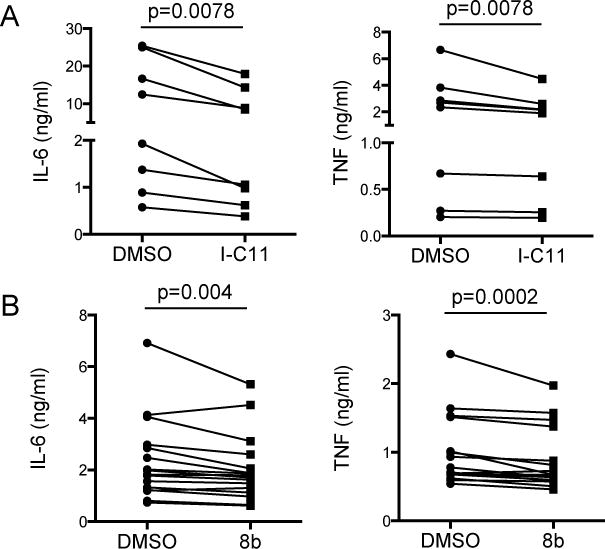
CD14+ monocytes were isolated from freshly thawed PBMC and rested for 2 hours before addition of A) Lyp inhibitor I-C11 (5 μM) or B) 8b (1 μM) or DMSO control. After 30 minutes monocytes were treated with 0.1 ng/ml LPS (S. minnesota R595, List Biological Laboratories, Campbell, CA, USA) and supernatants were collected after 16 hours. TNF and IL-6 were measured by ELISA (eBioscience, San Diego, CA, USA). These inhibitors did not cause cytotoxicity during the overnight culture as measured by LDH release assay (J. Pottle and J.A. Hamerman, unpublished observations). Statistical significance was determined using a paired Wilcoxon matched pairs signed rank test.
We next investigate the role of the Lyp620W variant in controlling TLR responses in monocytes. We purified CD14+ monocytes from PBMC from healthy subjects with no first-degree relatives with autoimmunity bearing the PTPN22-1858C/C or PTPN22-1858C/T genotype and treated with non-saturating doses of LPS, CpG DNA or PAM3CSK4. Monocytes from PTPN22-1858C/T subjects produced significantly more IL-6 than monocytes from PTPN22-1858C/C subjects in response to both LPS and CpG-A (Fig. 3A and B). This difference was less pronounced at higher doses, where it did not reach statistical significance (Fig. 3C and D). Interestingly, we saw only a small increase in CpG-induced TNF secretion from PTPN22-1858C/T subjects, and no difference in LPS-induced TNF secretion between groups (Fig. 3A–D). This is consistent with the stronger effect of the I-C11 Lyp inhibitor on IL-6 secretion than TNF secretion (Fig. 2A). We saw similar trends with TLR2/1 responses to PAM3CSK4, though we had few subjects after excluding TLR1 genotypes that result in TLR2/1 unresponsiveness(69) (J. Pottle and J.A. Hamerman, unpublished observations). We then asked if healthy subjects with the PTPN22-1858C/T genotype had increased systemic signs of innate immune activation compared to PTPN22-1858C/C subjects by examining serum cytokine levels. Interestingly, we found a significant increase in serum IL-8 in subjects heterozygous for the PTPN22-1858C/T autoimmunity risk allele, consistent with increased basal inflammation in these otherwise healthy subjects, potentially through increased TLR responses (Fig. 4). Therefore, the expression of the Lyp620W variant associated with increased risk of many autoimmune diseases causes increased inflammatory responses in vitro and in vivo.
Figure 3. Monocytes from PTPN22C/T subjects have increased TLR4 and TLR9-induced cytokine production.
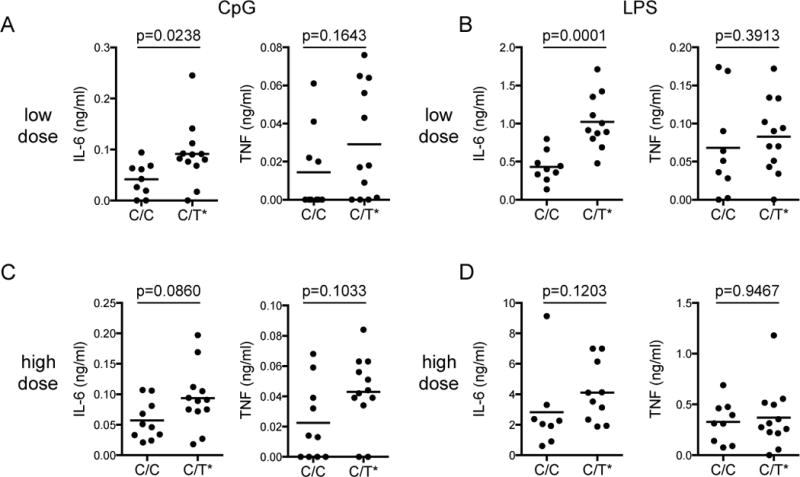
CD14+ monocytes were isolated from freshly thawed PBMC and rested for 2 hours before addition of CpG-A (Invivogen, San Diego, CA, USA) or LPS (S. minnesota R595, List Biological Laboratories). Supernatants were collected after 16 hours and TNF and IL-6 measured by ELISA. Doses were as follows: A) Low dose CpG-A 1 μM, B) Low dose LPS 0.025 ng/ml, C) High dose CpG-A 5 μM, D) High dose LPS 0.1 ng/ml. Statistical significance was determined using the Mann-Whitney test. Healthy subjects were recruited through the Benaroya Research Institute immune-mediated disease registry. The research protocols were approved by the institutional review board at the Benaroya Research Institute.
Figure 4. Healthy subjects with PTPN22C/T genotype have higher serum IL-8 than PTPN22C/C subjects.
