

Abstract
White matter degeneration is a pathological hallmark of neurodegenerative diseases including Alzheimer's. Age remains the greatest risk factor for Alzheimer's and the prevalence of age-related late onset Alzheimer's is greatest in females. We investigated mechanisms underlying white matter degeneration in an animal model consistent with the sex at greatest Alzheimer's risk. Results of these analyses demonstrated decline in mitochondrial respiration, increased mitochondrial hydrogen peroxide production and cytosolic-phospholipase-A2 sphingomyelinase pathway activation during female brain aging. Electron microscopic and lipidomic analyses confirmed myelin degeneration. An increase in fatty acids and mitochondrial fatty acid metabolism machinery was coincident with a rise in brain ketone bodies and decline in plasma ketone bodies. This mechanistic pathway and its chronologically phased activation, links mitochondrial dysfunction early in aging with later age development of white matter degeneration. The catabolism of myelin lipids to generate ketone bodies can be viewed as a systems level adaptive response to address brain fuel and energy demand. Elucidation of the initiating factors and the mechanistic pathway leading to white matter catabolism in the aging female brain provides potential therapeutic targets to prevent and treat demyelinating diseases such as Alzheimer's and multiple sclerosis. Targeting stages of disease and associated mechanisms will be critical.
Abbreviations: WM, white matter; AD, Alzheimer's disease; MTL, medial temporal lobe; H2O2, hydrogen peroxide; cPLA2, cytosolic phospholipase A2; CC, corpus callosum; APO-ε4, apolipoprotein ε4; FDG-PET, 2-[18F]fluoro-2-deoxy-d-glucose; CMRglu, cerebral glucose metabolic rate; PCC, posterior cingulate; PDH, pyruvate dehydrogenase; COX, complex IV cytochrome c oxidase; ABAD, Aβ-binding-alcohol-dehydrogenase; BBB, blood brain barrier; MCT1, monocarboxylate transporter 1; WT, wild type; MIB, mitochondrial isolation buffer; OCR, oxygen consumption rate; RCR, respiratory control ratio; GFAP, glial fibrillary acidic protein; PEI, polyethyleneimine; TLDA, TaqMan low density array; PCR, polymerase chain reaction; MBP, myelin basic protein; HADHA, hydroxyacyl-CoA dehydrogenase; ABAD, Aβ-binding alcohol dehydrogenase; CPT1, carnitine palmitoyltransferase 1; PB, phosphate buffer; LC MS, liquid chromatography mass spectrometer; Cyp2j6, arachidonic acid epoxygenase; ACER3, alkaline ceramidase; MAG, myelin associated glycoprotein; MOG, myelin oligodendrocyte glycoprotein; Erbb3, Erb-B2 receptor tyrosine kinase 3; Cldn11, claudin 11; Olig2, oligodendrocyte transcription factor; NEFA, nonesterified fatty acids; S1P, sphingosine; DHA, docosahexaesnoic acid; ROS, reactive oxygen species; APP, amyloid precursor protein; BACE1, beta-secretase 1; HK, hexokinase
Keywords: White matter, Mitochondria, Neurodegeneration, Alzheimer's disease, Aging oxidative stress, Ketone bodies, Cytosolic phospholipase A2
Highlights
-
•
Mitochondrial dysfunction activates mechanisms for catabolism of myelin lipids to generate ketone bodies for ATP production.
-
•
Mechanisms leading to ketone body driven energy production in brain coincide with stages of reproductive aging in females.
-
•
Sequential activation of myelin catabolism pathway during aging provides multiple therapeutic targets and windows of efficacy.
The mechanisms underlying white matter degeneration, a hallmark of multiple neurodegenerative diseases including Alzheimer's, remain unclear. Herein we provide a mechanistic pathway, spanning multiple transitions of aging, that links mitochondrial dysfunction early in aging with later age white matter degeneration. Catabolism of myelin lipids to generate ketone bodies can be viewed as an adaptive survival response to address brain fuel and energy demand. Women are at greatest risk of late-onset-AD, thus, our analyses in female brain address mechanisms of AD pathology and therapeutic targets to prevent, delay and treat AD in the sex most affected with potential relevance to men.
1. Introduction
Age-related myelin breakdown occurs during normal aging and in major neurodegenerative diseases including Alzheimer's (Bartzokis, 2004, Decarli et al., 1995, Erten-Lyons et al., 2013, Ge et al., 2002, Lu et al., 2013, Lebel et al., 2012, Zhang et al., 2007, Tang et al., 1997). White matter degeneration in Alzheimer's disease (AD) worsens with progression of disease and is predictive of cognitive decline (Bartzokis, 2004, Decarli et al., 1995, Erten-Lyons et al., 2013, Ge et al., 2002, Lu et al., 2013, Lebel et al., 2012, Zhang et al., 2007, Tang et al., 1997). Brain regions that myelinate late in brain development and which are populated by small, thinly myelinated are most vulnerable to breakdown and degeneration in AD. Late-myelinating regions include cortical association areas such as fronto-parietal tracts, the genu of the corpus callosum (CC), the uncinate fasiculus, and the superior longitudinal fasiculus (Brickman et al., 2012, Marner et al., 2003). Further, the afferent targets of these fiber systems, which include the hippocampus and subiculum, also show white matter (WM) hyperintensities (Di Paola et al., 2010, Brickman et al., 2012, Marner et al., 2003). Many of these vulnerable fiber tracts, such as the superior longitudinal fasiculus, the CC, internal capsule, corona radiata, and parahippocampal WM also have a high degree of heritability (Sprooten et al., 2014, Jahanshad et al., 2013). Myelin breakdown manifests earlier in apolipoprotein ε4 (APO-ε4) carriers, a major genetic risk factor for AD (Bartzokis et al., 2006). Regions most vulnerable to WM degeneration map onto regions preferentially affected in the pathological trajectory of AD (Bartzokis, 2004), suggesting a possible link between mechanistic pathways affected early in AD progression, and late stage WM degeneration and cognitive deficits.
The bioenergetic system of the brain is compromised early in the progression of AD and is evident during the prodromal (preclinical) stage of the disease (Mosconi, 2005, Mosconi et al., 2006, Mosconi et al., 2009b, Reiman et al., 1996, Reiman et al., 2004, Yao et al., 2009, Yao et al., 2011b, Brinton et al., 2015, Moreira et al., 2006, Lin and Beal, 2006). Glucose hypometabolism (Mosconi, 2005, Mosconi et al., 2006, Mosconi et al., 2009b, Reiman et al., 1996, Reiman et al., 2004) and a compensatory shift to an alternative fuel substrate, ketone bodies, have been established as a metabolic phenotype characteristic of the AD brain in both clinical and preclinical studies (Yao et al., 2009, Ding et al., 2013, Hoyer, 1991, Brinton et al., 2015, Yao et al., 2009). Reduction in cerebral metabolic rate of glucose utilization, particularly in the entorhinal cortex and hippocampus, correlate with cognitive deficits over time and most accurately predict future cognitive decline in normal individuals as well as conversion to mild cognitive impairment (De Leon et al., 2001, Herholz, 2010, Jagust et al., 2007, Mosconi, 2005). Multiple clinical studies have identified cerebral glucose hypometabolism in persons with AD as well as in those at increased risk for AD, such as APO-ε4 carriers and women (Mosconi, 2005, Mosconi et al., 2005, Mosconi et al., 2006, Mosconi et al., 2008a, Mosconi et al., 2008b, Mosconi et al., 2008c, Mosconi et al., 2009a, Mosconi et al., 2009b, Mosconi et al., 2010, Mosconi et al., 2011, Reiman et al., 1996, Reiman et al., 2004, Silverman et al., 2011, Herholz, 2010, Vlassenko et al., 2010, Brinton et al., 2015). Further, persons with incipient AD exhibit a utilization ratio of 2:1 glucose to alternative fuel, whereas comparably aged controls exhibit a ratio of 29:1, whereas young controls exclusively use glucose as with a ratio of 100:0 (Hoyer et al., 1991).
Loss of estrogenic control of glucose metabolism in brain during menopause can lead to decreased glucose utilization, diminished aerobic glycolysis and altered oxidative phosphorylation, which together generate a hypometabolic phenotype (Yao et al., 2009, Yao et al., 2010, Yao et al., 2012, Ding et al., 2013, Nilsen et al., 2007, Yin et al., 2015). Clinical positron emission tomography with 2-[18F]fluoro-2-deoxy-d-glucose (FDG-PET) analyses indicate a significant decline in cerebral glucose metabolic rate (CMRglu) in the posterior cingulate (PCC) in postmenopausal women (Rasgon et al., 2005) and a decline in cognition during the menopausal transition (Weber et al., 2014, Weber et al., 2013). These clinical findings are recapitulated in studies from animal models of female endocrine aging and AD, which demonstrate a decline in mitochondrial bioenergetics and generalized shift from glycolytic energy production toward use of an alternative fuel, ketone bodies, during the transition to reproductive senescence and early in AD pathology progression (Yao et al., 2009, Yao et al., 2010, Yao et al., 2012, Nilsen et al., 2007). Development of Alzheimer's pathology is accompanied by a decrease in expression and activity of enzymes involved in mitochondrial bioenergetics and glucose metabolism, including pyruvate dehydrogenase (PDH) and complex IV cytochrome c oxidase (COX) (Blass et al., 2000, Federico et al., 2012, Gibson et al., 1998, Gibson et al., 2005, Gibson and Huang, 2005, Yao et al., 2009, Yao et al., 2010, Yao et al., 2012, Nilsen et al., 2007). Mitochondrial dysfunction leads to decreased mitochondrial respiration, increased oxidative stress, and increased mitochondrial Aβ load and Aβ-binding-alcohol-dehydrogenase (ABAD) expression (Chou et al., 2011, Du et al., 2010, Yao et al., 2009). Basic science discovery analyses indicate that glucose hypometabolism and decline in bioenergetic capacity in brain is associated with generation of hallmark pathologies of AD (Yao et al., 2009, Yao et al., 2011b, Ding et al., 2013, Kadish et al., 2009, Blalock et al., 2003).
Under conditions of diminished glucose availability, the brain will progressively utilize circulating fatty acids as a ketogenic energy source (Morris, 2005, Guzman and Blazquez, 2004). Utilization of liver-derived ketone bodies by brain is well established under two conditions: during breast feeding of high lipid diet and during periods of starvation (Morris, 2005). During these states, the ketone bodies acetoacetate and β-hydroxybutyrate supply up to 60% of the human brain's energy requirements (Veech et al., 2001, Cahill, 2006). Ketone bodies derived from liver metabolism of lipids cross the blood brain barrier (BBB) through the monocarboxylic acid transporter (MCT1) (Ding et al., 2013, Morris, 2005). However, as with glucose transporters, MCT1 expression can decline with age and AD (Ding et al., 2013). A potential compensatory response to decline in peripherally derived ketone bodies is the utilization of brain-derived sources of fatty acids to generate ketone bodies (Yao et al., 2011b). Herein, we provide evidence from the aging female brain indicating that endogenous brain lipids can serve as a source of ketone bodies. Specifically, we provide a mechanistic pathway for myelin catabolism initiated by mitochondrial H2O2 activation of the cPLA2-acid sphingomyelinase pathway that leads to loss of myelin integrity, lipid droplet accumulation, fatty acid metabolism, and ketone body generation.
2. Materials and Methods
2.1. Animals
Animal studies were performed following National Institutes of Health guidelines on use of laboratory animals; protocols were approved by the University of Southern California Institutional Animal Care and Use Committee. Five cohorts of female aged (3, 6, 9, 12, 15 and 18 months) wild type (WT) mice were used to investigate the H2O2 induced PLA2-sphingomyelinase pathway for the catabolism of the myelin sheath. Female mice were used to interrogate the effects of the menopausal transition on WM degeneration in brain due to the translational comparability of the reproductively aging female rodent to the reproductively aging female human. Age changes in reproductive cycles begin relatively early in the lifespan of all mammals because of ovarian senescence (Finch et al., 1984). In lab rodents, cycle regularity and fertility decline begin after 6 months, the age of ‘retired breeders’. The human perimenopause is characterized by the menopausal transition as a ‘regularly irregular’ process, with marked cycle-to-cycle variability (Prior, 1998, Santoro, 2005, Burger et al., 2007, Burger et al., 2008). This feature of the perimenopause is closely matched in laboratory rodents. Increasing cycle irregularity is characteristic of laboratory rodents of most genotypes after 8 months and presents a convenient model for the irregular cycles of human perimenopause (Yin et al. 2015). By age 12 months, some mice have fewer than the minimum 100 follicles required to maintain cyclicity (Gosden et al., 1983). Therefore four groups in varying stages of reproductive functionality were defined for this study: 3–6 month old mice were designated reproductively competent, 9 months reproductively irregular, 12 months reproductively incompetent and 15–18 months mice were termed aged.
