

Abstract
Dysregulated expression of MYC family genes is a hallmark of many malignancies. Unfortunately, these proteins are not amenable to blockade by small molecules or protein-based therapeutic agents. Therefore, we must find alternative approaches to target MYC-driven cancers. Amplification of MYCN, a MYC family member, predicts high-risk neuroblastoma (NB) disease. We have shown that R9-caPep blocks the interaction of PCNA with its binding partners and selectively kills human NB cells, especially those with MYCN amplification, and we now show the mechanism. We found elevated levels of DNA replication stress in MYCN-amplified NB cells. R9-caPep exacerbated DNA replication stress in MYCN-amplified NB cells and NB cells with an augmented level of MYC by interfering with DNA replication fork extension, leading to Chk1 dependence and susceptibility to Chk1 inhibition. We describe how these effects may be exploited for treating NB.
Keywords: Proliferating Cell Nuclear Antigen (PCNA), MYC, MYCN, Replication stress, Neuroblastoma, DNA damage response, Chk1
Highlights
-
•
MYCN-amplified neuroblastoma (NB) cells contain a high level of ATR/Chk1 signaling and DNA replication stress
-
•
R9-caPep, a PCNA-derived cell permeable peptide interferes with DNA replication fork extension and causes Chk1 dependence
-
•
R9-caPep works synergistically with Chk1 inhibitors to kill NB cells that have MYCN amplification or enhanced MYC expression
Enhanced expression of MYC proteins, including MYC, MYCN, and MYCL, is a hallmark of many cancers. These functionally redundant oncoproteins aren't amenable to blockade by traditional therapeutics. MYCN amplification is a prominent genetic marker for high-risk neuroblastoma. We discovered the presence of chronic DNA replication stress and the dependence on ATR/Chk1-mediated DNA damage response as crucial vulnerabilities in MYCN-amplified neuroblastoma cells. Induction of replication stress by PCNA-derived R9-caPep works synergistically with Chk1 inhibition to kill MYCN-amplified cells. Our study provides proof-of-concept evidence that targeting a well defined region of PCNA may lead to an effective therapy for treating MYC-driven cancers.
1. Introduction
Dysregulated expression of the MYC family oncogenes, including MYC, MYCN, and MYCL, is a hallmark of malignancy and is involved in cancer initiation and maintenance (Dang, 2012, Meyer and Penn, 2008). The MYC family proteins are structurally similar and functionally equivalent (K.C. et al., 2014) transcription factors that promote S-phase entry and facilitate DNA replication during stress (Dominguez-Sola and Gautier, 2014, Dominguez-Sola et al., 2007, Felsher et al., 2000, Mai et al., 1996), thereby conferring a proliferation and survival advantage to cancer cells. Proof-of-principle studies demonstrated that blocking MYC protein function is beneficial to cancer treatment (Soucek et al., 2008, Soucek et al., 2013). However, finding small molecule or biologic inhibitors of MYC has proven to be a difficult task. MYC is localized within nuclei (Smith et al., 2004) and has no deep surface binding pocket (Nair and Burley, 2003). Therefore, MYC is not amenable to blockade by small molecules or accessible to neutralization by antibodies. It is also difficult to manipulate MYC target genes. The target genes regulated by MYC are from a broad range of functional categories and are often cell type dependent (Lin et al., 2012, Nie et al., 2012). As a result, the downstream gene(s) that truly mediates MYC's tumorigenic activities remains elusive. Despite more than a decade of effort, no MYC inhibitor has reached the clinic. Exploiting functional vulnerabilities caused by MYC dysregulation may provide a viable alternative to translate the wealth of MYC-related research into clinical benefit.
Amplification of the MYCN oncogene occurs in 20–25% of all neuroblastomas (NBs) and correlates with a particularly poor prognosis; it is the most prominent genetic marker for high-risk NB disease (Maris, 2010). Furthermore, by studying the expression signature of MYC target genes, Fredlund et al. found that the combined pathway activity of all MYC family proteins correlated with clinical risk and poor prognosis in NB patients independent of MYCN amplification (Fredlund et al., 2008). Current treatment for high-risk NB patients consists of induction treatment (conventional chemotherapy and surgery with or without radiotherapy), high-dose chemotherapy and autologous stem cell transplantation (HDCT/autoSCT) as a consolidation treatment, and 13-cis-retinoic acid treatment to reduce relapse from minimal residual disease. Up to 20% of the high-risk NB patients are refractory to initial chemotherapy (Bhatnagar and Sarin, 2012). Of the high-risk population that does respond to induction chemotherapy, a substantial portion relapses, and relapse occurs despite intensive consolidation and maintenance therapies (Aaltomaa et al., 1993, Park et al., 2010).
In an effort to develop a novel therapy for treating high-risk NB, we designed a cell permeable peptide, R9-caPep (Gu et al., 2014), which contains the L126-Y133 sequence of proliferating cell nuclear antigen (PCNA). PCNA, through its interaction with more than a dozen of proteins, plays an essential role in DNA synthesis and repair (Maga and Hubscher, 2003). Inhibition of PCNA is viewed as an effective way to suppress tumor growth (Stoimenov and Helleday, 2009). The L126-Y133 peptide region, located within the PCNA's interconnector domain that mediates the interaction of PCNA with many of its binding partners (Krishna et al., 1994), is differentially modified between cancerous and non-malignant cells (Hoelz et al., 2006, Malkas et al., 2006). Therefore, this peptide region, essential to PCNA function, provides a structural base for selectively targeting cancer cells. We previously reported that R9-caPep blocks the interaction of PCNA with flap structure-specific endonuclease 1 (Fen1), DNA ligase I (LIGI), and DNA polymerase δ in vitro (Smith et al., 2015) and in vivo (Gu et al., 2014). It selectively kills NB cells with minimal toxicity to human peripheral blood mononuclear cells (PBMC) or neural crest stem cells (Gu et al., 2014). Importantly, we found that MYCN-amplified NB cell lines are twice as sensitive to R9-caPep treatment as non-MYCN-amplified lines (Gu et al., 2014).