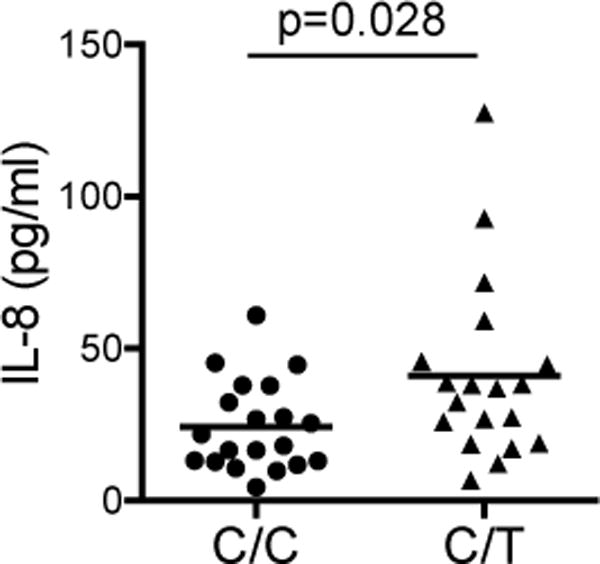
Serum IL-8 was measured by Bio-plex assay (Bio-Rad, Hercules, CA, USA) in 20 subjects of each genotype. Statistical significance was determined using a Mann-Whitney test.
Our Lyp inhibitor data indicates that both Lyp620W and Lyp620R can function to promote TLR responses in monocytes, as we saw reduced LPS-induced cytokines after inhibitor treatment in both PTPN22-1858C/C and PTPN22-1858C/T subjects (J.Pottle and J.A. Hamerman, unpublished observations). Furthermore, the fact that PTPN22-1858C/T subjects secrete more LPS and CpG DNA-induced IL-6 compared to PTPN22-1858C/C subjects demonstrates that the Lyp620W variant associated with enhanced risk of autoimmunity has an increased ability to promote TLR responses in comparison with the Lyp620R protein. These data suggest Lyp may dephosphorylate and thereby turn off a negative regulator of TLR signaling in monocytes. The Lyp620W variant may have increased activity against this TLR negative regulator compared to Lyp620R. Alternatively, Lyp620W may dephosphorylate a unique inhibitor of TLR signaling that is not dephosphorylated by Lyp620R, consistent with the evidence discussed above that the Lyp620W variant has altered phosphatase function in lymphocytes compared to Lyp620R.
While there are many potential substrates through which Lyp may promote TLR responses in monocytes by turning off an inhibitory pathway, many of which are likely yet to be identified, we favor a model in which Lyp dephosphorylates Syk or the DAP12 ITAM tyrosines. In T cells, Lyp dephosphorylates the Syk family kinase Zap-70 and the CD3 ITAM tyrosines in T cells(51). Syk has also been identified as a Lyp target(70). This would lead to reduced ITAM-mediated inhibition of TLR responses, which depend upon ITAM tyrosine phosphorylation and Syk(63), and higher TLR-induced cytokine secretion when Lyp activity is increased, and conversely increased ITAM-mediated inhibition of TLR responses and lower TLR-induced cytokine secretion when Lyp activity is reduced (Fig. 5). Alternatively, Lyp and Lyp620W may act upon other TLR inhibitory molecule or molecules yet to be identified.
Figure 5. Potential model for regulation of TLR signaling by Lyp downstream of the TREM2/DAP12 inhibitory pathway.
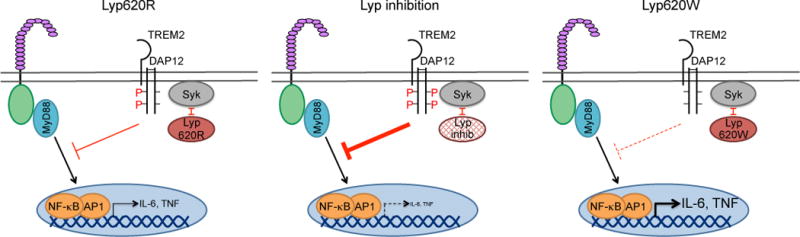
Signaling through TREM2/DAP12 inhibits TLR responses through the DAP12 ITAM tyrosines, Syk and Btk/Tec. A) We hypothesize that the Lyp620R variant has the capacity to dephosphorylate Syk and/or the DAP12 ITAM tyrosines, resulting in partial phosphorylation of the DAP12 ITAM and/or Syk. This causes a reduction in the function of the TREM2/DAP12 inhibitory pathway in monocytes and macrophages, thereby moderating TLR-induced NF-kB-dependent inflammatory cytokine production. B) We propose that when Lyp activity is inhibited, there is increased phosphorylation of DAP12 or increased signaling through Syk, resulting in a stronger inhibitory signal through TREM2/DAP12 and reduced IL-6 and TNF secretion. C) We propose that Lyp620W has a greater capacity to dephosphorylate DAP12 or Syk. This results in reduced DAP12 or Syk phosphorylation and a weaker inhibitory signal through TREM2/DAP12, leading to increased IL-6 and TNF secretion. Alternatively, Lyp620W may dephosphorylate a unique, distinct inhibitor than Lyp620R, thereby promoting TLR-induced IL-6 and TNF secretion.
Our data support the findings of Zhang et al. discussed above in which expression of the mouse variant analogous to Lyp620W (Lyp619R) causes increased DC TLR inflammatory responses in comparison with the wild-type Lyp allele(56). Zhang et al. did not assess monocyte or macrophage TLR-induced inflammatory cytokines, but we would predict they would also see increased responses as these are similarly regulated as DC IL-12 secretion. Our data are distinct from those of Wang et al., who show no difference in TLR-induced inflammatory cytokine production using bone marrow-derived DC from wild-type and Ptpn22−/− mice, DC from Ptpn22−/− mice with BAC transgenes expressing the Lyp620W and Lyp620R proteins, and PBMC-derived DC from PTPN22-1858C/C subjects(61). This could be due to differences between the monocytes used in our studies and the DC used by Wang et al., but we find it more likely that the difference is due to the high dose of TLR agonists used in their study—100 ng/ml LPS for mouse cells and 1 μg/ml LPS for human cells—that may overcome the Lyp-mediated regulation of inflammatory cytokine production. We did not measure IFNβ or IFNα mRNA or protein expression in our experiments, but it is interesting to note that there is often a reciprocal effect of regulators on NF-κB-dependent production of inflammatory cytokines, such as TNF, IL-6 and IL-12, and IRF3/7-dependent production of type I IFN. Therefore, decreased type I IFN production is often accompanied by increased inflammatory cytokines.