The number of animals included in mitochondrial, genomic, electron microscopy and lipidomics analyses was determined based on the 95% chance to detect changes in 30–50% of animals. Variance in the number of animals per group was due to three variables: 1.) the number of mice available per age group in the mouse colony, 2.) technical mishap e.g. tubes cracking during ultra-centrifugation, and 3.) limitation in assay format e.g. number of wells per plate. Animal randomization was not possible because age was the variable determining group assignment. Mice within each age group were assigned a randomization number to ensure that analyses were conducted blind to age group.
2.2. Replicates
This program of research was designed to sequentially investigate activation of the mechanistic pathway underlying mitochondrial dysfunction to WM degeneration. Key aspects of the hypothesized mechanistic pathway leading to WM degeneration were confirmed i.e. replicated through multiple analytic strategies. For example, cPLA2 activity was determined by enzyme activity assay, immunohistochemical detection and by arachidonic acid production.
2.3. Inclusion and Exclusion Criteria
Data were not eliminated. Standard errors are indicative of the full range of data. In some instances data exhibited high variability and thus did not reach significance.
2.4. Statistics
All statistical analyses were conducted using One-way ANOVA and were corrected for multiple comparisons followed by a Holmes–Sidak post hoc analysis. All statistical computations were carried out using Prism (Graphpad Software). Center and dispersion were defined as the mean and standard of the mean respectively.
2.5. Brain Tissue Preparation and Mitochondrial Isolation
Mice were anesthetized and sedated, followed by perfusion with PBS and formalin for immunohistochemistry (Tiwari-Woodruff et al., 2007). Prior to cryostat slicing and staining whole brains were embedded in a 7.5% gelatin and 15% sucrose solution in water. The gelatin block was then fixed and placed in a 20% sucrose solution for 2 days or until ready to slice.
For biochemical, genetic and mitochondrial isolation mice were euthanized and the brains were rapidly dissected on ice. Cerebellum and brain stem were excluded. Both cortex and hippocampus were harvested and frozen in − 80 °C for subsequent biochemical and genetic analyses, while mitochondria were isolated from whole brain. During mitochondrial isolation mitochondria, cytosol and tissue homogenate were collected. Whole brain mitochondria were isolated as previously described (Yao et al., 2009, Yao et al., 2010) Briefly, the brain was rapidly minced and homogenized at 4 °C in mitochondrial isolation buffer (MIB) (pH 7.4), containing sucrose (320 mM), EDTA (1 mM), Tris–HCl (10 mM), and Calbiochem's Protease Inhibitor Cocktail Set I (AEBSF-HCl 500 μM, aprotonin 150 nM, E-64 1 μM, EDTA disodium 500 μM, leupeptin hemisulfate 1 μM). Single-brain homogenates were then centrifuged at 1500 g for 5 min. The pellet was re-suspended in MIB, re-homogenized, and centrifuged again at 1500 g for 5 min. The post-nuclear supernatants from both centrifugations were combined, and crude mitochondria were pelleted by centrifugation at 21,000 g for 10 min. The resulting mitochondrial pellet was re-suspended in 15% Percoll made in MIB, layered over a preformed 23% and 40% Percoll discontinuous gradient, and centrifuged at 31,000 g for 10 min. The purified mitochondria were collected at the 23% and 40% interface and washed with 10 ml MIB by centrifugation at 16,700 g for 13 min. The loose pellet was collected and transferred to a micro-centrifuge tube and washed in MIB by centrifugation at 9000 g for 8 min. The resulting mitochondria samples were used immediately for respiratory measurements and hydrogen peroxide production or stored at − 80 °C for later protein and enzymatic assays. During mitochondrial purification, aliquots were collected for confirmation of mitochondrial purity and integrity following a previously established protocol (Irwin et al., 2008).
2.6. Respiratory Measurement
Mitochondrial respiration was determined using the Seahorse XF-96 metabolic analyzer following established protocol (Rogers et al., 2011). Briefly, 4 μg of mitochondrial samples in 25 μl 1 × MAS buffer were seeded into each assay well. Mitochondrial samples were spun down at 2000 g for 10 min and then supplemented with 75 μl 1 × MAS buffer with substrate (glutamate and malate 5 mM). Metabolic flux cartridges were loaded with the following reagents in 1 × MAS: port A: ADP (20 mM); port B: Oligomycin (30 μg/ml); port C FCCP (28 μM; and port D: Rotenone (16 μM) + Antimycin (70 μM). Mitochondrial respiration was determined sequentially in a coupled state with substrate present (basal respiration, state 4 respiration), followed by ADP stimulated state 3 respiration after port A injection, (phosphorylating respiration in the presence of ADP and substrates). Injection of oligomycin in port B induced state 40 respiration and the subsequent injection of FCCP in port C induced uncoupler-stimulated respiration state 3u while the final injection of rotenone and antimycin in port D resulted in non-oxidative phosphorylation related residual oxygen consumption OCRresidual. Respiratory control ratio value was calculated as the ratio of oxygen consumption rate (OCR) at state 3 respiration over OCR at state 4 respiration:respiratory control ratio (RCR) = OCRstate 4/OCRstate 3.
2.7. Hydrogen Peroxide (H2O2) Production, Enzyme Activity Assays and β-Hydroxybutyrate (Ketone Body) Measurements
The rate of hydrogen peroxide production by isolated mitochondria (20 μg), was determined by the Amplex Red Hydrogen Peroxide or Peroxidase Assay kit (Invitrogen) following the manufacturer's instructions with the presence of 5 mM glutamate and malate but not ADP (state 4). cPLA2 activity was determined in tissue homogenate obtained during the mitochondrial isolation and hippocampal tissue homogenate using the arachidonoyol thio-PC substrate method for determining cPLA2 activity (Cayman Chemical). A coupled enzymatic reaction was used to monitor sphingomyelinase activity (Cayman chemical). Brain ketone body level was determined using the Liquid Color β-Hydroxybutyrate assay kit (Cayman Chemical) following the manufacturer's instruction.
2.8. Arachidonic Acid Production
Lipids were extracted from thawed tissue homogenate (1 ml) using a modified Bligh and Dyer procedure as previously described (Fonteh et al., 2014, Bligh and Dyer, 1959). The lipid extract in CHCl3 was dried under a stream of N2 and free arachidonic acid and [2H8]-arachidonic acid internal standard were converted to pentafluorobenzyl esters (Quehenberger et al., 2008). Derivatized arachidonic acid was re-dissolved in 50 μl dodecane and then transferred into GC–MS vials. Carboxylate arachidonic acid ions (m/z = 303) and deuterated internal standard (2H8-AA, m/z = 311) were detected by injecting 1 μl derivatized extracts onto a 7890A GC System coupled to a 7000 MS Triple Quad (Agilent Technologies). Gas chromatography was performed using a Phenomenex Zebron ZB-1MS Capillary GC Column (30 m length, 0.25 mm I.D., 0.50 μm film thickness) as previously described (please reference Fonteh et al., 2014). The temperature of the ion source was 200 °C, and the temperature of the quadrupoles was 300 °C. Single ion monitoring was used to determine m/z for arachidonic acid and deuterated internal standards in all samples. An Agilent MassHunter Workstation Software was used to analyze GC–MS data. A calibration curve was acquired prior to sample analysis and quality control standards were analyzed for calibration and retention time reference. Data for arachidonic acid analyzed in triplicates had a consistent QC Accuracy within 80–120% of expected concentrations. Peak integration was automatic and manual integration was used for selected cases when the arachidonic acid peaks were too broad.
2.9. Immunohistochemistry
Frozen, embedded brains were sliced 30 μm thick and then processed for immunohistochemistry using a standard protocol (Tiwari-Woodruff et al., 2007). Sections were immunostained using antibodies directed against astrocytes and PLA2 (glial fibrillary acidic protein (GFAP), 1:1000 4 °C overnight, Millipore; PLA2, 1:200 4 °C overnight) followed by secondary antibody fluorescein goat anti-mouse and fluorescein goat anti-rabbit (1:500, 1 h at room temperature; Life Technologies). Myelin area was assessed using the BrainStain™ Imaging Kit from (Invitrogen) according to the manufacturer's instructions. Sections were mounted with anti-fade mounting medium with DAPI (Vector Laboratories). Fluorescent images were taken using a fluorescent microscope, normalized, and analyzed with the slide book software (Intelligent Imaging Innovations). Masks were created over the CC, anterior commissure and hippocampal fimbria using the slide book software to determine astrocyte reactivity and myelin area.
2.10. Neuron and Enriched Astrocyte Culture, and H2O2 Treatment
Primary neuron and enriched astrocyte cultures were prepared according to a previously established protocol (Yao et al., 2011a, Gilmore et al., 2007). Briefly hippocampal neurons and glia from day 18 (E18) embryos of female Sprague–Dawley rats were cultured in 60 × 15 mm polystyrene petri dishes and T75 flasks respectively. Neurons were grown in Neurobasal Medium + B27 supplement for 10 days prior to H2O2 treatment. Mixed glia were grown in growth media (DMEM:F12 (1:1) + 10% FBS) for 14 days or until confluent. 24 h prior to the experiment the mixed glia were placed onto an orbital shaker for 4 h to shake off the majority of microglia and yield enriched astrocyte cultures. Astrocytes were then plated on polystyrene petri dishes coated with polyethyleneimine (PEI) and allowed to grow overnight. Neurons and enriched astrocytes were treated with doses of H2O2. Media and cells were collected and stored at − 80 °C to be later analyzed for cPLA2 activity.
2.11. Custom WM Gene Array
Low density gene expression array was performed as previously described (Zhao et al., 2012). Custom 384-well TaqMan® low density array (TLDA) cards were designed with the help of Dr. Liqin Zhao and was used to investigate the WM degeneration profile. Total mRNA from mouse hippocampus was extracted with TRIzol reagent (Gibco BRL) according to manufacturer's instructions. Total RNA (1–5 μg) was used to create the first strand cDNA using reverse transcriptase superscript III (Invitrogen). cDNA was amplified by polymerase chain reaction (PCR) using SYBERGreen PCR master mix (BioRad) on an iCycler real-time PCR machine (Bio-RAD).
Data were analyzed using the RQ Manager Version 1.2 and DataAssist Version 2.0 (Applied Biosystems). Relative gene expression levels or fold changes relative to the reference group were calculated by the comparative Ct (ΔΔCt) method using iCycler software (BioRad), with Ct denoting threshold cycle (Schmittgen and Livak, 2008). Selection of the endogenous control gene for normalization was based on the control stability measure (M), which was calculated using the geNom algorithm; genes with the lowest M values have the most stable expression. Four samples per group were included in the analysis. For each sample, average Ct for each target gene was calculated as the mean of 2 technical replicates; ΔCt was calculated as the difference in average Ct of the target gene and the endogenous control gene. Fold change was then calculated as mean 2− ΔCt (comparison group)/mean 2− ΔCt (reference group). The 2− ΔCt values for each target gene were statistically analyzed by ANOVA followed by pairwise comparisons using Student's t-test. The statistical significance was indicated by p ≤ 0.05; p-values were not adjusted for multiple testing as discussed previously (Zhao et al., 2012).
2.12. Western Blot Analysis
Protein concentrations were determined using the BCA protein assay kit (Pierce). For Western blot analysis, equal amounts of protein (25–40 μg/well) was loaded in a 12% SDS-PAGE gel, electrophoresed with a Tris glycine running buffer, and transferred to a polyvinylidine difluoride membrane. Blots were probed overnight at 4 °C with myelin basic protein (MBP, Millipore 1:200), hydroxyacyl-CoA dehydrogenase 3-ketoacyl-CoA thiolase enoyl-CoA hydratase (trifunctional protein) alpha subunit (HADHA, Abcam 1:5000), Aβ-binding alcohol dehydrogenase (ABAD, Abcam 1:500), and carnitine palmitoyltransferase (CPT-1, Abcam 1:1000) followed by incubation with an HRP-conjugated secondary antibody. Antigen–antibody complexes were visualized with enhanced chemiluminescence. Band intensities were determined using the VersaDoc System and software (Bio-Rad). All band intensities were quantified using Un-Scan-it.