To dissect the mechanism that makes MYCN-amplified NB cells vulnerable to R9-caPep, we analyzed the expression of biological markers indicative of replication stress (RS) and DNA damage response (DDR) (Chanoux et al., 2009, Wang et al., 2006) and found that MYCN-amplified NB cells and tumors contain higher levels of markers indicative of ATR/Chk1 signaling and RS than non-MYCN-amplified ones, indicating the presence of chronic RS. This finding also suggests that the MYCN-amplified NB cells depend on ATR/Chk1-mediated DDR to proliferate despite high levels of RS. This observation raises the possibility that MYCN-amplified NB cells are intrinsically sensitive to the induction of additional RS and are susceptible to Chk1 inhibition. Here, we have demonstrated that R9-caPep interferes with DNA replication fork extension and induces RS in MYCN-amplified cells. In addition, R9-caPep treatment works synergistically with Chk1 inhibition to exacerbate RS and kill MYCN-amplified NB cells and other NB cells that express an augmented level of MYC. Based on these observations, we propose that chronic RS in MYCN-amplified NB cells is a weak link, and the RS-induced dependence of MYCN-amplified NB cells on ATR/Chk1-mediated DDR is a promising target for treating this devastating disease. Our study also demonstrated the therapeutic potential of R9-caPep to attack this weak link for treating MYCN-amplified NB disease and other high-risk NB disease that expresses an enhanced level of MYC.
2. Materials & Methods
2.1. Reagents and Cell Lines
The cell permeable peptide, R9-caPep (RDRDRDRDRDRDRDRDRDCCLGIPEQEY), was created by fusing the L126-Y133 sequence of human PCNA to the C-terminus of a nine D-arginine sequence (R9) through a spacer of two cysteines (CC). The peptide was synthesized by AnaSpec (Fremont, CA). The Chk1 inhibitors, MK-8776 and UCN-01, were purchased from Selleckchem (Houston, TX) and EMD Millipore (Billerica, MA), respectively. siRNAs targeting MYCN and non-targeting siRNAs were purchased from GE Healthcare (Pittsburgh, PA).
The human NB cell lines, SK-N-DZ, SK-N-BE(2)c, BE(2)c, CHP-212, IMR-32, SK-N-AS, SK-N-SH, and SH-SY5Y were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Cells were maintained in DMEM with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin in the presence of 5% CO2 at 37 °C. The non-malignant HCN1-A cortical neuronal cell line and bone marrow-derived Mesenchymal Stem Cells (hBM-MSCs) were obtained from the ATCC as well and were cultured according to the ATCC instructions. The human embryonic progenitor cell line 7SM0032 was acquired from Millipore (Billerica, MA) and grown in the hEPM-1 Media Kit purchased from the same company.
2.2. siRNA Transfection
SK-N-BE(2)c cells were reverse transfected with siRNAs targeting MYCN or non-targeting siRNA by Lipofectamine 2000 (Thermo Fisher Scientific,) according to the manufacturer's instructions. The transfected cells were plated at 2.5 × 105 cells/cm2 in a cell culture dish. 48 h after the initial transfection, cells were detached and reverse transfected again with the same siRNA. Transfected cells were directly seeded at 1 × 105 cells/cm2 into a 96-well plate. After being allowed to attach overnight, cells were treated with various concentrations of R9-caPep for an additional 72 h. The relative abundance of cells was measured by a CellTiter-Glo assay (Promega, Madison, WI).
2.3. Gene Expression Profile Analysis
A well-annotated microarray dataset consisting of gene expression profiles of 478 NB patient tumor samples was previously published by Oberthuer, et al. and is accessible through ArrayExpress (E-MTAB-179). Total cellular RNAs were extracted from 5 MYCN-amplified NB cell lines, 3 non-MYCN-amplified NB cell lines, and 2 non-malignant cell lines in triplicate, using the RNeasy kit purchased from Qiagen (Valencia, CA). The gene expression profiles of each cell line were analyzed by hybridizing the labeled cDNA to the Affymetrix Human Gene Array 1.0. The microarray data were analyzed by the Partek Genomics Suite 6.6 software (Partek, Saint Louis, MO). Briefly, raw data were normalized and converted into the expression measurements by the RMA algorithm. The ANOVA model was used to identify genes that were differentially expressed between the sample groups.
2.4. Western Blot
Cell extracts were prepared by dissolving cell pellets directly into the Laemmli sample buffer with 5% DTT. After ultrasonication and boiling, samples were resolved in a 4–12% SDS polyacrylamide gel and blotted onto a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk and incubated with an antibody specific to γH2A.X, total H2A.X, Chk1, phospho-Chk1 (Ser345), Rad17, cleaved caspase-3, cleaved PARP1, or β-actin in the blocking buffer. All antibodies used in western analyses, except for γH2A.X (EMD Millipore) and Rad17 (LifeSpan Biosciences, Seattle, WA), were purchased from Cell Signaling Technology (Danvers, MA). After incubation with peroxidase-conjugated secondary antibodies, the proteins of interest were detected by chemiluminescence, using an ECL kit purchased from GE Healthcare (Pittsburgh, PA).
2.5. DNA Combing Analysis
A DNA combing assay was performed to measure the effect of AOH1160 on replication fork extension, as described (Frum et al., 2013). Briefly, synchronized S-phase cells were first pulsed with CldU, a modified thymidine analog, in the absence of AOH1160 for 10 min. After washing away the unincorporated CIdU, we added another modified thymidine analog, IdU, to the cells with or without 100 μM of R9-caPep. After 20 min of incubation, the cells were collected, spotted on microscope slides, and lysed. The incorporation of CldU and IdU into DNA was visualized under a fluorescence microscope after the cells were stained by monoclonal antibodies that are specific to CldU and IdU respectively and are linked to different fluorophores: green for CIdU and red for IdU. The rate of DNA replication fork extension before and after R9-caPep treatment was estimated by measuring the relative length of green- and red-stained DNA segments respectively, using the ImageJ software (National Institute of Health, Bethesda, MD).
To measure the effect of UCN-01 and R9-caPep on activation of replication origins, synchronized cells were treated with each or both reagents for 12 h and were sequentially labeled by CldU and IdU for 15 min each. Origins were identified as the center point of CldU (green)-labeled regions surrounded on both sides by IdU (red)-labeled regions. The relative distance between adjacent origins on individual fibers was measured using the ImageJ software.
2.6. Immunohistochemistry
The study was carried out in accordance with City of Hope's and Vanderbilt University's policies governing the use of patient specimens and PDX tissues, respectively. 5-μm-thick serial sections were made from formalin-fixed, paraffin-embedded patient tumors or PDX tumors. The γH2A.X-specific antibody was purchased from Cell Signaling Technology. Antibodies specific to Chk1 and phosphorylated Rad17 were purchased from LifeSpan Biosciences. The staining was detected by the EnVision + horseradish peroxidase system purchased from Dako (Carpinteria, CA). Counterstaining was with 50% Mayer's hematoxylin (DAKO) for 3 min. Slides were visualized and images were acquired on an Aperio® CS2 slide scanner under a 20 × objective lens (Leica, Buffalo Grove, IL).