In conclusion, our work in human monocytes along with the work of Zhang et al. in mouse DC, suggest a novel function for Lyp in promoting TLR responses. As expression of the Lyp620W variant encoded by the PTPN22-1858T allele is associated with risk of many autoimmune diseases, this suggests that Lyp may have a broad role in promoting immune responses, not only in lymphocytes, but also in myeloid cells. The association of Lyp620W with increased TLR responses may lead to an increased inflammatory state with every infection, and in response to endogenous host-derived or microbiota-derived TLR ligands, in subjects carrying the PTPN22-1858T allele, thereby cooperating with dysregulated T and B cell responses and promoting autoimmunity. Our findings suggest that Lyp inhibitors may be efficacious in blunting inflammation in autoimmunity through their effects on myeloid cells.
Further investigation of the role of Lyp in TLR responses is needed as at the moment there are more questions than answers. These include: 1) What is(are) the target(s) of Lyp620R and Lyp620W required for inhibition of TLR-induced inflammatory cytokines in monocytes? We propose Lyp dephosphorylates the DAP12 ITAM tyrosines, Syk or other downstream mediators of the TREM2/DAP12 inhibitory pathway, but Lyp may have a variety of targets that influence TLR signaling. 2) What is the relationship between Lyp inhibition of TLR-induced type I IFN and Lyp promotion of TLR-induced inflammatory cytokines? It is intriguing that Lyp both inhibits TLR-induced type I IFN and promotes TLR-induced inflammatory cytokines, suggesting Lyp may alter the balance of activation between the NF-κB and IRF3/7 pathways, potentially by regulating the trafficking of TLR. 3) Does Lyp620W function in macrophages or DC promote autoimmunity? Myeloid-specific expression of the Lyp620W protein in mouse models are critical to dissect the cell-intrinsic contribution of the variant in autoimmune disease. 4) Does Lyp regulate signaling pathways in addition to TLR responses in monocytes? Interestingly, two recent papers suggest that Lyp regulates IFNγR signaling in macrophages(71,72), and Lyp may regulate other important pathways in these cells such as Fc receptor signaling, TNF signaling and IL-1R signaling. TREM2/DAP12 signaling can inhibit not only TLR signaling, but also FcR signaling in macrophage(64). 5) What is the role of Lyp in neutrophils? Neutrophil responses are important in some autoimmune diseases, particularly in RA and SLE. In neutrophils, one study implicated Lyp620W in increased protein citrullination and increased NETosis, whereas another study implicated this risk variant in increased neutrophil adhesion and reactive oxygen species (ROS) production(73,74).
TLR inhibition by PI3-kinase signaling
In addition to the activation of the NF-κB, IRF3/7 and MAPK pathways, TLR ligation also leads to activation of the lipid kinase phosphatidylinositol 3-kinase (PI3K) and its downstream signaling mediators. Whereas the NF-κB and MAPK pathways are pro-inflammatory, many studies have found that class I PI3K signaling serves to negatively regulate TLR signaling in macrophages and dendritic cells. Class IA PI3K consists of a catalytic subunit (p110α, β, or δ) and a regulatory subunit, the most abundant of which is p85α. The catalytic p110 subunit and regulatory p85 subunit are constitutively found in the cytoplasm in an inactive PI3K complex(75). The p85α subunit contains two SH2 domains that bind with high affinity to phosphotyrosines within YxxM sequences found in a variety of proteins. The SH2-dependent binding to p-YxxM within proteins causes a conformational change in the p85 subunit, resulting in increased catalytic activity of the associated p110 protein, and recruitment of the PI3K complex to the plasma membrane where it can access its substrate PI(4,5)P2 resulting in PI(3,4,5)P3 production(76). This causes the recruitment and activation of PDK-1 and Akt and subsequent activation of the downstream signaling cascade. Thus, binding of p85 to phosphorylated-YxxM sequences is critical for initiating PI3K signaling.
There is abundant evidence in the literature that PI3K pathway activation limits TLR signaling in macrophages and DC. This is seen using pharmacological inhibitors of the PI3K pathway in human monocytes and macrophages(77–79) as well as in studies with macrophages or dendritic cells from mice modified genetically to have altered PI3K. This was first seen with mice lacking the p85α regulatory subunit, which displayed increased TLR-induced DC production of IL-12, which allowed Balb/c mice typically susceptible to Leishmania major infection to clear this pathogen due to enhanced IL-12 dependent Th1 responses(80). This has been extended with mice deficient in proteins activated by the PI3K pathway, including the key downstream kinases PDK-1(81) and Akt1(82,83) as well as pharmacological inhibition of Akt(78). Conversely, macrophages lacking PTEN, the phosphatase that counters PI3K activity by catalyzing the removal of the phosphate added by PI3K, have reduced TLR-induced cytokine production, and mice with macrophage-specific PTEN deficiency had delayed clearance of Leishmania major(84–86). Thus, the PI3K pathway is a potent negative regulator of downstream of TLR signaling.