2.13. Electron Microscopy
Mice were anesthetized and sedated with a single I.P. dose of ketamine (80 mg/kg)/xylazine (10 mg/kg) prior to surgery. An incision was made up the midline through the abdomen and chest cavity to expose the heart. A small incision was made in the right atrium of the heart so mice can be transcardially perfused with PBS for 5 min. Whole brains were harvested and fixed using Trump's fixative (4% paraformaldehyde with 1% glutaraldehyde in phosphate buffered saline, pH 7.2). Whole brains were then sent to the Mayo Clinic, Rochester, MN, to be processed for EM analysis.
Tissue prepared for EM was cut into flat slices approximately 2 × 4 × 2 mm (keeping the slices flat helped to maintain correct orientation during processing). Tissue was immersed in Trump's fixative (4% paraformaldehyde with 1% glutaraldehyde in phosphate buffered saline, pH 7.2) and stored at 4 °C until processed (12 h minimum). Further processing steps were performed using a laboratory microwave oven (Pelco Biowave 3450, Ted Pella, Inc., Redding, CA). Tissue was rinsed with 0.1 M phosphate buffer (PB) and secondarily fixed in 1% osmium tetroxide followed by 2% aqueous uranyl acetate. Dehydration was performed in a sequential ethanol series followed by 100% acetone. Tissue was embedded in Embed 812 Araldite epoxy resin and polymerized 24 h at 60 °C. Ultrathin sections (0.1 μM) were post-stained with 2% lead citrate. Micrographs were obtained using a JEOL 1400 TEM at 80 kV.
2.14. Metabolomics (Trushina et al., 2013)
Mice were sacrificed using cervical dislocation. Brains were rapidly removed and flash frozen in liquid nitrogen and tissue pulverized under liquid nitrogen. Pulverized tissue was then sent to the Mayo Clinic, Rochester, MN, to undergo targeted lipidomic analysis. Brain tissue was homogenized in 1 × PBS after adding 5 ml of PBS to 1 mg of tissue and prior to dividing into 4 different aliquots for various the following measurements.
Ceramides: Ceramides were extracted from 25 ml of tissue homogenate after the addition of internal standards and sonication. The extracts were measured against a standard curve on the Thermo TSQ Quantum Ultra mass spectrometer (West Palm Beach, FL) coupled with a Waters Acquity UPLC system (Milford, MA) as previously described (Blachnio-Zabielska et al., 2012).
Non-esterified free fatty acids (NEFA): Fatty acids were measured against a standard curve on an Agilent 6460 triple quadrupole liquid chromatography mass spectrometer (LC MS) as previously described (Persson et al., 2010). Briefly, 50 ml of homogenate was spiked with internal standard prior to extraction. The extracts were dried down and brought up in running buffer prior to injecting on the LCMS. Data acquisition was performed under negative electrospray ionization condition.
TCA: TCA analytes were quantitated via GC–MS as previously published with a few modifications (Koek et al., 2006). Briefly, 100 ml of internal standard solution containing 10 ng/ml each of 13C2-lactate, 13C4-fumarate, 13C4-succinate, 13C4-malate, 13C5-ketoglutarate, and 13C6-citrate was added to 100 ml of homogenate. The samples were quenched by adding 400 μl of chilled methanol and acetonitrile (1:1) solution and kept at − 20 °C for 2 h prior to spinning down in a centrifuge. The supernatant was dried down in the speed vac before derivatizing for GC–MS analysis. Concentrations of lactic acid (m/z 261.2), fumaric acid (m/z 287.1), succinic acid (m/z 289.1), ketoglutaric acid (m/z 360.2), malic acid (m/z 419.3), citric acid (m/z 591.4), and isocitric acid (m/z 591.4) were measured against 12-point calibration curves that underwent the same derivatization.
3. Results
3.1. Pathway of Mitochondrial Deficits, H2O2 Production and cPLA2 Activation in the Aging Female Brain
Three key initiating elements required for activation of the proposed cPLA2- sphingomyelinase pathway for WM catabolism are: 1) dysregulated mitochondrial respiration, 2) a source for and generation of H2O2 and 3) activation of cPLA2. If there is a direct relationship between mitochondrial generated H2O2 and subsequent cPLA2 activation then an increase in cPLA2 activity should be coincident with mitochondrial dysfunction and H2O2 generation.
To investigate the impact of female brain aging on mitochondrial function and subsequent activation of the cPLA2, whole forebrain mitochondria were isolated from female mice at three ages associated with endocrine aging: 1) reproductively competent; 2) reproductively irregular and 3) reproductively incompetent to aged. Results obtained from reproductively incompetent and aged mice were indistinguishable in magnitude and were not statistically different (Fig. 1, A. p = 0.67, B. p = 0.72, C. p = 0.56) and were therefore pooled. Respiration was determined using the respiratory control ratio (RCR = OCRstate 4/OCRstate 3).
Fig. 1.
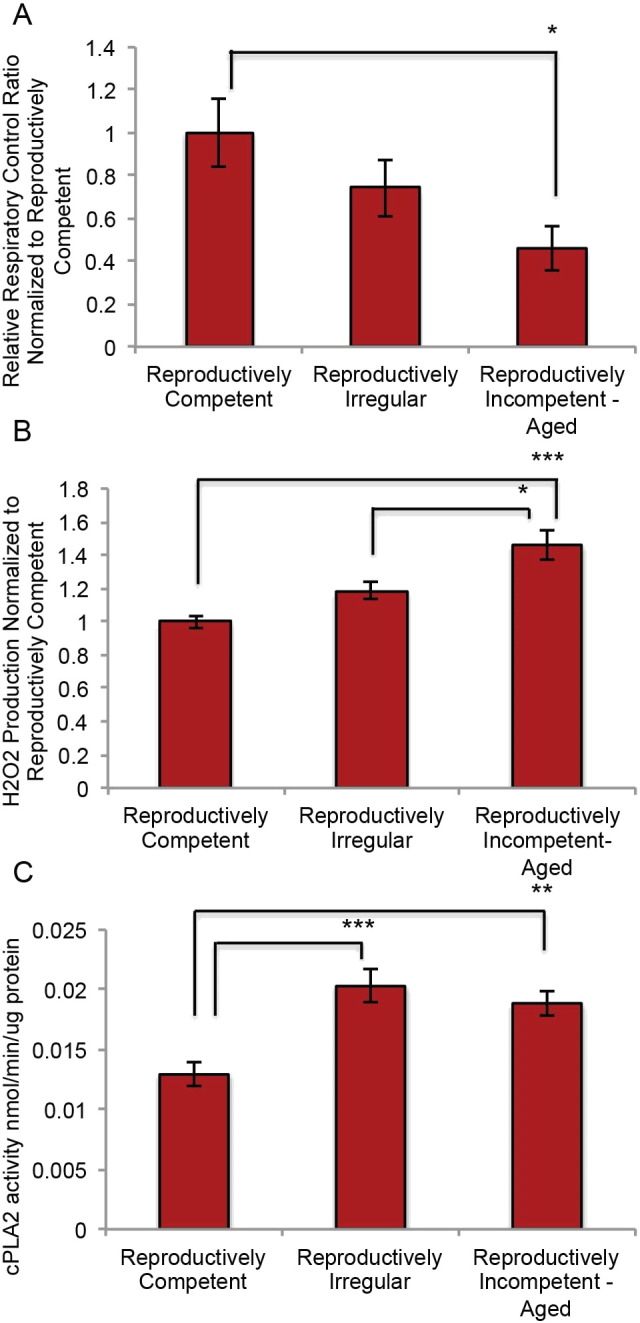
Mitochondrial function and cytoplasmic phospholipase A2 activity in reproductively aging female mice: A. Respiratory control ratio (RCR = OCRstate 4/OCRstate 3) and B. H2O2 production in isolated whole brain mitochondria from reproductively aging female mice. C. cPLA2 enzyme activity in whole brain tissue homogenate obtained during mitochondrial isolation preparation. (A. N = 6–8; B. N = 6–7; C. N = 5–7.) (*p < 0.05, **p < 0.005 and ***p < 0.0005).
Age-related decline in mitochondrial respiration was first evident in reproductively irregular mice and declined significantly in reproductively incompetent-aged mice (Fig. 1A; F (3, 18) = 2.772, p = 0.0714). Decline in mitochondrial respiratory efficiency would predict a rise in oxidative stress and the generation of H2O2. H2O2 production was determined in the same preparation of isolated mitochondria using the Amplex Red H2O2 assay. An inverse relationship was evident in H2O2 production relative to mitochondrial respiration such that an age-related increase in H2O2 production paralleled the decline in mitochondrial respiration (Fig. 1B; F (3, 16) = 7.031, p = 0.0031). A statistically significant increase in H2O2 production was evident in reproductively incompetent and aged mice when compared to reproductively competent animals (Fig. 1B). Further a statistically significant increase in H2O2 production occurred in the reproductively incompetent-aged mice relative to the reproductively irregular. H2O2 production levels during aging ranged from 300 nM/min/mg to 1 μM/min/mg. To investigate H2O2 dependent activation of the cPLA2, cPLA2 enzyme activity was determined in brain tissue homogenate generated from the same preparation used for mitochondrial isolation. Relative to reproductively competent mice, a significant increase in cPLA2 activity was evident in reproductively irregular animals and remained significantly elevated in reproductively incompetent-aged (Fig. 1C; F (3, 15) = 7.726, p = 0.0024).
Collectively these data indicate that decline in mitochondrial respiration was associated with a concomitant rise in H2O2 production which was coincident with a sustained activation of cPLA2 enzyme activity in the aging mammalian female brain. These findings indicate an association between the decline in mitochondrial respiration, an increase in mitochondrial H2O2 production and cPLA2 activation.
3.2. cPLA2-sphingomyelinase Pathway Activation in White Matter Astrocytes During Reproductive Senescence
If decline in mitochondrial respiration, H2O2 over-production and cPLA2 activation are precipitating events in myelin degeneration, then activation of the cPLA2-sphingomyelinase pathway should be evident. Three critical components of the cPLA2-sphingomyelinase pathway are cPLA2, arachidonic acid and acid sphingomyelinase. To investigate activation of this pathway during the most critical transition in reproductive aging—perimenopause, cPLA2 and acid sphingomyelinase enzyme activity, and arachidonic acid production were investigated in female brains from reproductively competent, reproductively irregular and reproductively incompetent animals in hippocampal lysates and tissue homogenate from the same cohort of mice used in mitochondria analyses. cPLA2 and sphingomyelinase activity were determined using enzyme activity assays and arachidonic acid measurements were determined by GC–MS. A significant increase in cPLA2 activity (Fig. 2A; F (2, 10) = 25.99, p < 0.0001) and arachidonic acid production (Fig. 2B; p = 0.02) occurred in reproductively incompetent mice relative to reproductively irregular. In tandem, a trend toward an increase in acid sphingomyelinase activity was evident in the reproductively incompetent mice (Fig. 2C; F (2, 25) = 0.5370, p = 0.5911).
Fig. 2.
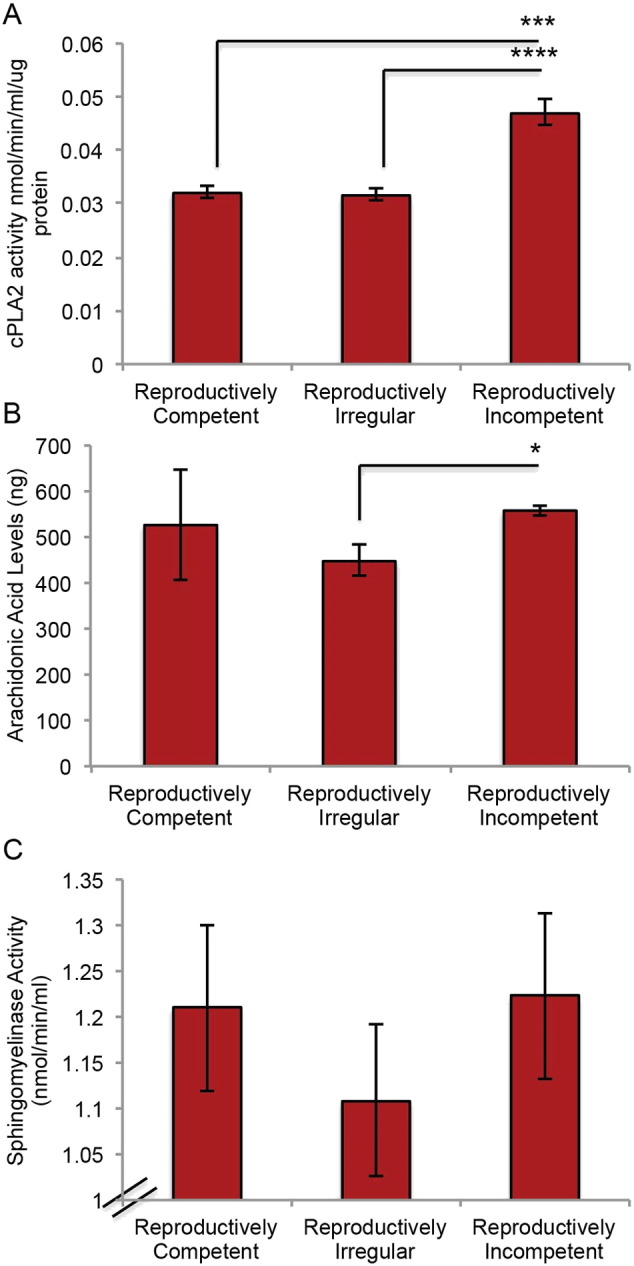
Cytoplasmic phospholipase A2-sphingomyelinase pathway activation during reproductive senescence: A. cPLA2 enzyme activity in hippocampal tissue homogenate. B. Arachidonic acid production determined by GC-MS in tissue homogenate obtained during mitochondrial isolation preparation. B. Acid sphingomyelinase enzyme activity in hippocampal tissue homogenate. (A. N = 4–11; B. N = 3,-4; C. N = 7–12.) (*p < 0.05, ***p < 0.0005, ****p < 0.00005).