2.7. Growth Inhibition and Synergism Analysis
Cells were seeded at 1.5 × 103–6 × 103/well depending on the growth rate of each cell line in a 96-well plate. After being allowed to attach overnight, cells were treated with various concentrations of the R9-caPep and/or Chk1 inhibitors for 72 h. Cell growth was measured by the CellTiter-Glo assay according to the manufacturer's instructions. The synergism between the peptide and a Chk1 inhibitor was analyzed according to the median-effect principle by the CalcuSyn software (Biosoft, Cambridge, UK).
3. Results
3.1. Enhanced RS and Chk1 Signaling in MYCN-amplified NB
To determine the mechanism that makes MYCN-amplified NB cells sensitive to R9-caPep, we analyzed the gene expression profiles of 5 MYCN-amplified NB cell lines, 3 non-MYCN-amplified NB cell lines, and 2 non-malignant human cell lines. Raw microarray data are deposited in the Gene Expression Omnibus database (accession no. GSE66586). We found that NB cells express a higher level of Chk1 mRNA than non-malignant human cells and the Chk1 mRNA levels correlate strongly with MYCN expression levels and amplification status in NB cell lines (Fig 1a). Western analysis confirmed that MYCN-amplified cells express a higher level of Chk1 protein than non-MYCN-amplified cells and non-malignant cells (Fig 1b). Protein complexes containing Rad17 assembled at the DNA lesion site are required for ATR/Chk1 phosphorylation and activation (Cimprich and Cortez, 2008, Nam and Cortez, 2011, Zou et al., 2002). MYCN-amplified cell lines also express higher levels of phosphorylated Chk1 (p-Chk1) and Rad17, indicating that ATR/Chk1 signaling is enhanced in MYCN-amplified NB cells (Fig. 1b). All the non-MYCN-amplified NB cell lines included in this study express an augmented level of MYC (Gumireddy et al., 2003, Vandesompele et al., 2003), relative to MYCN-amplified cell lines. Although the levels of Chk1 signaling components in these non-MYCN-amplified cells are lower than those in MYCN-amplified cells, they are much higher than the levels in non-malignant neural crest stem cells (Fig. 1a & b). This result is consistent with the functional overlaps between MYCN and MYC and with the observation that the total pathway signaling of MYC family proteins, determined by the expression of MYC target genes, are stronger in MYCN-amplified NB cell lines than in non-MYCN-amplified lines (Chayka et al., 2015). To determine if the enhanced Chk1 expression observed in the MYCN-amplified cell lines also occurs in patient specimens, we analyzed a microarray dataset published by Oberthuer et al. (Oberthuer et al., 2010) consisting of expression data from 472 NB patients with known MYCN amplification status. Consistent with our observations in NB cell lines, Chk1 expression is significantly higher in MYCN-amplified human NB specimens than in non-MYCN-amplified specimens (Fig. 1c). We also compared Chk1 mRNA levels between MYCN-amplified tumors and the high and low-risk subsets of non-MYCN-amplified tumors (Fig. 1c). Although, in the population of non-MYCN-amplified tumors, the high-risk subset expresses a significantly higher level of Chk1 mRNA than the low-risk subset (Fig. 1c), Chk1 mRNA levels are higher in MYCN-amplified tumors than in the high-risk non-MYCN-amplified tumors. In the population of non-MYCN-amplified tumors, Chk1 mRNA level also correlates with MYC expression (analysis not shown). These observations are consistent with previous studies of human NB specimens (Cole et al., 2011).
Fig. 1.

MYCN amplification is associated with high Chk1 signaling and RS. a) Total RNA extracted from MYCN-amplified NB cell lines (blue: SK-N-DZ, SK-N-BE(2)c, BE(2)c, CHP-212, and IMR-32), non-MYCN-amplified NB cell lines (green: SK-N-AS, SK-N-SH, and SH-SY5Y), and non-malignant human cell lines (red: HCN1-A and hBM-MSCs) were analyzed by microarray analysis. The Log2 transformed Chk1 (X-axis) and MYCN (Y-axis) mRNA levels in these cell lines were graphed. The correlation between relative Chk1 and MYCN mRNA levels in these cell lines were analyzed by linear regression (r = 0.886). b) Total cell extracts from the indicated cell lines were analyzed by Western blotting to determine the levels of p-Chk1, Chk1, and Rad17. c) A microarray dataset of NB patient specimens (ArrayExpress: E-MTAB-179) was analyzed. The relative expression of Chk1 mRNA in patient specimens with or without MYCN amplification was graphed with the bottom and top ends of the whiskers representing the 5th and 95th percentiles of the expression levels respectively. “All” represents all non-MYCN-amplified NB samples. The “low-risk” subset includes patients with stage 1, 2, or 4S non-MYCN-amplified NB and under the age of 18 months. The “high-risk” subset includes patients with stage 4 non-MYCN-amplified tumors and above the age of 18 months. The p-value was determined by an unpaired t-test. Serial sections made from d) patient NB specimens and e) PTX NB tumors were stained with the indicated antibodies, as described in the Materials and Methods. The left two columns in each panel box are serial sections made from two MYCN-amplified tumors and the right two columns are sections made from two non-MYCN-amplified tumors.
The enhanced signaling of the ATR/Chk1 pathway suggests that chronic RS may be present in MYCN-amplified NB cells and that the cells may depend on ATR/Chk1-mediated DDR to deal with such RS. To determine whether RS levels and Chk1 signaling are elevated in MYCN-amplified NB tumors, we analyzed intracellular markers of RS and Chk1 signaling in 27 clinical NB specimens (of which 2 are MYCN-amplified and 25 are non-MYCN-amplified) and in 9 patient-derived xenograft (PDX) tumors (of which 3 are MYCN-amplified and 6 are non-MYCN-amplified) by immunohistochemistry. Strong nuclear staining of Chk1 was seen in all 5 MYCN-amplified tumors that we analyzed (Fig. 1d and e). In contrast, of the 31 non-MYCN-amplified tumors (25 patient specimens and 6 PDX), only 7 had Chk1 staining in nuclei, although many non-MYCN-amplified tumors had weak Chk1 staining in the cytoplasm (Fig. 1d and e). On average, MYCN-amplified tumors also had a higher level of γH2A.X (a DNA damage marker) and phosphor-Rad17 (p-Rad17) than non-MYCN-amplified tumors (Fig. 1d and e). Collectively, these results indicate that RS levels are higher and ATR/Chk1 signaling is more active in MYCN-amplified tumors.