Several pathways have been implicated downstream of PI3K in inhibition of TLR responses in macrophages and DC, including induction of IL-10 via mTOR, activation of GSK3β, and phosphorylation and nuclear export of Foxo1(87–91). IL-10 is a potent anti-inflammatory cytokine produced by macrophages and DC in response to TLR agonists that signals through STAT3 to inhibit inflammatory responses, and dysregulated IL-10 production has been implicated in inflammatory diseases. Several studies have shown that PI3K pathway mediated activation of the mTOR complex promoted IL-10 secretion in response to LPS, and identified the mTORC2 complex that includes Rictor as specifically regulated by PI3K(88,90,92). Therefore, one mechanism by which PI3K activation inhibits inflammatory responses is via mTORC2 induction of IL-10 secretion, which then feeds back on macrophages by activating STAT3. Two groups also showed that there are IL-10 independent mechanisms by which PI3K inhibits TLR-induced TNF, IL-6 and IL-12 production that depend upon activation of GSK3β(88,93). Lastly, Foxo1, a member of the forkhead family of transcription factors whose activity is regulated by the PI3K pathway, has been implicated downstream of TLR-induced PI3K signaling in macrophages. In the absence of PI3K activation, Foxo1 is found in the nucleus bound to Foxo1 consensus DNA sequences. After activation of the PI3K pathway, active Akt, phsophorylated by PDK1 and mTORC2, can phosphorylate Foxo1 on three amino acids (Thr24/Ser256/Ser319) resulting in the export of Foxo1 from the nucleus into the cytoplasm, causing the termination of Foxo1-dependent transcription(94). Nuclear export of Foxo1 has been implicated in terminating transcription of several genes downstream of LPS-induced PI3K, Akt1 and mTORC2 activation in macrophages, including expression of IL-12 p40, IL-6 and IL-1β(89–91). Therefore, there are likely several different mechansims by which PI3K pathway activation inhibits TLR-induced inflammatory responses.
The studies described above show a clear inhibitory function of the PI3K pathway on TLR responses, but the exact mechanism by which PI3K is activated downstream of TLR ligation was not clear. Several studies showed that TLR2, TLR3 and TLR5, and TIRAP and MyD88, can directly bind the p85 subunit of PI3K after tyrosine phosphorylation in over-expression studies, many of which were performed in non-hematopoietic cells(95–99). Additionally, all TLR can activate the PI3K pathway, suggesting there may be additional mechanisms for PI3K activation. As binding of the p85 PI3K subunit to phosphorylated YxxM sequences is a general mechanism for activating class I PI3K, we hypothesized there is a myeloid-expressed YxxM protein linking TLR ligation to PI3K activation. One candidate was B cell adapter for PI3K (BCAP), a protein first identified in B cells(100), but with broad expression in hematopoietic cells including B cells, NK cells, neutrophils, monocytes, macrophages, and DC. BCAP is a large cytosolic protein with four YxxM motifs that are tyrosine phosphorylated after BCR crosslinking and subsequently recruit and activate the p85/p110 complex and downstream signals(100,101). BCAP also participates in PI3K activation in NK cells(102). Because of the high expression of BCAP in macrophages, we investigated if BCAP links TLR signaling to activation of the PI3K pathway and therefore serves as an inhibitor of TLR responses(103).
The first clue that BCAP was the elusive YxxM-containing adapter between TLR and PI3K activation came from an examination of TLR-induced cytokine production in BCAP-deficient (Pik3ap1−/−) mice. We found increased TNF, IL-6 and IL-12 p40 secretion in response to TLR4, TLR7 and TLR9 agonists in Pik3ap1−/− macrophages compared to wild-type macrophages, and increased serum IL-12 p40 after LPS injection in Pik3ap1−/− mice compared to wild-type mice(103). Thus, BCAP is a negative regulator of TLR responses in mice, and macrophages lacking BCAP phenocopy deficiency in the PI3K p85 regulatory subunit or macrophages treated with PI3K inhibitors. We next specifically examined the PI3K pathway activation by measuring phosphorylation of two sites on Akt required for its activation, Thr308, the site directly phosphorylated by PDK-1, and Ser473, a site phosphorylated by mTORC2(76). Pik3ap1−/− macrophages had reduced LPS-induced phosphorylation at both sites on Akt compared to wild-type macrophages, showing that as in B and NK cells, BCAP mediates activation of the PI3K pathway. This was specific to TLR signaling as TNF-mediated Akt phosphorylation was no different between wild-type and Pik3ap1−/− macrophages. Importantly, we showed that BCAP’s four YxxM tyrosines were required for the ability of BCAP to inhibit TLR responses in macrophages, proving that the mechanism by which BCAP inhibits TLR responses requires PI3K p85 binding. Interestingly, we found that BCAP was constitutively tyrosine phosphorylated and bound to p85 PI3K in macrophages and this phosphorylation did not change in response to LPS treatment, but instead BCAP was recruited to membranes after LPS signaling(103). Therefore, BCAP may function to bring the active p85/p110 complex to the membrane where it can access its substrate PI(4,5)P2, thereby generating PI(3,4,5)P3 that can recruit and activate PDK-1 initiating the PI3K signaling cascade.
Simultaneous to our work, two other groups identified this inhibitory role for BCAP in macrophages(104,105). Troutman et al. came to study BCAP in a unique manner, through a bioinformatics search for new TIR-containing proteins(104). They found that BCAP contains a TIR homology domain in its previously uncharacterized N-terminus, and can bind to both MyD88 and TIRAP through this interaction, though this was only shown in a transfection system in 293T cells. Importantly, Troutman et al. also demonstrated the anti-inflammatory effects of BCAP in vivo using two models(104). They found an increase in TNF-producing monocytes in the spleen early after infection with Salmonella Typhimurium and an increase in severity of DSS-mediated colitis in Pik3ap1−/− mice. Though these in vivo studies are consistent with increased TLR responses in the absence of BCAP, many innate pathways in addition to TLR can contribute to inflammation in these models.