Because age is the greatest risk factor for AD (Alzheimer's, 2015) and because cPLA2 immunoreactivity is well documented to be elevated in astrocytes of AD brain (Stephenson et al., 1996), brain slices from the aging mouse model containing anterior commissure, fimbria, cingulum and Schaffer collateral WM tracts were co-labeled for GFAP and cPLA2. Four sections per group per brain region were quantitatively analyzed blind to group identifiers for astrocyte reactivity and cPLA2 localization. Outcomes of these analyses indicated that reproductive incompetence was accompanied by a statistically significant increase in quantitative immuno-fluorescent astrocyte reactivity in multiple WM tracts in brain. Immunoreactivity for astrocyte reactivity was statically significant in the fimbria and cingulum (Fig. 3A; F (2, 9) = 21.58, p = 0.0004 and B; F (2, 8) = 9.541, p = 0.0076). Astrocyte reactivity was greatest in the fimbria and was unique in its aging profile of increasing reactivity with each stage of aging. This is in contrast to the cingulum profile where astrocyte reactivity is greatest during the transition to reproductive incompetence with a return to the reactivity of the reproductively competent animals in the aged population (Fig. 3B). The profile of the Schaffer collateral pathway (Fig. 3C; F (2, 9) = 4.417, p = 0.0461) is consistent with an onset of elevated astrocyte reactivity during reproductive incompetence that is sustained during later aging. The astrocytes of the anterior commissure (Fig. 3D; F (2, 8) = 2.57, p = 0.1374) were the least reactive and maintain a steady low level of reactivity across the ages we investigated. The reactive astrocyte phenotype co-localized with cPLA2 immunofluorescence (Fig. 3E). The pattern of astrocyte reactivity and coexpression of cPLA2 predicts that the afferent and efferent white matter tracts of the hippocampus are most vulnerable to catabolism which would contribute to disconnecting the hippocampus from the frontal and association cortices, the limbic system and sensory integrative pathways.
Fig. 3.

Co-localization of immunoreactivity of reactive astrocytes and cytoplasmic phospholipase A2 in brain slices from the aging female mouse model: white matter tracts were co-labeled with GFAP (FITC) and cPLA2 (CY3) antibodies, and assessed for astrocyte reactivity and cPLA2 cellular localization in the A. fimbria, B. cingulum, C. the Schaffer collateral pathway, and D. anterior commissure during reproductive aging. E. Representative images depicting GFAP (FITC) and cPLA2 (CY3) labeling in the cingulum of reproductively incompetent female mice. (A, B, C, D. N = 4 per group) (*p < 0.05, and ***p < 0.0005).
To investigate cPLA2-sphingomyelinase pathway activation in astrocytes, primary hippocampal neurons and astrocytes in culture were treated with H2O2 (50 μM, 100 μM and 200 μM) for 30 min and cPLA2 activity determined (Fig. 4). Hippocampal neurons exhibited no change in cPLA2 activity in response to pharmacological concentrations of H2O2, consistent with previous reports (Fig. 4B) (Sun et al., 2004). In contrast, astrocytes exhibited a steep dose dependent increase in cPLA2 activity, reaching maximal activation at 200 μM H2O2 (Fig. 4A). To investigate whether H2O2 concentrations produced by mitochondria derived from reproductively senescent female brains increased cPLA2 activity, cultured astrocytes were treated with 100 nM, 1 μM and 5 μM H2O2 (Table 1). A dose dependent increase in cPLA2 activity was observed in response to mitochondrial relevant doses of H2O2, with approximately a 50% increase in cPLA2 activity at 1 μM (0.0378 nmol/min/ml/μg protein) and 5 μM (0.0370 nmol/min/ml/μg protein) compared to control (0.0214 nmol/min/ml/μg protein) (Fig. 4C). Collectively these data demonstrate that elevations in astrocyte reactivity appear to be preferentially localized to WM tracts surrounding the para-hippocampal formation, consistent with earliest development of AD pathology in the medial temporal lobe (MTL). In vitro data confirm that H2O2 induced cPLA2 activation is astrocyte mediated.
Fig. 4.

Cytoplasmic phospholipase A2 enzyme activity in cultured astrocytes and neurons following H2O2 exposure: embryonic hippocampal neurons and astrocytes in culture were treated with H2O2 and assessed for cPLA2 activation using an enzyme activity assay. cPLA2 enzyme activity normalized to untreated cells in cultured A. astrocytes and B. neurons. C. cPLA2 enzyme activity normalized to untreated cells in cultured astrocytes following exposure to physiologically relevant concentrations of H2O2. H2O2 concentrations were determined by the levels of H2O2 produced by whole brain mitochondria isolated from reproductively incompetent female mice. D. Levels of cPLA2 enzyme activation in astrocytes and neurons (nmol/min/ml/mg of protein).
Table 1.
H2O2 production across endocrine aging in female mouse brain mitochondria. Values are presented as nm/min/mg of mitochondrial protein. Range of values across all endocrine states spanned from 300 to 1500. (N = 5–8 per group).
| Age group | Average H2O2 production by group (nM/min/mg) | Range (nM/min/mg) |
|---|---|---|
| Reproductively competent | 434 | ≅ 300–1500 |
| Reproductively irregular | 515 | |
| Reproductively incompetent | 760 | |
| Aged | 810 |
3.3. Investigation of White Matter Gene Expression Profile During Reproductive Senescence
To determine whether enzymatic activation of the cPLA2-sphingomyelinase pathway observed at the protein level was coincident with associated gene expression and to gain a systems level view of adaptive responses, we developed a hypothesis driven targeted custom gene array to assess the impact of reproductive senescence on WM degeneration. Consistent with findings from biochemical assays, gene expression analyses indicated upregulation of genes associated with the cPLA2-sphingomyelinase myelin degradation pathway during the transition to reproductive senescence between the reproductively irregular and reproductively incompetent stages (Fig. 5). Significant upregulation of arachidonic acid epoxygenase (Cyp2j6) (p = 0.004) is consistent with an increase in arachidonic acid generation. Alkaline ceramidase (Acer3), the gene encoding the enzyme required for the breakdown of ceramide into sphingosine and fatty acids, was significantly up-regulated in reproductively incompetent animals (p = 0.01). Further, up-regulation of the gene encoding the tri-functional protein hydroxyacyl-CoA dehydrogenase (Hadha) occurred in reproductively incompetent mice (p = 0.06), consistent with an increase in the metabolism of very long and long chain fatty acids. Upregulation of genes involved in myelin synthesis included myelin-associated glycoprotein (Mag), myelin oligodendrocyte glycoprotein (Mog), Erb-B2 receptor tyrosine kinase 3 (Erbb3), claudin 11 (Cldn11) and oligodendrocyte transcription factor (Olig2) in reproductively irregular mice. These genes were subsequently down-regulated in reproductively incompetent mice. Analysis of gene expression in aged mice indicated a pattern of down regulation of myelin synthesis genes, while myelin degradation genes remained up-regulated, suggestive of long-term activation of pathways involved in WM degeneration. An endocrine aging-related upregulation in inflammatory genes was also observed, consistent with previously published data in the perimenopausal rat model (Yin et al., 2015). The gene expression profile of reproductively senescent mice favors WM degeneration, exhibiting up-regulation in genes associated with the cPLA2-sphingomyelinase pathway and fatty acid metabolism.
Fig. 5.

Myelin and fatty acid metabolism, myelin generation and repair, and inflammation related gene expression in reproductively aging female mice: A. Table depicting the fold change and p-value differences in expression of white matter related genes between reproductively irregular and reproductively incompetent female mice. Red p-value is indicative of upregulation while a green p-value indicates down regulation. Boxes outlined in red reached statistical significance. B. Heatmaps of gene expression organized by function: myelin and fatty acid metabolism, myelin generation and repair, and inflammation. Each gene was normalized and colored based on its relative expression level across 4 reproductive aging groups. This method allows genes that have different magnitude of signal intensity but which belong to the same functional group and share similar expression patterns to be displayed on the same heatmap. (N = 5 per group.)
3.4. Ultra Structural Analysis of Myelin Sheath During Reproductive Senescence
Based on biochemical and genomic evidence supporting activation of an astrocyte mediated myelin degradation pathway; immunohistochemistry, MBP protein expression analysis and electron microscopy were conducted to determine age-related ultra-structural changes of myelin sheath in the anterior commissure, CC and hippocampus. Initial immunohistochemical mapping of MBP fluorescence indicated an unexpected increase in total WM area in the corpus callosum (Fig. 6A) between 9 (reproductively irregular) and 12 (reproductively incompetent) months, followed by a precipitous decline between 12 (reproductively incompetent) and 15 (aged) months of age which was also evident in anterior commissure and hippocampal fimbria (data not shown). Overall, myelin basic protein declined across reproductive status and age (Fig. 6C; F (3, 15) = 3.5, p = 0.04). Myelin basic protein (MBP) expression did not differ between reproductively competent (6 months of age), and reproductively irregular (9 months of age). A statistically significant decline occurred between reproductively competent MBP expression and reproductively incompetent and aged mice (Fig. 6C; p = 0.004 and 0.01 respectively). MBP expression in hippocampus of reproductively incompetent and aged females were both decreased to the same magnitude (Fig. 6C). While the area and protein expression data were consistent for the aged 15–18 month old females, the MBP area and protein expression data were not consistent between reproductively irregular and reproductively competent. We therefore conducted analyses to investigate myelin integrity at the electron microscopic level of analysis.
Fig. 6.

Immunohistochemical and Western blot analysis of white matter integrity in the aging female mouse model: A. Immunohistochemical mapping of myelin basic protein area in the corpus callosum of the aging female mouse model. B. Representative immunohistochemical images mapping myelin basic protein area in the anterior commissure and corpus callosum. Mask of corpus callosum generated via slide book software is highlighted in purple. C. Myelin basic protein expression during reproductive aging in the female mouse. (C. N = 4–10 per group). (*p < 0.05).
Two possibilities could account for the increase in WM area between reproductively irregular and reproductively incompetent: 1) an increase in WM volume; or, alternatively 2) expansion of the white matter zone due to a loss in compactness of myelin integrity. To investigate these two possibilities, electron microscopy was performed to assess WM structural organization at critical transition ages, reproductively irregular (9 months of age), reproductively incompetent (12 months of age) and aged (15–18 months of age) (Fig. 7A, B, C). Electron microscopic imaging of the anterior commissure revealed that at reproductively irregular stage (9 months of age) myelin integrity is compact with no detectable signs of irregularity (Fig. 7A). At the reproductively incompetent stage (12 months of age) expansion of myelin area, disintegration of myelin compactness, unraveling of the myelin sheath and abnormal structural integrity were evident (Fig. 7B). These same indicators of myelin disintegration were greater in magnitude in aged females (15 months of age) and were compounded by a severe decline in myelin sheath density (Fig. 7C). Comparable patterns of myelin disintegration were also observed in the Schaffer collateral pathway and corpus callosum white matter tracks (Fig. 7D and F). Electron microscopic analyses indicate that the increased white matter area detected by myelin histochemistry (Fig. 6), is due to an expansion of the white matter zone induced by disintegration of myelin integrity and compactness of myelin sheaths.
Fig. 7.