3.2. MYCN Amplification Causes RS and Confers Sensitivity to R9-caPep
To determine whether MYCN amplification and the resulting protein overexpression are responsible for chronic RS and enhanced sensitivity to R9-caPep, we transfected MYCN-amplified SK-N-BE(2)c NB cells with a siRNA (siMYCN) targeting the human MYCN mRNA and examined the effect on intracellular γH2A.X levels. Cells transfected with a non-targeting siRNA (siCTL) were used as control. R9-caPep treatment induced γH2A.X levels in SK-N-BE(2)c cells transfected by siCTL (Fig. 2a), suggesting that the peptide can cause RS. Downregulation of MYCN by siMYCN reduced basal γH2A.X levels by about 5 fold in SK-N-BE(2)c cells (Fig. 2a), indicating that a high level of MYCN expression is responsible for the chronic RS and DNA lesions in MYCN-amplified cells. R9-caPep treatment increased γH2A.X level by about 3 fold in cells transfected by siMYCN. However, the total γH2A.X level in siMYCN transfected cells was about a third of the level in control cells under the same treatment (Fig. 2a), indicating a meaningful contribution of MYCN to the total RS and DNA lesion in MYCN-amplified cells before and after R9-caPep treatment. Consistent with the effect of MYCN on intracellular γH2A.X levels, downregulation of MYCN expression by siMYCN significantly increased the IC50 value of R9-caPep in a cell growth assay (Fig. 2b), indicating that MYCN overexpression confers sensitivity to R9-caPep. Western analysis confirmed a near depletion of MYCN protein in cells twice transfected with siMYCN compared with the level in cells transfected with siCTL over the full experimental time frame (Fig. 2c).
Fig. 2.

MYCN overexpression confers sensitivity to R9-caPep. SK-N-BE(2)c cells were transfected by a siRNA (siMYCN) targeting MYCN mRNA or a non-targeting control siRNA (siCTL). 48 h after the initial transfection, cells were reverse transfected again by the same siRNA. a) SK-N-BE(2)c cells transfected by siMYCN or siCTL were incubated in the presence of 20 μM R9-caPep for 12 h. Intracellular MYCN, γH2A.X, and total H2A.X levels were determined by Western blot. The density levels of γH2A.X and total H2A.X were quantified by TotalLab Quant (Tyne and Wear, England) and were presented at the bottom of the panel. b) After the second transfection, cells were seeded into a 96-well plate directly and allowed to attach overnight. The cells were then treated by various concentration of R9-caPep for 72 h, before their growth was measured by a CellTiter-Glo assay. The R9-caPep IC50 values from three independently transfected cell populations were averaged and graphed ± standard deviation (S.D.). c) MYCN protein levels in cell lysates extracted from aliquots of cells at 0, 72, and 144 h after the first siRNA transfection were examined by Western blot.
3.3. R9-caPep Interferes with DNA Replication Fork Extension
The presence of chronic RS in MYCN-amplified NB cells raises the possibility that these cells are more sensitive to additional RS induced by R9-caPep than non-MYCN-amplified NB cells. To understand the mechanism by which R9-caPep induces RS, we measured the effects of R9-caPep on DNA replication fork extension. In this experiment, synchronized S-phase cells were first pulsed with CldU, a modified thymidine analog, in the absence of R9-caPep. After washing away the unincorporated CldU, cells were incubated with another modified thymidine analog, IdU, in the presence or absence of 100 μM of R9-caPep. The rate of DNA replication fork extension before and after R9-caPep treatment was estimated by measuring the relative length of CldU-incorporated DNA strands and adjacent IdU-incorporated DNA strands, respectively. The DNA replication forks extended at similar rates in the control and experimental cells before R9-caPep treatment, as indicated by the similar average lengths of the CldU-incorporated DNA strands (Fig 3). In contrast, cells treated by R9-caPep contain significantly shorter IdU-incorporated DNA strands than the control cells, indicating that R9-caPep interferes with the extension of preexisting DNA replication forks (Fig 3).
Fig. 3.
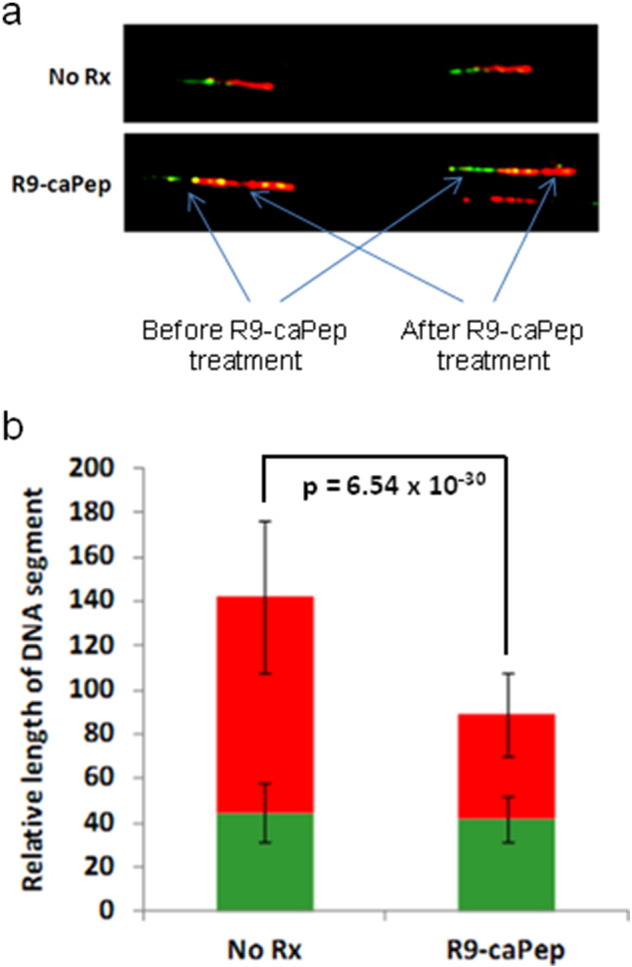
R9-caPep interferes with replication fork extension. Synchronized SK-N-BE(2)c cells were sequentially treated with two nucleotide analogs, CldU and IdU, before and after R9-caPep treatment. Cells sequentially treated with the same two nucleotide analogs without R9-caPep treatment were used as controls. The incorporation of these two nucleotides, representing replication fork extension before and after R9-caPep treatment respectively, was visualized by a fluorophore-coupled monoclonal antibody specific to CldU (green) or IdU (red). a) Shown are representative images of the labeled DNA strands from cells treated with or without R9-caPep. b) The lengths of IdU (red bars) and CldU (green bars) incorporated DNA segments measured from more than 30 independent DNA strands from cells treated with or without R9-caPep were averaged and graphed ± S.D.