From these studies, we are left with a model in which BCAP is recruited to the forming myddosome, potentially by interactions with MyD88 and TIRAP. This may explain the membrane recruitment of BCAP we see shortly after LPS treatment(103). BCAP brings the p85/p110 complex to its lipid substrate in membranes, where it can catalyze the formation of PI(3,4,5)P3 that recruits and activates PDK1, thereby initiating the downstream activation of Akt and downstream pathways. There are several outstanding questions about how BCAP functions to activate TLR signaling. 1) What kinase(s) phosphorylate BCAP in macrophages? In B cells, Syk and Btk have been shown to participate in BCR-mediated BCAP phosphorylation and Lyn in CD19-mediated BCAP phosphorylation. c-Abl has also been shown to tyrosine phosphorylate BCAP in K562 human CML cell line(106). In macrophages, we found that Syk-deficient macrophages have increased, not decreased BCAP phosphorylation(103), ruling out Syk as the BCAP kinase. Future studies will examine other kinases, both those implicated in BCAP phosphorylation and related kinases. What is the role of BCAP in myeloid cells in vivo? Because of the broad expression of BCAP in hematopoietic cells, cell-type specific deletion of BCAP is critical to investigate its function in vivo, both in macrophages and conventional DC. BCAP is also highly expressed in pDC and neutrophils and therefore may have important functions in these cells. What pathways in addition to TLR-induced PI3K can BCAP regulate? BCAP does not affect TNF-induced Akt phosphorylation(103,104), but BCAP likely inhibits IL-1R-induced PI3K activation and may also regulate other key signaling pathways in macrophages. Therefore, a comprehensive analysis of what pathways BCAP can regulate is warranted. What are the downstream pathways used by BCAP and PI3K to inhibit TLR-induced inflammatory responses? As discussed above, several pathways downstream of PI3K have been implicated in inhibiting TLR responses. We found no difference in IL-10 secretion between LPS-treated wild-type and BCAP-deficient macrophages, suggesting differences in IL-10 are not responsible. Our preliminary analyses show that levels of LPS-induced phosphorylated Foxo1 (Ser256) in the nucleus are dramatically reduced in BCAP-deficient macrophages compared to wild-type macrophages (M. Ni and J.A. Hamerman, unpublished observations), suggesting that BCAP promotes Akt-mediated phosphorylation and nuclear export of Foxo1, thereby terminating Foxo1-dependent transcription. Future studies will further investigate if activation of Foxo1 is required for BCAP-mediated inhibition of TLR responses.
Lastly, we ask, What is the impact of BCAP and PI3K-mediated inhibition of TLR responses on autoimmune disease? No SNPs in the PIK3AP1 locus have been associated with autoimmune diseases, but this many not be surprising because loss of BCAP function may reduce B cell survival(101), in addition increasing inflammatory responses. Therefore, how BCAP functions in macrophages and DC in autoimmunity will require cell-type specific deletion mouse models. Particularly interesting will be to assess models associated with increased TLR signaling, including models of SLE, RA and IBD. We predict that specific deletion of BCAP in macrophages and/or DC will cause an increase in severity of these diseases. Interestingly, the impact of myeloid PI3K signaling has not been investigated thoroughly in mouse models of autoimmunity, though this could be done using an existing conditional allele of the phosphatase Pten crossed to either CD11c-Cre or LysM-Cre strains, which would have decreased TLR signaling in targeted cells due to increased PIP3 levels. One study used Lysm-Cre/Ptenfl/fl mice to determine the role of PI3K in two arthritis models. The authors found the Lysm-Cre/Ptenfl/fl mice were protected from collagen-induced arthritis, but not from K/BxN serum transfer arthritis(107). Clearly, more studies of this kind are needed both using existing conditional PTEN mice with increased PI3K activity, as well as development of models with cell type specific and inducible reductions in PI3K activity.
Understanding the impact of macrophage and DC PI3K inhibition is a relevant question for autoimmunity because pan or selective class I PI3K inhibitors are being tested in clinical trials or are in pre-clinical development for treatment of autoimmune diseases, such as RA and psoriasis, inflammatory diseases such as asthma and COPD, and various forms of cancer(108–110). Some of these inhibitors have shown efficacy in mouse models of autoimmune disease, particularly models of SLE and RA, but a p110δ inhibitor has been tested in a phase 2 clinical trial in RA, but did not have efficacy(108). While these inhibitors are being developed and used based upon the idea that PI3K signaling promotes immune responses in B and T cells or promotes survival and growth of malignant cells, the impact of these inhibitors on the anti-inflammatory functions of PI3K in macrophages and DC needs to be considered. This is particularly important because of their likely long-term use if used in treatment of asthma and COPD, as well as in autoimmune diseases(111). Supporting this idea, mice with targeted inactivation of the catalytic PI3K subunit p110δ develop spontaneous colitis(112,113), and colitis, among other side effects, was caused by treatment of patients with a selective p110δ inhibitor for follicular B-cell non-Hodgkin lymphoma or relapsed small lymphocytic lymphoma(114,115). Therefore, a better understanding of the role of class I PI3K in both in innate and adaptive immune cells is warranted, including inhibitory roles of this pathway.
Concluding remarks
Here, we have highlighted only three of many pathways that have been shown to inhibit TLR responses in macrophages and DC, and have discussed implications of each of these pathways for autoimmune disease pathogenesis and treatment. There are TLR negative regulators that have been investigated in autoimmunity that we were not able to cover here, as well as many that have not yet been studied in the context of autoimmunity. Two proteins we discuss, A20 and Lyp, have risk variants broadly associated with autoimmunity, suggesting they have general roles in promoting immune responses, but by distinct mechanisms. In macrophages and DCs, A20 inhibits TLR signaling directly, whereas we propose that Lyp is involved in turning off inhibitory pathways through its phosphatase function, potentially through TREM2 and DAP12. No variants in genes encoding BCAP or the class I PI3K pathway have been associated with autoimmunity to date, however contrasting activating and inhibitory functions of these pathways in lymphocytes and myeloid cells may complicate this type of analysis. In fact, all three of the inhibitory pathways discussed here have in common broad expression amongst hematopoietic cells, regulation of multiple signaling pathways, and complex functions, making further study of each pathway in inhibiting TLR responses critical to better understanding both productive and pathogenic immune responses.
Acknowledgments
The authors acknowledge the following sources of funding that supported this work. J.A. Hamerman was supported by NIH grants R01AI113325, R01AI081948 and a pilot grant awarded under U19AI050864. Z-Y Zhang was supported by NIH grant R01CA69202 and J.H. Buckner was supported by R01AI083455.