Electron microscopic analysis of the structural integrity of white matter in reproductively aging female mice: representative electron microscopy images of myelinated axons in the anterior commissure of A. reproductively irregular, B. reproductively incompetent and C. aged female mice. Quantitative analysis of the percentage of compromised axons in the D. Schaffer collateral pathway, E. anterior commissure and F. corpus callosum in reproductively aging female mice. (A, B, C. N = 3–4; D. N = 4; E. N = 4; F. N = 3). (*p < 0.05) and **p < 0.005).
Qualitative observations of myelin integrity were quantitatively confirmed by calculating the percentage of compromised axons in a given WM tract (number of compromised axons/number of total axons). Axons of the Schaffer collateral pathway, anterior commissure and CC had 63% (244/387 axons; Fig. 7D), 71% (266/440 axons; Fig. 7E) and 55% (348/616 axons; Fig. 7F) compromised axons respectively in reproductively incompetent animals, and 61% (247/403 axons; Fig. 7D), 83% (253/305 axons; Fig. 7E) and 61% (259/418 axons; Fig. 7F) respectively in aged animals (Fig. 7A, B, C).
If myelin is being catabolized for ketone body generation, then evidence indicating accumulation of lipids should be present. Analysis of EM images for lipid droplets revealed the presence of lipid droplets throughout multiple WM tracks (Fig. 8A, B, C). A statistically significant increase of intracellular lipid droplets was evident in the anterior commissure (Fig. 8D) and CC (Fig. 8E) of reproductively incompetent animals. The increase in lipid droplets was time limited. A decline in lipid droplets was evident in aged animals indicative of a release of myelin fatty acids from temporary storage in lipid droplets (Fig. 8D, E). These observations made at the EM level of analysis are consistent with MBP protein expression indicating a decline in MBP expression in reproductively incompetent and aged animals (Fig. 6A, C). The rise in lipid droplets is consistent with an increase in free fatty acid storage for ketone body generation (Cabodevilla et al., 2013).
Fig. 8.

Electron microscopic analysis of lipid droplet accumulation in reproductively aging female mice: representative electron microscopy images of lipid droplet accumulation in the anterior commissure of A. reproductively irregular, B. reproductively incompetent and C. aged female mice. Average number of lipid droplets per cell body in the D. anterior commissure and E. corpus callosum. (A, B. N = 3–4; C. N = 4; D. N = 4; E. N = 3). (*p < 0.05).
3.5. Analysis of the Lipid Profile of Brain During the Transition to Reproductive Senescence
Biochemical, genomic and ultra structural analyses provide evidence indicative of metabolism of myelin following reproductive senescence. If myelin metabolism is occurring then there should be evidence of lipid byproducts of myelin breakdown. The myelin sheath is composed of two main lipids, sphingomyelin and galactocerebroside. A series of enzymatic reactions catabolize sphingomyelin into ceramide, and subsequently into sphingosine and fatty acids (Fig. 9A). To investigate the impact of reproductive aging on the lipid profile of brain tissue, a targeted metabolomic analysis was conducted in whole brain minus cerebellum and olfactory bulb. Lipidomics panels included ceramides and nonesterified free fatty acids (NEFA). Metabolites in the TCA cycle were also assessed as indicators of mitochondrial function.
Fig. 9.

Lipid profile of female brain during reproductive aging: A. Sequence of catabolic events that occur during myelin breakdown. B. Levels of brain lipids in reproductively irregular, reproductively incompetent and aged female mice. Lipids were separated into three categorical panels: ceramides, non-esterified fatty acids and TCA metabolites. Red values are indicative of the peak level of a particular lipid and green values indicate statistical significance. (N = 4–6 per group.)
Lipidomic analysis indicated that levels of ceramides were greatest in reproductively incompetent animals with peak ceramide levels occurring within reproductively incompetent and aged groups. The rise in ceramide levels was followed by a global age-related increase in NEFA levels in aged brain, specifically docosahexaesnoic acid (DHA) (p = 0.052), arachidonic acid (p = 0.023), palmitic acid (p = 0.009) and oleic acid (p = 0.031) (Fig. 9 B). The pattern of lipid metabolism is consistent with breakdown of myelin at age of reproductive incompetence (Fig. 9B Ceremide Panel) followed by metabolism into fatty acids in aged brain (Fig. 9B NEFA Panel). These data derived from lipidomic analyses are consistent with electron micrographs showing loss in structural integrity and density of myelin first evident at the reproductively incompetent stage and greatly increased in the aging brain (Fig. 7A–F).
Metabolomic analysis of the TCA cycle indicated few age-associated alterations, with only two metabolites exhibiting significant changes: succinate (p = 0.043) and citrate (p = 0.001) (Fig. 9B TCA panel). Peak levels of TCA metabolites were greatest in the aged brain (Fig. 9B TCA Panel) consistent with an increased level of acetyl-CoA availability. Further, the rise in TCA metabolites indicates that in the aged brain the catalytic machinery for mitochondrial energy production was largely preserved. These data are consistent with an activated mitochondrial beta-oxidation pathway, required to utilize fatty acids to generate TCA metabolites and elevated mitochondrial H2O2 production. Neurons are incapable of beta-oxidation whereas astrocytes are capable of beta-oxidation of fatty acids. The endocrine age-dependent alterations in the lipidomic profile of the reproductively senescent female brain are consistent with biochemical, genomic and structural data demonstrating activation of the cPLA2-sphingomyelinase pathway and degeneration of WM in reproductively incompetent mice. The release of myelin lipids in aged animals creates an environment in which fatty acids can readily be used as a substrate in ketogenesis.
3.6. Fatty Acid Metabolism and Ketone Generation Following the Transition to Reproductive Senescence
Data derived from lipidomic and mitochondrial metabolic analyses indicated the generation of fatty acids derived from myelin lipids and a mitochondrial phenotype consistent with beta-oxidation of fatty acids. A critical link in the pathway to utilize fatty acids is their transport into mitochondria and subsequent enzymatic generation of medium and short chain fatty acids to ultimately generate ketone bodies. We therefore investigated the expression of: 1) CPT I, the key enzyme in the carnitine-dependent transport of long-chain fatty acids across the mitochondrial inner membrane, 2) hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (aka trifunctional protein; HADHA) which catalyzes the last three steps of mitochondrial beta-oxidation of long chain fatty acids; and 3) Amyloid β-peptide-binding alcohol dehydrogenase (ABAD) (aka hydroxysteroid (17-beta) dehydrogenase 10; aka SCHAD, short-chain-3-hydroxyacyl-CoA dehydrogenase) mitochondrial protein that catalyzes the oxidation of a wide variety of fatty acids and steroids, and is a subunit of mitochondrial ribonuclease P, which is involved in tRNA maturation. In mitochondria in which respiration declined and H2O2 production was increased (Fig. 1A and B), CPT1, HADHA and ABAD protein expression was significantly increased in aged female brain (Fig. 10A; F (3, 18) = 8.128, p = 0.0012, B; F (3, 18) = 8.779, p = 0.0008 and C; F (3, 18) = 7.892, p = 0.0014 respectively). These data indicate that within the aged female brain key elements necessary to transport and metabolize fatty acids are significantly increased.
Fig. 10.
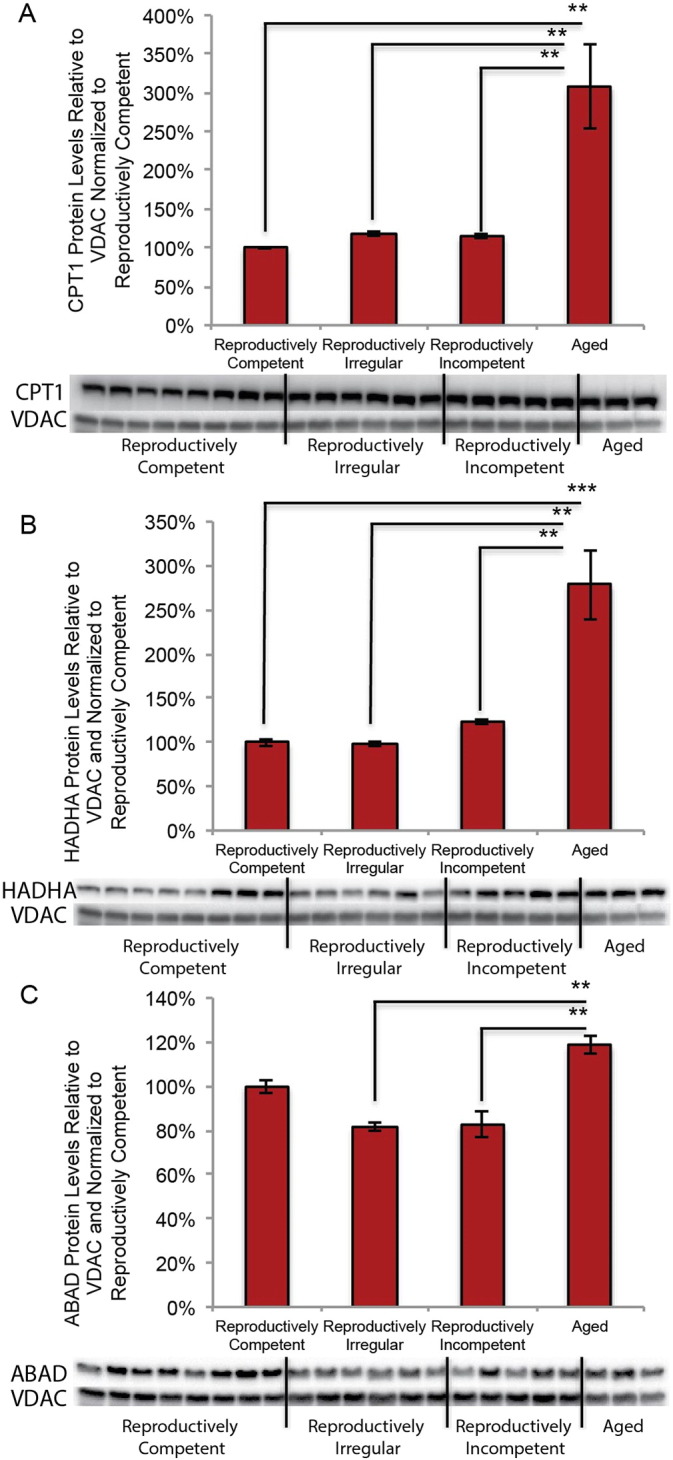
Expression of proteins involved in fatty acid transport and metabolism in the aging female mouse model: A. CPT1 protein expression in isolated whole brain mitochondria. B. HADHA protein expression in isolated whole brain mitochondria. C. ABAD protein expression in isolated whole brain mitochondria (A, B, C. N = 3–8). (**p < 0.005 and ***p < 0.0005).
To determine whether fatty acids derived from myelin lipids and metabolized within the mitochondrial would lead to a rise in ketone bodies to fuel ATP generation, analysis of brain and plasma levels of the principle ketone β-hydroxybutyrate was conducted. Within hippocampus an incremental rise in ketone body level occurred during the course of reproductive aging with significant elevation the aged brain (Fig. 11A; F (3, 12) = 9.185, p = 0.002). In cortex, the level of ketone bodies was significantly elevated at the earliest stage of reproductive aging (irregular) and was sustained at the significantly elevated level thereafter (Fig. 11B; F (3, 20) = 6.416, p = 0.0032). In parallel to the rise in brain level of ketone bodies, plasma level of ketone bodies underwent a precipitous decline at age of reproductive incompetence and remained at a low level thereafter (Fig. 11C; F (3, 16) = 5.982, p = 0.0062). These data are consistent with elevation in brain ketone bodies due to brain derived ketone body generation and not with ketone bodies derived from the periphery.
Fig. 11.