3.4. R9-caPep Works Synergistically with Chk1 Inhibitors
The sensitivity of MYCN-amplified NB cells to R9-caPep-induced RS and the possible dependence of these cells on Chk1-mediated DDR to deal with RS strongly suggest a synergism between R9-caPep treatment and Chk1 inhibition. To test this, we performed a synergism assay based on the median-effect principle (Chou and Talalay, 1984). In the experiment, cell growth was measured after treatment with 2-fold serial dilutions of R9-caPep and a Chk1 inhibitor [UCN-01 (Graves et al., 2000) or MK8776 (Guzi et al., 2011)] at a fixed concentration ratio that matches their respective IC50s. Cells treated by each agent alone were used as controls. The combination of R9-caPep with UCN-01 or MK8776 significantly inhibited the growth of two MYCN-amplified NB cancer cell lines using doses of R9-caPep and Chk1 inhibitors that did not significantly affect cell growth (Fig. 4a–c). These growth data were analyzed by the method developed by Chou et al. (Chou and Talalay, 1984) to determine synergism. As shown in the bottom panels (Fig. 4a–c), combination dose pairs ranging from ED50 to ED90 are all plotted well below their respective additive isoboles with combination indices (CI) between 0.17 and 0.55, indicating synergism between the combined agents.
Fig. 4.

R9-caPep works synergistically with Chk1 inhibitors. Sk-N-BE(2)c (Panels a and c), SK-N-DZ (Panel b), and SH-SY5Y (Panel d) NB cells were treated with 2-fold serial dilutions of R9-caPep and UCN-01 (triangles in panels a, b, and d) or R9-caPep and MK-8776 (triangles in panel c) at a fixed concentration ratio. Cells treated with the same concentrations of R9-caPep (squares), UCN-01 (circles), or MK-8776 (circles) alone were used as control. Cells were cultured in the presence of the reagents for 3 days and their growth was determined by a CellTiter-Glo assay (Promega). Top: the relative abundance of cells in triplicates under each treatment conditions was averaged and graphed ± S.D. Bottom: conservative isobolograms were graphed and CI values were calculated for each study by CalcuSyn. Blue: ED90; green: ED75; and red: ED50.
Since MYC expression is a risk factor in non-MYCN-amplified high-risk NB and is associated with augmented Chk1 signaling, we sought to determine whether R9-caPep can induce RS and confer sensitivity to Chk1 inhibition in non-MYCN-amplified cells that express a high level of MYC. We performed a similar synergism study in SH-SY5Y cells (Gumireddy et al., 2003, Vandesompele et al., 2003). Although this non-MYCN-amplified cell line is less sensitive to treatment by R9-caPep alone than MYCN-amplified cell lines (Gu et al., 2014), combined treatment by R9-caPep and UCN-01 worked synergistically to inhibit its growth (Fig. 4d).
Chk1-mediated DDR can enable cells to survive RS by stabilizing existing replication forks and preventing excessive firing of new replication origins (Mcneely et al., 2010, Seiler et al., 2007). In addition, Chk1 signaling plays a critical role in regulating the initiation of normal DNA replication (Maya-Mendoza et al., 2007). Unscheduled replication initiation causes DNA lesions and genome instability. To determine the mechanism by which R9-caPep and Chk1 inhibitors work synergistically to kill cancer cells, we sought to determine their effects on the regulation of replication origin firing. The average distance between neighboring replication origins is significantly shorter in both MYCN-amplified SK-N-BE(2)c and MYC-expressing SH-SY5Y cells treated by UCN-01 and R9-caPep than in the corresponding cells treated by either agent alone, indicating that these two agents work synergistically to dysregulate the control of replication origin firing (Fig. 5b and c). The amount of DNA lesions indicated by the intracellular γH2A.X levels is much higher in cells treated by both R9-caPep and UCN-01 than by each agent alone (Fig 5d), indicating that interference with replication by R9-caPep leads to stalling of replication forks and accumulation of double-stranded DNA break in the absence of Chk1 signaling. Combined treatment by R9-caPep and UCN-01 also caused higher levels of cleaved caspase-3 and PARP1, indicating activation of caspase-3-mediated apoptosis (Fig 5d).
Fig. 5.

Dysregulation of replication initiation and induction of DNA damage and apoptosis by R9-caPep and Chk1 inhibitors. b) SK-N-BE(2) cells or c) SH-SY5Y cells were incubated for 12 h with 7.8 nM UCN-01, 30 μM R9-caPep, or 7.8 nM UCN-01 and 30 μM R9-caPep for 12 h. Untreated corresponding cells were used as controls. Cells were sequentially labeled with CldU and IdU for 15 min each. Following incubation with the nucleotide analogs, the cells were stained by fluorophore-coupled monoclonal antibodies to visualized CIdU and IdU incorporation. The relative distance between two neighboring replication origins was measured as illustrated in panel a. The distances between two neighboring origins from at least 15 DNA segments from cells under each treatment condition were graphed. d) SK-N-BE(2)c (left) or SH-SY5Y (right) cells were treated by 7.8 nM UCN-01, 30 μM R9-caPep, or both for 12 h. The intracellular levels of the indicated proteins were determined by Western blot.
4. Discussion
Numerous studies have shown that the dysregulation of MYC family proteins underlies the pathogenesis and aggressiveness of many malignancies. It is well established that inhibition of MYC protein function is beneficial to cancer treatment. In the case of NB, stable downregulation of MYCN mRNA by lentivirus-based RNAi inhibits the growth of human MYCN-amplified NB cells in vitro and in vivo (Jiang et al., 2011). However, targeting MYC proteins directly has yet to be successful as a therapeutic approach in spite of efforts for more than a decade. We previously reported that MYCN-amplified NB cells are particularly sensitive to R9-caPep (Gu et al., 2014). Here, we have demonstrated that MYCN overexpression is responsible for conferring sensitivity to R9-caPep in NB cells. We have also observed that MYCN-amplified or MYC-amplified small cell lung cancer (SCLC) cells are more sensitive to R9-caPep than SCLC cells containing no amplification of any MYC family genes (data will be published separately). This raises the possibility that R9-caPep may selectively kill a broad range of cancers that is driven by the MYC family proteins. Based on the published crystal structure, the L126-Y133 peptide region of PCNA forms part of a pocket suitable for binding by small molecules (Punchihewa et al., 2012). The effectiveness of R9-caPep in inhibiting MYCN-amplified NB cells not only shows the therapeutic potential of this peptide, but also provides proof-of-concept evidence that targeting a well defined region of PCNA may lead to an effective therapy for treating MYC-driven cancers.