References
- 1.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998 Dec 11;282(5396):2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 2.Ewald SE, Barton GM. Nucleic acid sensing Toll-like receptors in autoimmunity. Curr Opin Immunol. 2011 Feb;23(1):3–9. doi: 10.1016/j.coi.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. (2011) 2011 Sep;243(1):61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29(1):185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 2010 May;11(5):373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006 May;13(5):816–25. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 7.Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med. 2015 Jul;21(7):730–8. doi: 10.1038/nm.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. 2013 Mar;9(3):154–63. doi: 10.1038/nrrheum.2013.8. [DOI] [PubMed] [Google Scholar]
- 9.Elkon KB, Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol. 2012 Sep;24(5):499–505. doi: 10.1097/BOR.0b013e3283562c3e. [DOI] [PubMed] [Google Scholar]
- 10.Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008 May;17(5):394–9. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lauwerys BR, Ducreux J, Houssiau FA. Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 2014 Aug;53(8):1369–76. doi: 10.1093/rheumatology/ket403. [DOI] [PubMed] [Google Scholar]
- 12.Cai X, Chiu Y-H, Chen ZJ. Mol Cell. 2. Vol. 54. Elsevier; 2014. Apr 24, The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling; pp. 289–96. [DOI] [PubMed] [Google Scholar]
- 13.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–41. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S, Fitzgerald KA, Cancro MP, Marshak-Rothstein A. Nucleic Acid-Sensing Receptors: Rheostats of Autoimmunity and Autoinflammation. J Immunol American Association of Immunologists. 2015 Oct 15;195(8):3507–12. doi: 10.4049/jimmunol.1500964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deane JA, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. (2007) 2007 Nov;27(5):801–10. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. (2006) 2006 Jun 16;312(5780):1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 17.Shen N, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci USA. 2010 Sep 7;107(36):15838–43. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng Y, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 2013;9(2):e1003336. doi: 10.1371/journal.pgen.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C-M, et al. Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. 2014;4:3792. doi: 10.1038/srep03792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawasaki A, et al. TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther. 2011;13(2):R41. doi: 10.1186/ar3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enevold C, et al. Single nucleotide polymorphisms in genes encoding toll-like receptors 7, 8 and 9 in Danish patients with systemic lupus erythematosus. Mol Biol Rep. 2014 Sep;41(9):5755–63. doi: 10.1007/s11033-014-3447-4. [DOI] [PubMed] [Google Scholar]
- 22.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007 Dec;37(12):3582–6. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 23.Guiducci C, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. Journal of Experimental Medicine. (2010) 2010 Dec 20;207(13):2931–42. doi: 10.1084/jem.20101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goh FG, Midwood KS. Rheumatology (Oxford) 1. Vol. 51. Oxford University Press; 2012. Jan, Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis; pp. 7–23. [DOI] [PubMed] [Google Scholar]
- 25.Santegoets KCM, van Bon L, van den Berg WB, Wenink MH, Radstake TRDJ. FEBS Lett. 23. Vol. 585. Elsevier; 2011. Dec 1, Toll-like receptors in rheumatic diseases: are we paying a high price for our defense against bugs? pp. 3660–6. [DOI] [PubMed] [Google Scholar]
- 26.Abdollahi-Roodsaz S, van de Loo FAJ, van den Berg WB. Arthritis Res Ther. 2. Vol. 13. BioMed Central Ltd; 2011. Trapped in a vicious loop: Toll-like receptors sustain the spontaneous cytokine production by rheumatoid synovium; p. 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdollahi-Roodsaz S, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008 Jan;118(1):205–16. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, van den Brand BT, van de Loo FAJ, van den Berg WB. Am J Pathol. 5. Vol. 175. Elsevier; 2009. Nov, Local interleukin-1-driven joint pathology is dependent on toll-like receptor 4 activation; pp. 2004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdollahi-Roodsaz S, et al. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007 Sep;56(9):2957–67. doi: 10.1002/art.22848. [DOI] [PubMed] [Google Scholar]
- 30.Liew FY, Xu D, Brint EK, O’Neill LAJ. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005 Jun;5(6):446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 31.Kondo T, Kawai T, Akira S. Trends Immunol. 9. Vol. 33. Elsevier; 2012. Sep, Dissecting negative regulation of Toll-like receptor signaling; pp. 449–58. [DOI] [PubMed] [Google Scholar]
- 32.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012 Nov;12(11):774–85. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catrysse L, Vereecke L, Beyaert R, van Loo G. Trends Immunol. 1. Vol. 35. Elsevier; 2014. Jan, A20 in inflammation and autoimmunity; pp. 22–31. [DOI] [PubMed] [Google Scholar]
- 34.Musone SL, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adrianto I, et al. Nature Genetics. 3. Vol. 43. Nature Publishing Group; 2011. Mar 1, Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus; pp. 253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majumdar I, Paul J. The deubiquitinase A20 in immunopathology of autoimmune diseases. Autoimmunity. 2014 Aug;47(5):307–19. doi: 10.3109/08916934.2014.900756. [DOI] [PubMed] [Google Scholar]
- 37.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000 Sep 29;289(5488):2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011 Sep;43(9):908–12. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 39.Hammer GE, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011 Dec;12(12):1184–93. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kool M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011 Jul 22;35(1):82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. 2014 Oct;10(10):602–11. doi: 10.1038/nrrheum.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burn GL, Svensson L, Sanchez-Blanco C, Saini M, Cope AP. FEBS Lett. 23. Vol. 585. Elsevier; 2011. Dec 1, Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? pp. 3689–98. [DOI] [PubMed] [Google Scholar]