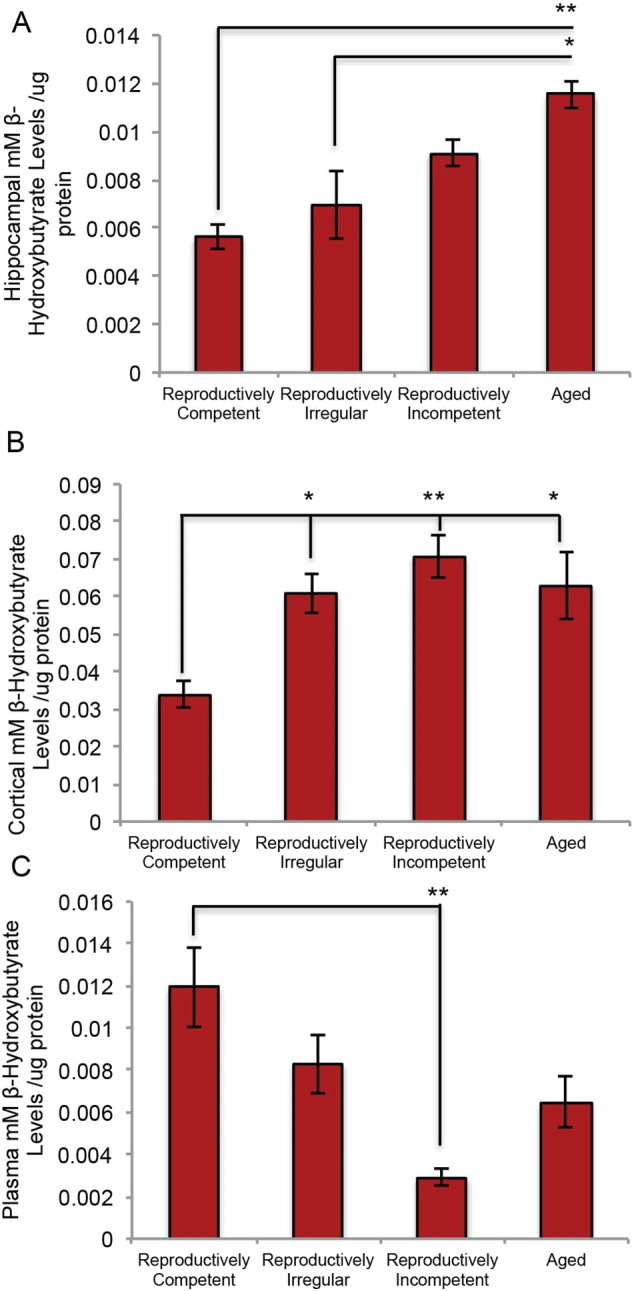
Hippocampal, cortical and plasma ketone body levels in the aging female mouse: A. Hippocampal levels of ketone bodies. B. Cortical levels of ketone bodies. C. Plasma levels of ketone bodies. (A. N = 4 per group; B. N = 5–6; C. N = 3–7). (*p < 0.05 and **p < 0.005).
Biochemical, genomic, ultra structural and lipidomic data indicated metabolism of the myelin sheath and release of myelin lipids in reproductively senescent females. Findings indicated that the aging female brain undergoes an alteration in lipid profile of brain following WM degeneration that is concomitant with an increase in fatty acid transport and metabolic capabilities. This is paralleled by an increase in ketone body generation indicating that myelin derived lipids can ultimately be used in ketogenesis to fuel ATP demands of the bioenergetically compromised brain.
4. Discussion
Age remains the greatest risk factor for developing AD (Hansson et al., 2006, Alzheimer's, 2015). Thus, investigation of transitions in the aging brain is a reasoned strategy for elucidating mechanisms and pathways of vulnerability for developing AD. Aging, while typically perceived as a linear process, is likely composed of dynamic transition states, which can protect against or exacerbate vulnerability to AD (Brinton et al., 2015). An aging transition unique to the female is the perimenopausal to menopausal conversion (Brinton et al., 2015). The bioenergetic similarities between the menopausal transition in women and the early appearance of hypometabolism in persons at risk for AD make the aging female a rational model to investigate mechanisms underlying risk of late onset AD.
Findings from this study replicate our earlier findings that age of reproductive senescence is associated with decline in mitochondrial respiration, increased H2O2 production and shift to ketogenic metabolism in brain (Yao et al., 2010, Ding et al., 2013, Yin et al., 2015). These well established early age-related changes in mitochondrial function and shift to ketone body utilization in brain, are now linked to a mechanistic pathway that connects early decline in mitochondrial respiration and H2O2 production to activation of the cPLA2-sphingomyelinase pathway to catabolize myelin lipids resulting in WM degeneration (Fig. 12). These lipids are sequestered in lipid droplets for subsequent use as a local source of ketone body generation via astrocyte mediated beta-oxidation of fatty acids. Astrocyte derived ketone bodies can then be transported to neurons where they undergo ketolysis to generate acetyl-CoA for TCA derived ATP generation required for synaptic and cell function (Fig. 12).
Fig. 12.

Schematic model of mitochondrial H2O2 activation of cPLA2-sphingomyelinase pathway as an adaptive response to provide myelin derived fatty acids as a substrate for ketone body generation: The cPLA2-sphingomyelinase pathway is proposed as a mechanistic pathway that links an early event, mitochondrial dysfunction and H2O2, in the prodromal/preclinical phase of Alzheimer's with later stage development of pathology, white matter degeneration. Our findings demonstrate that an age dependent deficit in mitochondrial respiration and a concomitant rise in oxidative stress activate an adaptive cPLA2-sphingomyelinase pathway to provide myelin derived fatty acids as a substrate for ketone body generation to fuel an energetically compromised brain.
Biochemical evidence obtained from isolated whole brain mitochondria confirms that during reproductive senescence and in response to estrogen deprivation brain mitochondria decline in respiratory capacity (Yao et al., 2009, Yao et al., 2010, Brinton, 2008a, Brinton, 2008b, Swerdlow and Khan, 2009). A well-documented consequence of mitochondrial dysfunction is increased production of reactive oxygen species (ROS), specifically H2O2 (Boveris and Chance, 1973, Beal, 2005, Yin et al., 2014, Yap et al., 2009). While most research focuses on the damage generated by free radicals, in this case H2O2 functions as a signaling molecule to activate cPLA2, the initiating enzyme in the cPLA2-sphingomyelinase pathway (Farooqui and Horrocks, 2006, Han et al., 2003, Sun et al., 2004). In AD brain, increased cPLA2 immunoreactivity is detected almost exclusively in astrocytes suggesting that activation of the cPLA2-sphingomyelinase pathway is localized to astrocytes in AD, as opposed to the neuronal or oligodendroglial localization that is observed during apoptosis (Sun et al., 2004, Malaplate-Armand et al., 2006, Di Paolo and Kim, 2011, Stephenson et al., 1996, Stephenson et al., 1999). In our analysis, cPLA2 (Sanchez-Mejia and Mucke, 2010) activation followed the age-dependent rise in H2O2 production and was sustained at an elevated level.
Direct and robust activation of astrocytic cPLA2 by physiologically relevant concentrations of H2O2 was confirmed in vitro. Astrocytic involvement in the cPLA2-sphingomyelinase pathway was also indicated by an increase in cPLA2 positive astrocyte reactivity in WM tracts of reproductively incompetent mice. These data are consistent with findings from brains of persons with AD that demonstrate the same striking localization of cPLA2 immunoreactivity within astrocytes, specifically in the hippocampal formation (Farooqui and Horrocks, 2004). While neurons and astrocytes contain endogenous levels of cPLA2, neuronal cPLA2 is activated by an influx of intracellular calcium, whereas astrocytic cPLA2 is directly activated by excessive generation of H2O2 (Sun et al., 2004, Xu et al., 2003, Tournier et al., 1997). Evidence of this cell type specific activation was confirmed by the activation of cPLA2 in astrocytes by H2O2 and the lack of activation in neurons. These data support that astrocytic, not neuronal, cPLA2 is the cellular mediator of the H2O2 dependent cPLA2-sphingomyelinase pathway activation and provide associative evidence supporting a role of astrocytic mitochondrial H2O2 in age-related WM catabolism.
The pattern of gene expression during the shift to reproductive senescence in the female mouse hippocampus recapitulates key observations in human AD brain tissue, specifically elevation in cPLA2, sphingomyelinase and ceramidase (Schaeffer et al., 2010, He et al., 2010, Li et al., 2014). Further, up-regulation of myelin synthesis, lipid metabolism and inflammatory genes in reproductively incompetent female mice is consistent with the gene expression pattern previously reported from aged male rodent hippocampus, aged female non-human primate hippocampus and human AD hippocampus (Blalock et al., 2003, Blalock et al., 2004, Blalock et al., 2010, Blalock et al., 2011, Kadish et al., 2009, Rowe et al., 2007). In these analyses of gene expression in aged male rodent hippocampus, aged female non-human primate hippocampus and human AD hippocampus down regulation of genes related to mitochondrial function, and up-regulation in multiple genes encoding for enzymes involved in ketone body metabolism occurred (Blalock et al., 2003, Blalock et al., 2004, Blalock et al., 2010, Blalock et al., 2011, Kadish et al., 2009, Rowe et al., 2007). The comparability across data derived from aging female mouse hippocampus reported herein and those derived from male rodent brain, female nonhuman brain and human AD brain strongly suggest that cPLA2-sphingomyelinase pathway activation, myelin sheath degeneration and fatty acid metabolism leading to ketone body generation is a metabolic adaptation that is generalizable across these naturally aging models and are evident in aged human AD brain. Collectively, these data support the translational relevance of findings reported herein.
Data obtained via immunohistochemistry, electron microscopy and MBP protein analyses demonstrated an age-related loss in myelin sheath integrity. Evidence for a loss of myelin structural integrity emerged in reproductively incompetent mice following activation of the cPLA2-sphingomyelinase pathway. The unraveling myelin phenotype observed following reproductive senescence and aging reported herein is consistent with the degenerative phenotype that emerges following exposure to the chemotherapy drug bortezomib which induces mitochondrial dysfunction and increased ROS generation (Carozzi et al., 2010, Cavaletti et al., 2007, Ling et al., 2003). In parallel to the decline in myelin integrity, lipid droplet density increased. In aged mice, accumulation of lipid droplets declined in parallel to the rise in ketone bodies consistent with the utilization of myelin-derived fatty acids to generate ketone bodies. Due to the sequential relationship between WM degeneration and lipid droplet formation, we posit that lipid droplets serve as a temporary storage site for myelin-derived fatty acids prior to undergoing β-oxidation in astrocytes to generate ketone bodies.
Microstructural alterations in myelin integrity were associated with alterations in the lipid profile of brain, indicative of WM degeneration resulting in release of myelin lipids. Sphingomyelin and galactocerebroside are two main lipids that compose the myelin sheath (Baumann and Pham-Dinh, 2001). Ceramide is common to both galactocerebroside and sphingomyelin and is composed of sphingosine coupled to a fatty acid. Ceramide levels increase in aging, in states of ketosis and in neurodegeneration (Filippov et al., 2012, Blazquez et al., 1999, Costantini et al., 2005). Specifically, ceramide levels are elevated at the earliest clinically recognizable stage of AD, indicating a degree of WM degeneration early in disease progression (Di Paolo and Kim, 2011, Han et al., 2002, Costantini et al., 2005). Sphingosine is statistically significantly elevated in the brains of AD patients compared to healthy controls; a rise that was significantly correlated with acid sphingomyelinase activity, Aβ levels and tau hyperphosphorylation (He et al., 2010). In our analyses, a rise in ceramides was first observed early in the aging process in reproductively incompetent mice. The rise in ceramides was coincident with the emergence of loss of myelin integrity consistent with the release of myelin ceramides from sphingomyelin via sphingomyelinase activation. Following the rise in ceramides, sphingosine and fatty acid levels increased. The temporal sequence of the lipid profile was consistent with gene expression indicating activation of ceramidase for catabolism of ceramide into sphingosine and fatty acid during reproductive senescence. Once released from ceramide, fatty acids can be transported into the mitochondrial matrix of astrocytes via CPT-1, where β-oxidation of fatty acids leads to the generation of acetyl-CoA (Glatz et al., 2010). It is well documented that acetyl-CoA cannot cross the inner mitochondrial membrane, thus posing a barrier to direct transport of acetyl-CoA generated by β-oxidation into neurons. In response, the newly generated acetyl-CoA undergoes ketogenesis to generate ketone bodies to fuel energy demands of neurons (Morris, 2005, Guzman and Blazquez, 2004, Stacpoole, 2012). Because astrocytes serve as the primary location of β-oxidation in brain they are critical to maintaining neuronal metabolic viability during periods of reduced glucose utilization (Panov et al., 2014, Ebert et al., 2003, Guzman and Blazquez, 2004).
Once fatty acids are released from myelin ceramides, they are transported into astrocytic mitochondria by CPT1 to undergo β-oxidation. The mitochondrial trifunctional protein HADHA catalyzes the last three steps of mitochondrial β-oxidation of long chain fatty acids, while mitochondrial ABAD (aka SCHAD—short chain fatty acid dehydrogenase) metabolizes short chain fatty acids. Concurrent with the release of myelin fatty acids in aged female mice, CPT1, HADHA and ABAD protein expression as well as ketone body generation increased significantly. These findings indicate that astrocytes play a pivotal role in the response to bioenergetic crisis in brain to activate an adaptive compensatory system that activates catabolism of myelin lipids and the metabolism of those lipids into fatty acids to generate ketone bodies necessary to fuel neuronal demand for acetyl-CoA and ATP.