We compared gene expression profiles from multiple NB cell lines and non-malignant cell lines and found that the expression of Checkpoint kinase 1 (Chk1) is significantly higher in MYCN-amplified cell lines than in non-MYCN-amplified cell lines or non-malignant human cells. Analysis of microarray data previously published by Oberthuer et al. (Oberthuer et al., 2010) also revealed a strong correlation between Chk1 expression and MYCN amplification in patient samples. In non-MYCN-amplified tumors, Chk1 expression is significantly higher in the high-risk subset than in the low-risk subset and correlates with MYC expression. Chk1, whose expression and activity correlates with RS and the ensuing DDR (Bartkova et al., 2005), plays a critical role in the ATR signaling pathway that senses RS and arrests the cell cycle, thereby allowing cells time to deal with RS and to resolve DNA lesions (Abraham, 2001, Smith et al., 2010). After DNA replication forks encounter lesions and stall, a protein complex, consisting of Rad17 and the Replication Factor C (RFC), recognizes junctions of single stranded and double-stranded DNAs and recruits the 9-1-1 checkpoint sliding clamp complex to the arrested forks (Bermudez et al., 2003, Ellison and Stillman, 2003). Whereas Rad17 may function as a tumor suppressor (Bric et al., 2009), suppression of Rad17 beyond a crucial threshold could also be deleterious to proliferation (Bric et al., 2009). In addition, depletion of Rad17 sensitizes pancreatic cancer cells to gemcitabine (Fredebohm et al., 2013), indicating a critical role of Rad17 in dealing with RS. Both the Rad17 checkpoint loading complex and the 9-1-1 complex are required for ATR activation (Zou et al., 2002). The ATR checkpoint signaling is transduced through phosphorylation of Chk1, which in turn activates a collection of Chk1 substrates that stabilize replication forks and arrest the cell cycle (Cimprich and Cortez, 2008, Nam and Cortez, 2011).
Mechanistically, R9-caPep is able to interfere with DNA replication fork extension and to induce RS in NB cells. It has been shown that MYC proteins, including MYCN, promote S-phase entry and inhibit G1 arrest after DNA damage. In the MYCN-amplified NB cell line, SK-N-BE(2)c, knockdown of MYCN expression by siRNA can restore DNA damage-induced G1 arrest, indicating a causal relationship between MYCN overexpression and dysregulation of the G1 checkpoint (Yu et al., 2008). Consequently, cells overexpressing MYCN are more likely to enter S-phase with unrepaired DNA damage. These cells are more like to experience RS and to be susceptible to further induction of RS by therapeutic agents. To verify this hypothesis and to exploit the vulnerability of MYCN-amplified NB for a therapeutic purpose, we analyzed the biomarkers indicative of RS in NB cell lines and tumor tissues and found that MYCN amplification is associated with a higher level of γH2A.X and more active Chk1 signaling. An augmented MYC expression is also associated with an enhanced Chk1 level in non-MYCN-amplified NB tumors. Collectively, these experiments revealed that the presence of RS and the dependence on Chk1 are crucial vulnerabilities in MYCN-amplified and MYC-expressing cells and can be used to target these cells for destruction.
Based on the current study, we propose a working model (Fig. 6), in which the MYC family oncoproteins dysregulate the G1/S checkpoint, inhibit G1 arrest after DNA damage, and promote S-phase entry. Consequently, NB cells with MYCN amplification or enhanced MYC expression are more likely to enter S-phase with unrepaired DNA damage and experience chronic RS. They are more dependent on the ATR/Chk1 pathway to resolve DNA lesions during replication. The cell-permeable R9-caPep can exacerbate chronic RS in these cells by interfering with DNA replication fork extension and HR-mediated DSB repair (Gu et al., 2014). We speculate that the presence of a high level of chronic RS makes these cells susceptible to additional RS induced by R9-caPep treatment. Additional RS induced by R9-caPep may overwhelm the DNA repair capacity and induce cell death through apoptosis. Alternatively, a low dose of R9-caPep may not kill cells directly, but makes cells more dependent on ATR/Chk1-mediated DDR and more susceptible to inhibition of the ATR/Chk1 pathway. Consistent with such a scenario, we demonstrated that R9-caPep works synergistically with Chk1 inhibitors to induce DNA lesions and kill cells with MYCN amplification or enhanced MYC expression. Exploiting the vulnerability associated with an augmented total MYC pathway activity in NB to interference with DNA replication and induction of RS, as well as Chk1 inhibition, may lead to an effective way of treating this type of cancer.
Fig. 6.
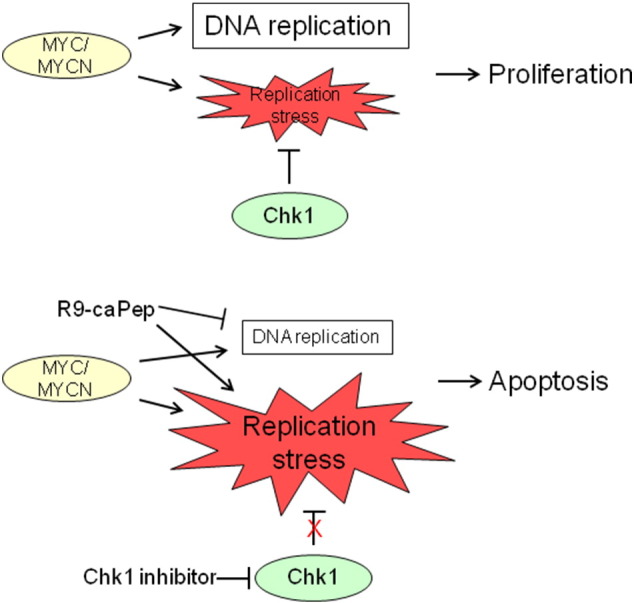
A working model for exploiting RS in MYCN-amplified or MYC-expressing NB cells. Cell cycle checkpoints play a critical role in maintaining genome integrity. Members of MYC family proto-oncogenes promote cell growth by dysregulating the G1/S checkpoint and inhibiting G1 arrest after DNA damage. Therefore, cells overexpressing MYC family proteins are more likely to enter S-phase with unrepaired DNA damages and to experience RS. R9-caPep induces additional RS by interfering with replication fork extension and by blocking HR-mediated DSB repair (Gu et al., 2014), making cells dependent on ATR/Chk1-mediated DDR to prevent the collapse of stalled replication forks and to resolve DNA lesions. By inducing RS and inhibiting DDR, R9-caPep and Chk1 inhibitors work synergistically in causing DSB and apoptosis. Such a synergism may provide an effective way of treating MYCN-amplified NB and other high-risk NB that expresses an augmented level of MYC.