- 43.Gomez LM, Anaya J-M, Martin J. Genetic influence of PTPN22 R620W polymorphism in tuberculosis. Hum Immunol. 2005 Dec;66(12):1242–7. doi: 10.1016/j.humimm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Boechat AL, Ogusku MM, Sadahiro A, Santos dos MC. Association between the PTPN22 1858C/T gene polymorphism and tuberculosis resistance. Infect Genet Evol. 2013 Jun;16:310–3. doi: 10.1016/j.meegid.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 45.Lamsyah H, et al. Association of PTPN22 gene functional variants with development of pulmonary tuberculosis in Moroccan population. Tissue Antigens. 2009 Sep;74(3):228–32. doi: 10.1111/j.1399-0039.2009.01304.x. [DOI] [PubMed] [Google Scholar]
- 46.Azarian M, et al. The PTPN22 R620W polymorphism is associated with severe bacterial infections after human leukocyte antigen geno-identical haematopoietic stem-cell transplantations. Transplantation. 2008 Jun 27;85(12):1859–62. doi: 10.1097/TP.0b013e31817729c4. [DOI] [PubMed] [Google Scholar]
- 47.Kouhpayeh H-R, et al. R620W functional polymorphism of protein tyrosine phosphatase non-receptor type 22 is not associated with pulmonary tuberculosis in Zahedan, southeast Iran. Genet Mol Res. 2012;11(2):1075–81. doi: 10.4238/2012.April.27.6. [DOI] [PubMed] [Google Scholar]
- 48.Bravo MJ, et al. PTPN22 C1858T polymorphism and human brucellosis. Scand J Infect Dis. 2009;41(2):109–12. doi: 10.1080/00365540802641864. [DOI] [PubMed] [Google Scholar]
- 49.Robledo G, González CI, Morillo C, Martin J, González A. Tissue Antigens. 3. Vol. 69. Blackwell Publishing Ltd; 2007. Mar, Association study of PTPN22 C1858T polymorphism in Trypanosoma cruzi infection; pp. 261–4. [DOI] [PubMed] [Google Scholar]
- 50.Montes-Cano MA, et al. PTPN22 C1858T polymorphism and the outcome of hepatitis C virus infection. Viral Immunol. 2008 Dec;21(4):491–4. doi: 10.1089/vim.2008.0040. [DOI] [PubMed] [Google Scholar]
- 51.Rawlings DJ, Dai X, Buckner JH. J Immunol. 7. Vol. 194. American Association of Immunologists; 2015. Apr 1, The role of PTPN22 risk variant in the development of autoimmunity: finding common ground between mouse and human; pp. 2977–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol. 2014;32(1):83–119. doi: 10.1146/annurev-immunol-032713-120249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. Science. 5658. Vol. 303. American Association for the Advancement of Science; 2004. Jan 30, PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells; pp. 685–9. [DOI] [PubMed] [Google Scholar]
- 54.Negro R, et al. Blood. 26. Vol. 119. American Society of Hematology; 2012. Jun 28, Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT; pp. 6278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai X, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013 May;123(5):2024–36. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011 Sep;43(9):902–7. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 57.Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. J Immunol. 7. Vol. 182. American Association of Immunologists; 2009. Apr 1, PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background; pp. 4093–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arechiga AF, et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. (2009) 2009 Mar 15;182(6):3343–7. doi: 10.4049/jimmunol.0713370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. (2007) 2007 Oct 1;179(7):4704–10. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- 60.Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Sci Signal Science Signaling. 2010;3(137):rs1–rs1. doi: 10.1126/scisignal.2000966. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, et al. Immunity. 1. Vol. 39. Elsevier; 2013. Jul 25, The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity; pp. 111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, et al. PTPN22 Variant R620W Is Associated With Reduced Toll-like Receptor 7-Induced Type I Interferon in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015 Sep;67(9):2403–14. doi: 10.1002/art.39211. [DOI] [PubMed] [Google Scholar]
- 63.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005 Jun;6(6):579–86. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006 Aug 15;177(4):2051–5. doi: 10.4049/jimmunol.177.4.2051. [DOI] [PubMed] [Google Scholar]
- 65.Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. 2012 Jan;42(1):176–85. doi: 10.1002/eji.201141679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tampella G, et al. The Tec Kinase-Regulated Phosphoproteome Reveals a Mechanism for the Regulation of Inhibitory Signals in Murine Macrophages. J Immunol. 2015 Jul 1;195(1):246–56. doi: 10.4049/jimmunol.1403238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu X, et al. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA. 2007 Dec 11;104(50):19767–72. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stanford SM, et al. Discovery of a novel series of inhibitors of lymphoid tyrosine phosphatase with activity in human T cells. J Med Chem. 2011 Mar 24;54(6):1640–54. doi: 10.1021/jm101202j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawn TR, et al. A common human TLR1 polymorphism regulates the innate immune response to lipopeptides. Eur J Immunol. 2007 Aug;37(8):2280–9. doi: 10.1002/eji.200737034. [DOI] [PubMed] [Google Scholar]
- 70.Martin VA, et al. Akt2 inhibits the activation of NFAT in lymphocytes by modulating calcium release from intracellular stores. Cell Signal. 2012 May;24(5):1064–73. doi: 10.1016/j.cellsig.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spalinger MR, et al. Loss of protein tyrosine phosphatase nonreceptor type 22 regulates interferon-γ-induced signaling in human monocytes. Gastroenterology. 2013 May;144(5):978–988.e10. doi: 10.1053/j.gastro.2013.01.048. [DOI] [PubMed] [Google Scholar]
- 72.Chang H-H, et al. J Immunol. 5. Vol. 191. American Association of Immunologists; 2013. Sep 1, PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis; pp. 2134–43. [DOI] [PubMed] [Google Scholar]
- 73.Bayley R, et al. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. 2015 Aug;74(8):1588–95. doi: 10.1136/annrheumdis-2013-204796. [DOI] [PubMed] [Google Scholar]
- 74.Chang H-H, Dwivedi N, Nicholas AP, Ho I-C. The W620 Polymorphism in PTPN22 Disrupts Its Interaction With Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol. 2015 Sep;67(9):2323–34. doi: 10.1002/art.39215. [DOI] [PubMed] [Google Scholar]
- 75.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. (2006) 2006 Aug;7(8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 76.Cantley LC. The phosphoinositide 3-kinase pathway. Science. (2002nd) 2002 May 31;296(5573):1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 77.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002 Aug 30;277(35):32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 78.Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003 Jul 15;171(2):717–25. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 79.Monick MM, et al. J Biol Chem. 2002nd. 36. Vol. 277. American Society for Biochemistry and Molecular Biology; 2002. Sep 6, Phosphatidylinositol 3-kinase activity negatively regulates stability of cyclooxygenase 2 mRNA; pp. 32992–3000. [DOI] [PubMed] [Google Scholar]