Collectively, these findings provide a mechanistic pathway that links mitochondrial dysfunction and H2O2 generation in brain early in the aging process to later stage white matter degeneration. Astrocytes play a pivotal role in providing a mechanistic strategy to address the bioenergetic demand of neurons in the aging female brain. While this pathway is coincident with reproductive aging in the female brain, it is likely to have mechanistic translatability to the aging male brain. Further, the mechanistic link between bioenergetic decline and WM degeneration has potential relevance to other neurological diseases involving white matter in which postmenopausal women are at greater risk, such as multiple sclerosis. The mechanistic pathway reported herein spans time and is characterized by a progression of early adaptive changes in the bioenergetic system of the brain leading to WM degeneration and ketone body production. Translationally, effective therapeutics to prevent, delay and treat WM degeneration during aging and Alzheimer's disease will need to specifically target stages within the mechanistic pathway described herein. The fundamental initiating event is a bioenergetic switch from being a glucose dependent brain to a glucose and ketone body dependent brain. It remains to be determined whether it is possible to prevent conversion to or reversal of a ketone dependent brain. Effective therapeutic strategies to intervene in this process require biomarkers of bioenergetic phenotype of the brain and stage of mechanistic progression. The mechanistic pathway reported herein may have relevance to other age-related neurodegenerative diseases characterized by white matter degeneration such as multiple sclerosis.
Conflicts and Interests
The authors of this manuscript have no conflicts of interest.
Acknowledgments
We thank Ms. H-M. T. Persson for contributions to metabolomics analyses, Dr. Seema Tiwari-Woodruff for guidance regarding WM degeneration and detection, and Dr. Enrique Cadenas for counsel regarding mitochondrial function and free radical biology. This research was supported by grants NIA P01AG026572 and NIA R01AG033288 (to RDB), and NIEHS R01ES020715 and NCATS UL1 TR000135 (to ET). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Alzheimer's A. 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 2015;11:332–384. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease. Neurobiol. Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. (author reply 49-62) [DOI] [PubMed] [Google Scholar]
- Bartzokis G., Lu P.H., Geschwind D.H., Edwards N., Mintz J., Cummings J.L. Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Arch. Gen. Psychiatry. 2006;63:63–72. doi: 10.1001/archpsyc.63.1.63. [DOI] [PubMed] [Google Scholar]
- Baumann N., Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Beal M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Blachnio-Zabielska A.U., Persson X.M., Koutsari C., Zabielski P., Jensen M.D. A liquid chromatography/tandem mass spectrometry method for measuring the in vivo incorporation of plasma free fatty acids into intramyocellular ceramides in humans. Rapid Commun. Mass Spectrom. 2012;26:1134–1140. doi: 10.1002/rcm.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock E.M., Buechel H.M., Popovic J., Geddes J.W., Landfield P.W. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer's disease. J. Chem. Neuroanat. 2011;42:118–126. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock E.M., Chen K.C., Sharrow K., Herman J.P., Porter N.M., Foster T.C., Landfield P.W. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock E.M., Geddes J.W., Chen K.C., Porter N.M., Markesbery W.R., Landfield P.W. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock E.M., Grondin R., Chen K.C., Thibault O., Thibault V., Pandya J.D., Dowling A., Zhang Z., Sullivan P., Porter N.M., Landfield P.W. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J. Neurosci. 2010;30:6058–6071. doi: 10.1523/JNEUROSCI.3956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass J.P., Sheu R.K., Gibson G.E. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 2000;903:204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- Blazquez C., Woods A., De Ceballos M.L., Carling D., Guzman M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999;73:1674–1682. doi: 10.1046/j.1471-4159.1999.731674.x. [DOI] [PubMed] [Google Scholar]
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boveris A., Chance B. The mitochondrial generation of hydrogen peroxide. general properties and effect of hyperbaric oxygen. Biochem. J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman A.M., Meier I.B., Korgaonkar M.S., Provenzano F.A., Grieve S.M., Siedlecki K.L., Wasserman B.T., Williams L.M., Zimmerman M.E. Testing the white matter retrogenesis hypothesis of cognitive aging. Neurobiol. Aging. 2012;33:1699–1715. doi: 10.1016/j.neurobiolaging.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton R.D. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease. Adv. Drug Deliv. Rev. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton R.D. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008;31:529–537. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton R.D., Yao J., Yin F., Mack W.J., Cadenas E. Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 2015;11:393–405. doi: 10.1038/nrendo.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger H.G., Hale G.E., Dennerstein L., Robertson D.M. Cycle and hormone changes during perimenopause: the key role of ovarian function. Menopause. 2008;15:603–612. doi: 10.1097/gme.0b013e318174ea4d. [DOI] [PubMed] [Google Scholar]
- Burger H., Woods N.F., Dennerstein L., Alexander J.L., Kotz K., Richardson G. Nomenclature and endocrinology of menopause and perimenopause. Expert. Rev. Neurother. 2007;7:S35–S43. doi: 10.1586/14737175.7.11s.S35. [DOI] [PubMed] [Google Scholar]
- Cabodevilla A.G., Sanchez-Caballero L., Nintou E., Boiadjieva V.G., Picatoste F., Gubern A., Claro E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled beta-oxidation of fatty acids. J. Biol. Chem. 2013;288:27777–27788. doi: 10.1074/jbc.M113.466656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- Carozzi V.A., Canta A., Oggioni N., Sala B., Chiorazzi A., Meregalli C., Bossi M., Marmiroli P., Cavaletti G. Neurophysiological and neuropathological characterization of new murine models of chemotherapy-induced chronic peripheral neuropathies. Exp. Neurol. 2010;226:301–309. doi: 10.1016/j.expneurol.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Cavaletti G., Gilardini A., Canta A., Rigamonti L., Rodriguez-Menendez V., Ceresa C., Marmiroli P., Bossi M., Oggioni N., D'incalci M., De Coster R. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp. Neurol. 2007;204:317–325. doi: 10.1016/j.expneurol.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Chou J.L., Shenoy D.V., Thomas N., Choudhary P.K., Laferla F.M., Goodman S.R., Breen G.A. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer's disease. J. Proteome. 2011;74:466–479. doi: 10.1016/j.jprot.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Costantini C., Kolasani R.M., Puglielli L. Ceramide and cholesterol: possible connections between normal aging of the brain and Alzheimer's disease. Just hypotheses or molecular pathways to be identified? Alzheimers Dement. 2005;1:43–50. doi: 10.1016/j.jalz.2005.06.004. [DOI] [PubMed] [Google Scholar]
- De Leon M., Bobinski M., Convit A., Wolf O., Insausti R. Usefulness of MRI measures of entorhinal cortex versus hippocampus in AD. Neurology. 2001;56:820–821. doi: 10.1212/wnl.56.6.820. [DOI] [PubMed] [Google Scholar]
- Decarli C., Murphy D.G., Tranh M., Grady C.L., Haxby J.V., Gillette J.A., Salerno J.A., Gonzales-Aviles A., Horwitz B., Rapoport S.I. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995;45:2077–2084. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- Di Paola M., Di Iulio F., Cherubini A., Blundo C., Casini A.R., Sancesario G., Passafiume D., Caltagirone C., Spalletta G. When, where, and how the corpus callosum changes in MCI and AD: a multimodal MRI study. Neurology. 2010;74:1136–1142. doi: 10.1212/WNL.0b013e3181d7d8cb. [DOI] [PubMed] [Google Scholar]
- Di Paolo G., Kim T.W. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat. Rev. Neurol. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F., Yao J., Rettberg J.R., Chen S., Brinton R.D. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H., Guo L., Yan S., Sosunov A.A., Mckhann G.M., Yan S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D., Haller R.G., Walton M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003;23:5928–5935. doi: 10.1523/JNEUROSCI.23-13-05928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erten-Lyons D., Woltjer R., Kaye J., Mattek N., Dodge H.H., Green S., Tran H., Howieson D.B., Wild K., Silbert L.C. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013;81:977–983. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui A.A., Horrocks L.A. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot. Essent. Fatty Acids. 2004;71:161–169. doi: 10.1016/j.plefa.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Farooqui A.A., Horrocks L.A. Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist. 2006;12:245–260. doi: 10.1177/1073858405285923. [DOI] [PubMed] [Google Scholar]
- Federico A., Cardaioli E., Da Pozzo P., Formichi P., Gallus G.N., Radi E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Filippov V., Song M.A., Zhang K., Vinters H.V., Tung S., Kirsch W.M., Yang J., Duerksen-Hughes P.J. Increased ceramide in brains with Alzheimer's and other neurodegenerative diseases. J. Alzheimers Dis. 2012;29:537–547. doi: 10.3233/JAD-2011-111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch C.E., Felicio L.S., Mobbs C.V., Nelson J.F. Ovarian and steroidal influences on neuroendocrine aging processes in female rodents. Endocr. Rev. 1984;5:467–497. doi: 10.1210/edrv-5-4-467. [DOI] [PubMed] [Google Scholar]
- Fonteh A.N., Cipolla M., Chiang J., Arakaki X., Harrington M.G. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer's disease. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y., Grossman R.I., Babb J.S., Rabin M.L., Mannon L.J., Kolson D.L. Age-related total gray matter and white matter changes in normal adult brain. Part II: quantitative magnetization transfer ratio histogram analysis. AJNR Am. J. Neuroradiol. 2002;23:1334–1341. [PMC free article] [PubMed] [Google Scholar]
- Gibson G.E., Huang H.M. Oxidative Stress in Alzheimer's disease. Neurobiol. Aging. 2005;26:575–578. doi: 10.1016/j.neurobiolaging.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Gibson G.E., Blass J.P., Beal M.F., Bunik V. The alpha-ketoglutarate–dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol. Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Gibson G.E., Sheu K.F., Blass J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- Gilmore S., Peakall R., Robertson J. Organelle DNA haplotypes reflect crop-use characteristics and geographic origins of Cannabis sativa. Forensic Sci. Int. 2007;172:179–190. doi: 10.1016/j.forsciint.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Glatz J.F., Luiken J.J., Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- Gosden R.G., Laing S.C., Flurkey K., Finch C.E. Graafian follicle growth and replacement in anovulatory ovaries of ageing C57BL/6J mice. J. Reprod. Fertil. 1983;69:453–462. doi: 10.1530/jrf.0.0690453. [DOI] [PubMed] [Google Scholar]
- Guzman M., Blazquez C. Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot. Essent. Fatty Acids. 2004;70:287–292. doi: 10.1016/j.plefa.2003.05.001. [DOI] [PubMed] [Google Scholar]
- Han X., Holtzman D.M., Mckeel D.W., Jr., Kelley J., Morris J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J. Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- Han W.K., Sapirstein A., Hung C.C., Alessandrini A., Bonventre J.V. Cross-talk between cytosolic phospholipase A2 alpha (cPLA2 alpha) and secretory phospholipase A2 (sPLA2) in hydrogen peroxide-induced arachidonic acid release in murine mesangial cells: sPLA2 regulates cPLA2 alpha activity that is responsible for arachidonic acid release. J Biol Chem. 2003;278:24153–24163. doi: 10.1074/jbc.M300424200. [DOI] [PubMed] [Google Scholar]
- Hansson O., Zetterberg H., Buchhave P., Londos E., Blennow K., Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- He X., Huang Y., Li B., Gong C.X., Schuchman E.H. Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol. Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz K. Cerebral glucose metabolism in preclinical and prodromal Alzheimer's disease. Expert. Rev. Neurother. 2010;10:1667–1673. doi: 10.1586/ern.10.136. [DOI] [PubMed] [Google Scholar]
- Hoyer S. Abnormalities of glucose metabolism in Alzheimer's disease. Ann. N. Y. Acad. Sci. 1991;640:53–58. doi: 10.1111/j.1749-6632.1991.tb00190.x. [DOI] [PubMed] [Google Scholar]
- Hoyer S., Nitsch R., Oesterreich K. Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J. Neural Transm. Park. Dis. Dement. Sect. 1991;3:1–14. doi: 10.1007/BF02251132. [DOI] [PubMed] [Google Scholar]