Numerous studies in recent years have validated Chk1 as an attractive anti-cancer target [reviewed in (Ma et al., 2011)]. Whereas the clinical development of the first Chk1 inhibitor, UCN-01, was hindered by its unfavorable pharmacokinetics, a new generation of selective Chk1 inhibitors is currently under clinical or preclinical development (Maugeri-Sacca et al., 2013). It has been shown that Chk1 inhibitors work synergistically with a broad range of DNA-damaging agents (Guzi et al., 2011, Karp et al., 2012, Zabludoff et al., 2008). However, all DNA-damaging agents, including chemotherapy and radiation therapy regimens, involve considerable harm to normal tissues. There is also a high risk of developing resistance to these drugs through mutations, which result from the genetic instability inherent to many cancers and from redundancy in DNA synthesis and repair pathways. Recent advancements in protein and peptide delivery technology have made peptide-based therapy feasible. Several protein and peptide-based drugs have successfully reached markets or are currently working their way through different stages of clinical trials (Stevenson, 2009). The ability of R9-caPep to target an essential cellular apparatus and to induce RS in cancer cells without significant toxicity to non-malignant cells (Gu et al., 2014, Lingeman et al., 2014, Smith et al., 2015) makes it a promising candidate to combine with Chk1 inhibitors in order to deliver synthetic lethality to cancer cells, especially those driven by the MYC family proteins.
Financial Support
This work was supported in part by research awards to LHM from the U.S. Department of Defense (W81XWH-11-1-0786), National Institutes of Health/National Cancer Institute (R01 CA121289), St. Baldrick's Foundation (www.stbaldricks.org), and the ANNA Fund (www.annafund.com). In addition, research reported in this publication was supported by National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
L.H.M, L.G., R.J.H., J.A.S., and G.B.F. conceived the project. L.H.M., L.G., and R.J.H. oversaw the overall performance of the project. L.H.M. and L.G. wrote this manuscript. P.C. and J.A.S. directed the immunohistochemistry study of patient specimens and PDX tissues, respectively. P.C. evaluated all the immunohistochemistry slides. R.L. performed the DNA combing assays. H.M. performed the microarray study. Z.L. and Y.C.Y. performed all bioinformatics analysis. L.G. and S.K. performed cell growth and Western blot studies.
Accession Numbers
The GEO accession number for the microarray data reported in this paper is GSE66586.
Acknowledgments
We thank the City of Hope Pathology Core for help with immunohistochemistry work. We are especially grateful to Dr. Nancy J. Linford for her editorial assistance in the preparation of this manuscript. The authors dedicate the work in memory of Anna Olivia Healey.
References
- Aaltomaa S., Lipponen P., Syrjanen K. Proliferating cell nuclear antigen (PCNA) immunolabeling as a prognostic factor in axillary lymph node negative breast cancer. Anticancer Res. 1993;13:533–538. [PubMed] [Google Scholar]
- Abraham R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., Orntoft T., Lukas J., Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bermudez V.P., Lindsey-Boltz L.A., Cesare A.J., Maniwa Y., Griffith J.D., Hurwitz J., Sancar A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S.N., Sarin Y.K. Neuroblastoma: a review of management and outcome. Indian J. Pediatr. 2012;79:787–792. doi: 10.1007/s12098-012-0748-2. [DOI] [PubMed] [Google Scholar]
- Bric A., Miething C., Bialucha C.U., Scuoppo C., Zender L., Krasnitz A., Xuan Z., Zuber J., Wigler M., Hicks J., Mccombie R.W., Hemann M.T., Hannon G.J., Powers S., Lowe S.W. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16:324–335. doi: 10.1016/j.ccr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanoux R.A., Yin B., Urtishak K.A., Asare A., Bassing C.H., Brown E.J. ATR and h2ax cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayka O., D'acunto C.W., Middleton O., Arab M., Sala A. Identification and pharmacological inactivation of the MYCN gene network as a therapeutic strategy for neuroblastic tumor cells. J. Biol. Chem. 2015;290:2198–2212. doi: 10.1074/jbc.M114.624056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Cimprich K.A., Cortez D. ATR: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K.A., Huggins J., Laquaglia M., Hulderman C.E., Russell M.R., Bosse K., Diskin S.J., Attiyeh E.F., Sennett R., Norris G., Laudenslager M., Wood A.C., Mayes P.A., Jagannathan J., Winter C., Mosse Y.P., Maris J.M. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 2011;108:3336–3341. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D., Gautier J. MYC and the control of DNA replication. Cold Spring Harb. Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D., Ying C.Y., Grandori C., Ruggiero L., Chen B., Li M., Galloway D.A., Gu W., Gautier J., Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- Ellison V., Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1 doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher D.W., Zetterberg A., Zhu J., Tlsty T., Bishop J.M. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10544–10548. doi: 10.1073/pnas.190327097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredebohm J., Wolf J., Hoheisel J.D., Boettcher M. Depletion of RAD17 sensitizes pancreatic cancer cells to gemcitabine. J. Cell Sci. 2013;126:3380–3389. doi: 10.1242/jcs.124768. [DOI] [PubMed] [Google Scholar]
- Fredlund E., Ringner M., Maris J.M., Pahlman S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 2008;105:14094–14099. doi: 10.1073/pnas.0804455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frum R.A., Deb S., Deb S.P. Use of the DNA fiber spreading technique to detect the effects of mutant p53 on DNA replication. Methods Mol. Biol. 2013;962:147–155. doi: 10.1007/978-1-62703-236-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves P.R., Yu L., Schwarz J.K., Gales J., Sausville E.A., O'connor P.M., Piwnica-Worms H. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J. Biol. Chem. 2000;275:5600–5605. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- Gu L., Smith S., Li C., Hickey R.J., Stark J.M., Fields G.B., Lang W.H., Sandoval J.A., Malkas L.H. A PCNA-derived cell permeable peptide selectively inhibits neuroblastoma cell growth. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumireddy K., Ikegaki N., Phillips P.C., Sutton L.N., Reddy C.D. Effect of 20-epi-1alpha,25-dihydroxyvitamin D3 on the proliferation of human neuroblastoma: role of cell cycle regulators and the Myc-Id2 pathway. Biochem. Pharmacol. 2003;65:1943–1955. doi: 10.1016/s0006-2952(03)00205-3. [DOI] [PubMed] [Google Scholar]