- 80.Fukao T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002 Sep;3(9):875–81. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 81.Chaurasia B, Mauer J, Koch L, Goldau J, Kock A-S, Brüning JC. Mol Cell Biol. 17. Vol. 30. American Society for Microbiology; 2010. Sep, Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-kappaB activation in macrophages; pp. 4354–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Androulidaki A, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. (2009) 2009 Aug 21;31(2):220–31. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arranz A, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA. 2012 Jun 12;109(24):9517–22. doi: 10.1073/pnas.1119038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao X, et al. J Immunol. 8. Vol. 172. American Association of Immunologists; 2004. Apr 15, The inositol 3-phosphatase PTEN negatively regulates Fc gamma receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages; pp. 4851–7. [DOI] [PubMed] [Google Scholar]
- 85.Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008 Mar 15;180(6):4218–26. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuroda S, et al. Effective clearance of intracellular Leishmania major in vivo requires Pten in macrophages. Eur J Immunol. 2008 May;38(5):1331–40. doi: 10.1002/eji.200737302. [DOI] [PubMed] [Google Scholar]
- 87.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J DENT RES. 2011 Apr;90(4):417–27. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohtani M, et al. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. (2008) 2008 Aug 1;112(3):635–43. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fan W, et al. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 2010 Dec 15;29(24):4223–36. doi: 10.1038/emboj.2010.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem. 2011 Dec 30;286(52):44295–305. doi: 10.1074/jbc.M111.258053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Su D, et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes. 2009 Nov;58(11):2624–33. doi: 10.2337/db09-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weichhart T, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008 Oct 17;29(4):565–77. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 93.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005 Aug;6(8):777–84. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eijkelenboom A, Burgering BMT. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013 Feb;14(2):83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 95.Arbibe L, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000 Dec;1(6):533–40. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 96.Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004 Nov;11(11):1060–7. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- 97.Rhee SH, Kim H, Moyer MP, Pothoulakis C. J Biol Chem. 2006. 27. Vol. 281. American Society for Biochemistry and Molecular Biology; 2006. Jul 7, Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells; pp. 18560–8. [DOI] [PubMed] [Google Scholar]
- 98.Santos-Sierra S, et al. EMBO J. 14. Vol. 28. EMBO Press; 2009. Jul 22, Mal connects TLR2 to PI3Kinase activation and phagocyte polarization; pp. 2018–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laird MHW, et al. J Leukoc Biol. 6. Vol. 85. Society for Leukocyte Biology; 2009. Jun, TLR4/MyD88/PI3K interactions regulate TLR4 signaling; pp. 966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity. 2000 Dec;13(6):817–27. doi: 10.1016/s1074-7613(00)00079-0. [DOI] [PubMed] [Google Scholar]
- 101.Yamazaki T, Kurosaki T. Contribution of BCAP to maintenance of mature B cells through c-Rel. Nat Immunol. 2003 Aug;4(8):780–6. doi: 10.1038/ni949. [DOI] [PubMed] [Google Scholar]
- 102.MacFarlane AWT, Yamazaki T, Fang M, Sigal LJ, Kurosaki T, Campbell KS. Enhanced NK-cell development and function in BCAP-deficient mice. Blood. (2008) 2008 Jul 1;112(1):131–40. doi: 10.1182/blood-2007-08-107847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ni M, MacFarlane AW, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc Natl Acad Sci USA. 2012 Jan 3;109(1):267–72. doi: 10.1073/pnas.1111957108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Troutman TD, et al. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc Natl Acad Sci USA. 2012 Jan 3;109(1):273–8. doi: 10.1073/pnas.1118579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsumura T, et al. Identification of BCAP-(L) as a negative regulator of the TLR signaling-induced production of IL-6 and IL-10 in macrophages by tyrosine phosphoproteomics. Biochem Biophys Res Commun. 2010 Sep 17;400(2):265–70. doi: 10.1016/j.bbrc.2010.08.055. [DOI] [PubMed] [Google Scholar]
- 106.Maruoka M, et al. Abi-1-bridged tyrosine phosphorylation of VASP by Abelson kinase impairs association of VASP to focal adhesions and regulates leukemic cell adhesion. Biochem J. 2011;441:889–899. doi: 10.1042/BJ20110951. [DOI] [PubMed] [Google Scholar]
- 107.Blüml S, et al. Arthritis Res Ther. 1. Vol. 17. BioMed Central Ltd; 2015. Phosphatase and tensin homolog (PTEN) in antigen-presenting cells controls Th17-mediated autoimmune arthritis; p. 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stark A-K, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol. 2015 Aug;23:82–91. doi: 10.1016/j.coph.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ball J, Archer S, Ward S. PI3K inhibitors as potential therapeutics for autoimmune disease. Drug Discov Today. 2014 Aug;19(8):1195–9. doi: 10.1016/j.drudis.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 110.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014 Feb;13(2):140–56. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. (2005) 2005 Mar;22(3):285–94. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 112.Okkenhaug K, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002 Aug 9;297(5583):1031–4. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 113.Uno JK, et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110δ. Gastroenterology. 2010 Nov;139(5):1642–53–1653.e1–6. doi: 10.1053/j.gastro.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Furman RR, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014 Mar 13;370(11):997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miller BW, et al. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015 Apr 1;21(7):1525–9. doi: 10.1158/1078-0432.CCR-14-2522. [DOI] [PubMed] [Google Scholar]