- Irwin R.W., Yao J., Hamilton R.T., Cadenas E., Brinton R.D., Nilsen J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology. 2008;149:3167–3175. doi: 10.1210/en.2007-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W., Reed B., Mungas D., Ellis W., Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–877. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- Jahanshad N., Kochunov P.V., Sprooten E., Mandl R.C., Nichols T.E., Almasy L., Blangero J., Brouwer R.M., Curran J.E., De Zubicaray G.I., Duggirala R., Fox P.T., Hong L.E., Landman B.A., Martin N.G., Mcmahon K.L., Medland S.E., Mitchell B.D., Olvera R.L., Peterson C.P., Starr J.M., Sussmann J.E., Toga A.W., Wardlaw J.M., Wright M.J., Hulshoff Pol H.E., Bastin M.E., Mcintosh A.M., Deary I.J., Thompson P.M., Glahn D.C. Multi-site genetic analysis of diffusion images and voxelwise heritability analysis: a pilot project of the ENIGMA-DTI working group. NeuroImage. 2013;81:455–469. doi: 10.1016/j.neuroimage.2013.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I., Thibault O., Blalock E.M., Chen K.C., Gant J.C., Porter N.M., Landfield P.W. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J. Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koek M.M., Muilwijk B., Van Der Werf M.J., Hankemeier T. Microbial metabolomics with gas chromatography/mass spectrometry. Anal. Chem. 2006;78:1272–1281. doi: 10.1021/ac051683+. [DOI] [PubMed] [Google Scholar]
- Lebel C., Gee M., Camicioli R., Wieler M., Martin W., Beaulieu C. Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage. 2012;60:340–352. doi: 10.1016/j.neuroimage.2011.11.094. [DOI] [PubMed] [Google Scholar]
- Li L., Ruau D.J., Patel C.J., Weber S.C., Chen R., Tatonetti N.P., Dudley J.T., Butte A.J. Disease risk factors identified through shared genetic architecture and electronic medical records. Sci. Transl. Med. 2014;6 doi: 10.1126/scitranslmed.3007191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Ling Y.H., Liebes L., Zou Y., Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- Lu P.H., Lee G.J., Tishler T.A., Meghpara M., Thompson P.M., Bartzokis G. Myelin breakdown mediates age-related slowing in cognitive processing speed in healthy elderly men. Brain Cogn. 2013;81:131–138. doi: 10.1016/j.bandc.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Malaplate-Armand C., Florent-Bechard S., Youssef I., Koziel V., Sponne I., Kriem B., Leininger-Muller B., Olivier J.L., Oster T., Pillot T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006;23:178–189. doi: 10.1016/j.nbd.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Marner L., Nyengaard J.R., Tang Y., Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol. 2003;462:144–152. doi: 10.1002/cne.10714. [DOI] [PubMed] [Google Scholar]
- Moreira P.I., Cardoso S.M., Santos M.S., Oliveira C.R. The key role of mitochondria in Alzheimer's disease. J. Alzheimers Dis. 2006;9:101–110. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- Morris A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005;28:109–121. doi: 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging. 2005;32:486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- Mosconi L., De Santi S., Brys M., Tsui W.H., Pirraglia E., Glodzik-Sobanska L., Rich K.E., Switalski R., Mehta P.D., Pratico D., Zinkowski R., Blennow K., De Leon M.J. Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry. 2008;63:609–618. doi: 10.1016/j.biopsych.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., De Santi S., Li J., Tsui W.H., Li Y., Boppana M., Laska E., Rusinek H., De Leon M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging. 2008;29:676–692. doi: 10.1016/j.neurobiolaging.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Mistur R., Switalski R., Brys M., Glodzik L., Rich K., Pirraglia E., Tsui W., De Santi S., De Leon M.J. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72:513–520. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Mistur R., Switalski R., Tsui W.H., Glodzik L., Li Y., Pirraglia E., De Santi S., Reisberg B., Wisniewski T., De Leon M.J. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Berti V., Glodzik L., Pupi A., De Santi S., De Leon M.J. Pre-clinical detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J. Alzheimers Dis. 2010;20:843–854. doi: 10.3233/JAD-2010-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., De Leon M., Murray J., Lezi E., Lu J., Javier E., Mchugh P., Swerdlow R.H. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer's disease. J. Alzheimers Dis. 2011;27:483–490. doi: 10.3233/JAD-2011-110866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Herholz K., Prohovnik I., Nacmias B., De Cristofaro M.T., Fayyaz M., Bracco L., Sorbi S., Pupi A. Metabolic interaction between ApoE genotype and onset age in Alzheimer's disease: implications for brain reserve. J. Neurol. Neurosurg. Psychiatry. 2005;76:15–23. doi: 10.1136/jnnp.2003.030882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Pupi A., De Leon M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann. N. Y. Acad. Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Sorbi S., De Leon M.J., Li Y., Nacmias B., Myoung P.S., Tsui W., Ginestroni A., Bessi V., Fayyazz M., Caffarra P., Pupi A. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer's disease. J. Nucl. Med. 2006;47:1778–1786. [PubMed] [Google Scholar]
- Nilsen J., Irwin R.W., Gallaher T.K., Brinton R.D. Estradiol in vivo regulation of brain mitochondrial proteome. J. Neurosci. 2007;27:14069–14077. doi: 10.1523/JNEUROSCI.4391-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov A., Orynbayeva Z., Vavilin V., Lyakhovich V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014;2014:472459. doi: 10.1155/2014/472459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson X.M., Blachnio-Zabielska A.U., Jensen M.D. Rapid measurement of plasma free fatty acid concentration and isotopic enrichment using LC/MS. J. Lipid Res. 2010;51:2761–2765. doi: 10.1194/jlr.M008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior J.C. Perimenopause: the complex endocrinology of the menopausal transition. Endocr. Rev. 1998;19:397–428. doi: 10.1210/edrv.19.4.0341. [DOI] [PubMed] [Google Scholar]
- Quehenberger O., Armando A., Dumlao D., Stephens D.L., Dennis E.A. Lipidomics analysis of essential fatty acids in macrophages. Prostaglandins Leukot. Essent. Fatty Acids. 2008;79:123–129. doi: 10.1016/j.plefa.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon N.L., Silverman D., Siddarth P., Miller K., Ercoli L.M., Elman S., Lavretsky H., Huang S.C., Phelps M.E., Small G.W. Estrogen use and brain metabolic change in postmenopausal women. Neurobiol. Aging. 2005;26:229–235. doi: 10.1016/j.neurobiolaging.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Reiman E.M., Caselli R.J., Yun L.S., Chen K., Bandy D., Minoshima S., Thibodeau S.N., Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman E.M., Chen K., Alexander G.E., Caselli R.J., Bandy D., Osborne D., Saunders A.M., Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc. Natl. Acad. Sci. U. S. A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A., Murphy A.N. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe W.B., Blalock E.M., Chen K.C., Kadish I., Wang D., Barrett J.E., Thibault O., Porter N.M., Rose G.M., Landfield P.W. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J. Neurosci. 2007;27:3098–3110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mejia R.O., Mucke L. Phospholipase A2 and arachidonic acid in Alzheimer's disease. Biochim. Biophys. Acta. 2010;1801:784–790. doi: 10.1016/j.bbalip.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro N. The menopausal transition. Am. J. Med. 2005;118(Suppl. 12B):8–13. doi: 10.1016/j.amjmed.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Schaeffer E.L., Da Silva E.R., Novaes B.D.E.A., Skaf H.D., Gattaz W.F. Differential roles of phospholipases A2 in neuronal death and neurogenesis: implications for Alzheimer disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2010;34:1381–1389. doi: 10.1016/j.pnpbp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Silverman D.H., Geist C.L., Kenna H.A., Williams K., Wroolie T., Powers B., Brooks J., Rasgon N.L. Differences in regional brain metabolism associated with specific formulations of hormone therapy in postmenopausal women at risk for AD. Psychoneuroendocrinology. 2011;36:502–513. doi: 10.1016/j.psyneuen.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprooten E., Knowles E.E., Mckay D.R., Goring H.H., Curran J.E., Kent J.W., Jr., Carless M.A., Dyer T.D., Drigalenko E.I., Olvera R.L., Fox P.T., Almasy L., Duggirala R., Kochunov P., Blangero J., Glahn D.C. Common genetic variants and gene expression associated with white matter microstructure in the human brain. NeuroImage. 2014;97:252–261. doi: 10.1016/j.neuroimage.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole P.W. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell. 2012;11:371–377. doi: 10.1111/j.1474-9726.2012.00805.x. [DOI] [PubMed] [Google Scholar]
- Stephenson D.T., Lemere C.A., Selkoe D.J., Clemens J.A. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer's disease brain. Neurobiol. Dis. 1996;3:51–63. doi: 10.1006/nbdi.1996.0005. [DOI] [PubMed] [Google Scholar]
- Stephenson D., Rash K., Smalstig B., Roberts E., Johnstone E., Sharp J., Panetta J., Little S., Kramer R., Clemens J. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia. 1999;27:110–128. doi: 10.1002/(sici)1098-1136(199908)27:2<110::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Sun G.Y., Xu J., Jensen M.D., Simonyi A. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J. Lipid Res. 2004;45:205–213. doi: 10.1194/jlr.R300016-JLR200. [DOI] [PubMed] [Google Scholar]
- Swerdlow R.H., Khan S.M. The Alzheimer's disease mitochondrial cascade hypothesis: an update. Exp. Neurol. 2009;218:308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Nyengaard J.R., Pakkenberg B., Gundersen H.J. Age-induced white matter changes in the human brain: a stereological investigation. Neurobiol. Aging. 1997;18:609–615. doi: 10.1016/s0197-4580(97)00155-3. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S., Morales L.B., Lee R., Voskuhl R.R. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C., Thomas G., Pierre J., Jacquemin C., Pierre M., Saunier B. Mediation by arachidonic acid metabolites of the H2O2-induced stimulation of mitogen-activated protein kinases (extracellular-signal-regulated kinase and c-Jun NH2-terminal kinase) Eur. J. Biochem. 1997;244:587–595. doi: 10.1111/j.1432-1033.1997.00587.x. [DOI] [PubMed] [Google Scholar]
- Trushina E., Dutta T., Persson X.M., Mielke M.M., Petersen R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer's disease using metabolomics. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech R.L., Chance B., Kashiwaya Y., Lardy H.A., Cahill G.F., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life. 2001;51:241–247. doi: 10.1080/152165401753311780. [DOI] [PubMed] [Google Scholar]
- Vlassenko A.G., Vaishnavi S.N., Couture L., Sacco D., Shannon B.J., Mach R.H., Morris J.C., Raichle M.E., Mintun M.A. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc. Natl. Acad. Sci. U. S. A. 2010;107:17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M.T., Maki P.M., Mcdermott M.P. Cognition and mood in perimenopause: a systematic review and meta-analysis. J. Steroid Biochem. Mol. Biol. 2014;142:90–98. doi: 10.1016/j.jsbmb.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M.T., Rubin L.H., Maki P.M. Cognition in perimenopause: the effect of transition stage. Menopause. 2013;20:511–517. doi: 10.1097/GME.0b013e31827655e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Yu S., Sun A.Y., Sun G.Y. Oxidant-mediated AA release from astrocytes involves cPLA2 and iPLA2. Free Radic. Biol. Med. 2003;34:1531–1543. doi: 10.1016/s0891-5849(03)00152-7. [DOI] [PubMed] [Google Scholar]
- Yao J., Chen S., Mao Z., Cadenas E., Brinton R.D. 2-Deoxy-d-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Hamilton R.T., Cadenas E., Brinton R.D. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys. Acta. 2010;1800:1121–1126. doi: 10.1016/j.bbagen.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Irwin R., Chen S., Hamilton R., Cadenas E., Brinton R.D. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiol. Aging. 2012;33:1507–1521. doi: 10.1016/j.neurobiolaging.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Irwin R.W., Zhao L., Nilsen J., Hamilton R.T., Brinton R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Rettberg J.R., Klosinski L.P., Cadenas E., Brinton R.D. Shift in brain metabolism in late onset Alzheimer's disease: implications for biomarkers and therapeutic interventions. Mol. Asp. Med. 2011;32:247–257. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap L.P., Garcia J.V., Han D., Cadenas E. The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 2009;61:1283–1298. doi: 10.1016/j.addr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Boveris A., Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 2014;20:353–371. doi: 10.1089/ars.2012.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Yao J., Sancheti H., Feng T., Melcangi R.C., Morgan T.E., Finch C.E., Pike C.J., Mack W.J., Cadenas E., Brinton R.D. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging. 2015;36:2282–2295. doi: 10.1016/j.neurobiolaging.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Dean D., Liu J.Z., Sahgal V., Wang X., Yue G.H. Quantifying degeneration of white matter in normal aging using fractal dimension. Neurobiol. Aging. 2007;28:1543–1555. doi: 10.1016/j.neurobiolaging.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Zhao L., Morgan T.E., Mao Z., Lin S., Cadenas E., Finch C.E., Pike C.J., Mack W.J., Brinton R.D. Continuous versus cyclic progesterone exposure differentially regulates hippocampal gene expression and functional profiles. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031267. [DOI] [PMC free article] [PubMed] [Google Scholar]