- Guzi T.J., Paruch K., Dwyer M.P., Labroli M., Shanahan F., Davis N., Taricani L., Wiswell D., Seghezzi W., Penaflor E., Bhagwat B., Wang W., Gu D., Hsieh Y., Lee S., Liu M., Parry D. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011;10:591–602. doi: 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- Hoelz D.J., Arnold R.J., Dobrolecki L.E., Abdel-Aziz W., Loehrer A.P., Novotny M.V., Schnaper L., Hickey R.J., Malkas L.H. The discovery of labile methyl esters on proliferating cell nuclear antigen by MS/MS. Proteomics. 2006;6:4808–4816. doi: 10.1002/pmic.200600142. [DOI] [PubMed] [Google Scholar]
- Jiang R., Xue S., Jin Z. Stable knockdown of MYCN by lentivirus-based RNAi inhibits human neuroblastoma cells growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2011;410:364–370. doi: 10.1016/j.bbrc.2011.06.020. [DOI] [PubMed] [Google Scholar]
- Karp J.E., Thomas B.M., Greer J.M., Sorge C., Gore S.D., Pratz K.W., Smith B.D., Flatten K.S., Peterson K., Schneider P., Mackey K., Freshwater T., Levis M.J., Mcdevitt M.A., Carraway H.E., Gladstone D.E., Showel M.M., Loechner S., Parry D.A., Horowitz J.A., Isaacs R., Kaufmann S.H. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012;18:6723–6731. doi: 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- K.C. W., Satpathy A.T., Tapaport A.S., Briseno C.G., Wu X., Albring J.C., Russler-Germain E.V., Kretzer N.M., Durai V., Persaud S.P., Edelson B.T., Loschko J., Cella M., Allen P.M., Nussenzweig M.C., Colonna M., Sleckman B.P., Murphy T.L., Murphy K.M. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature. 2014;507:243–247. doi: 10.1038/nature12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna T.S., Kong X.P., Gary S., Burgers P.M., Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lin C.Y., Loven J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingeman R.G., Hickey R.J., Malkas L.H. Expression of a novel peptide derived from PCNA damages DNA and reverses cisplatin resistance. Cancer Chemother. Pharmacol. 2014;74:981–993. doi: 10.1007/s00280-014-2574-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.X., Janetka J.W., Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga G., Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- Mai S., Hanley-Hyde J., Fluri M. c-Myc overexpression associated DHFR gene amplification in hamster, rat, mouse and human cell lines. Oncogene. 1996;12:277–288. [PubMed] [Google Scholar]
- Malkas L.H., Herbert B.S., Abdel-Aziz W., Dobrolecki L.E., Liu Y., Agarwal B., Hoelz D., Badve S., Schnaper L., Arnold R.J., Mechref Y., Novotny M.V., Loehrer P., Goulet R.J., Hickey R.J. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19472–19477. doi: 10.1073/pnas.0604614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri-Sacca M., Bartucci M., De Maria R. Checkpoint kinase 1 inhibitors for potentiating systemic anticancer therapy. Cancer Treat. Rev. 2013;39:525–533. doi: 10.1016/j.ctrv.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A., Petermann E., Gillespie D.A., Caldecott K.W., Jackson D.A. Chk1 regulates the density of active replication origins during the vertebrate s phase. EMBO J. 2007;26:2719–2731. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcneely S., Conti C., Sheikh T., Patel H., Zabludoff S., Pommier Y., Schwartz G., Tse A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- Meyer N., Penn L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Nair S.K., Burley S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Nam E.A., Cortez D. ATR signalling: more than meeting at the fork. Biochem. J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D.R., Tessarollo L., Casellas R., Zhao K., Levens D. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberthuer A., Juraeva D., Li L., Kahlert Y., Westermann F., Eils R., Berthold F., Shi L., Wolfinger R.D., Fischer M., Brors B. Comparison of performance of one-color and two-color gene-expression analyses in predicting clinical endpoints of neuroblastoma patients. Pharm. J. 2010;10:258–266. doi: 10.1038/tpj.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.R., Eggert A., Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol. Oncol. Clin. North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Punchihewa C., Inoue A., Hishiki A., Fujikawa Y., Connelly M., Evison B., Shao Y., Heath R., Kuraoka I., Rodrigues P., Hashimoto H., Kawanishi M., Sato M., Yagi T., Fujii N. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits dna replication. J. Biol. Chem. 2012;287:14289–14300. doi: 10.1074/jbc.M112.353201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler J.A., Conti C., Syed A., Aladjem M.I., Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- Smith K.P., Byron M., O'connell B.C., Tam R., Schorl C., Guney I., Hall L.L., Agrawal P., Sedivy J.M., Lawrence J.B. c-Myc localization within the nucleus: evidence for association with the PML nuclear body. J. Cell. Biochem. 2004;93:1282–1296. doi: 10.1002/jcb.20273. [DOI] [PubMed] [Google Scholar]
- Smith S.J., Gu L., Phipps E.A., Dobrolecki L.E., Mabrey K.S., Gulley P., Dillehay K.L., Dong Z., Fields G.B., Chen Y.R., Ann D., Hickey R.J., Malkas L.H. A Peptide mimicking a region in proliferating cell nuclear antigen specific to key protein interactions is cytotoxic to breast cancer. Mol. Pharmacol. 2015;87:263–276. doi: 10.1124/mol.114.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L., Whitfield J., Martins C.P., Finch A.J., Murphy D.J., Sodir N.M., Karnezis A.N., Swigart L.B., Nasi S., Evan G.I. Modelling Myc Inhibition as a Cancer Therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L., Whitfield J.R., Sodir N.M., Masso-Valles D., Serrano E., Karnezis A.N., Swigart L.B., Evan G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson C.L. Advances in peptide pharmaceuticals. Curr. Pharm. Biotechnol. 2009;10:122–137. doi: 10.2174/138920109787048634. [DOI] [PubMed] [Google Scholar]
- Stoimenov I., Helleday T. PCNA on the crossroad of cancer. Biochem. Soc. Trans. 2009;37:605–613. doi: 10.1042/BST0370605. [DOI] [PubMed] [Google Scholar]
- Vandesompele J., Edsjo A., De Preter K., Axelson H., Speleman F., Pahlman S. ID2 expression in neuroblastoma does not correlate to MYCN levels and lacks prognostic value. Oncogene. 2003;22:456–460. doi: 10.1038/sj.onc.1206148. [DOI] [PubMed] [Google Scholar]
- Wang X., Zou L., Lu T., Bao S., Hurov K.E., Hittelman W.N., Elledge S.J., Li L. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol. Cell. 2006;23:331–341. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Yu U.Y., Cha J.E., Ju S.Y., Cho K.A., Yoo E.S., Ryu K.H., Woo S.Y. Effect on cell cycle progression by N-Myc knockdown in SK-N-BE(2) neuroblastoma cell line and cytotoxicity with STI-571 compound. Cancer Res. Treat. 2008;40:27–32. doi: 10.4143/crt.2008.40.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabludoff S.D., Deng C., Grondine M.R., Sheehy A.M., Ashwell S., Caleb B.L., Green S., Haye H.R., Horn C.L., Janetka J.W., Liu D., Mouchet E., Ready S., Rosenthal J.L., Queva C., Schwartz G.K., Taylor K.J., Tse A.N., Walker G.E., White A.M. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Zou L., Cortez D., Elledge S.J. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]