

Abstract
In this issue, Lee et al. (2015) show that PKC directly phosphorylates the glucose transporter Glut1, in order to promote glucose uptake in response to growth factor signaling.
The ability of cells to access nutrients is critical for survival, growth, and function. Unlike single cellular organisms, which respond directly to nutrient availability, most essential nutrients are abundant in healthy and well-fed multicellular organisms. Thus regulation of cellular nutrient uptake and transport is a critical step and is highly regulated in nutrient access and cell metabolism of metazoans. Glucose is a central nutrient, and glucose uptake is correspondingly highly regulated through a family of glucose transporters, the SLC2 family or Gluts (Mueckler and Thorens, 2013). The best understood of this family is Glut4 (Slc2a4), which translocates rapidly from storage vesicles to the cell surface in response to insulin. Glut1 (Slc2a1), however, is ubiquitously expressed and contributes to both basal and growth factor-stimulated glucose uptake in a wide range of tissues (Wieman et al., 2007). Importantly, mutations in Glut1 lead to Glut1 deficiency syndrome (G1D), a disease characterized by neurologic defects and seizures (Gras et al., 2014). Defects in Glut1 expression or response to growth factor stimulation may contribute to G1D, but the molecular mechanisms that control Glut1 have been poorly understood. In this issue, Lee et al. (2015) describe identification of a protein kinase C (PKC) phosphorylation site on Glut1 that is critical for Glut1-mediated glucose uptake and is mutated in a subset of G1D patients. As such, they provide a new and direct signaling mechanism to link growth factors to increased glucose uptake.
Glut1 can be regulated by numerous mechanisms, including transcription, translation, trafficking, and activity. Slow induction of Glut1 is regulated on the transcriptional level and can be triggered by cellular stress such as hypoxia or induction of Myc upon mitogenic stimulation, while translational regulation occurs through pathways such as mTORC1 and HuR (MacIver et al., 2013). The rapid induction of glucose uptake through Glut1 within minutes of growth factor stimulation is less well understood. It has, however, been known for 30 years that PKC activation can rapidly initiate glucose uptake and may phosphorylate Glut1 to do so (Witters et al., 1985).
Using a mutagenesis and kinase screening strategy, Lee et al. identified serine 226 of Glut1 as a substrate for both conventional and novel PKCs (Lee et al., 2015). S226 lies in a conserved sequence in the large intracellular loop, which together with the C-terminal tail is thought to regulate Glut1 trafficking and activity, and is not present in other SLC2a transporters (Figure 1). In a series of experiments using phorbol esters to activate PKCs, Lee et al. (2015) show evidence of increased phosphorylation of Glut1 S226 in vitro. Importantly, mutation of S226 to alanine or treatment with PKC inhibitors abrogated the ability of phorbol esters to rapidly stimulate glucose uptake. Modeling more physiologic conditions, human umbilical vein endothelial cells and aortic endothelial cells were next stimulated with natural growth factors vascular endothelial growth factor (VEGF) and angiotensin II (AngII). Both increased Glut1 phosphorylation at S226 in a time-dependent manner. Furthermore, the effect of VEGF on glucose uptake was abrogated by treatment with a PKC inhibitor, thus supporting a key role for PKC-mediated Glut1 phosphorylation in rapid induction of glucose uptake.
Figure 1. Regulation of Glut1 Phosphorylation and Glucose Uptake.
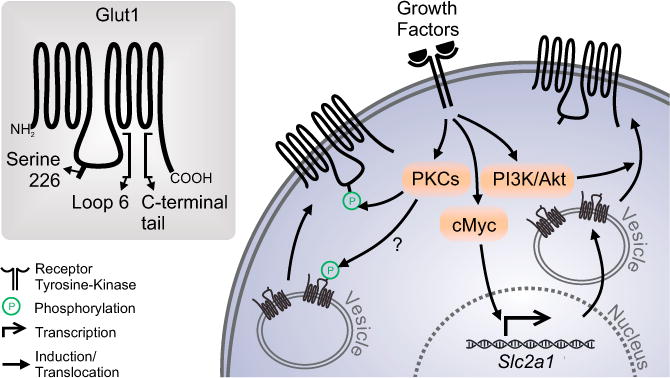
Growth factors stimulate rapid and slow increased glucose uptake and upregulation of Glut1. Slow Glut1 regulation is mediated by transcriptional induction, translation, and cell surface trafficking by Myc and the PI3K/Akt pathways. The rapid increase in glucose uptake, however, has been unclear. Lee et al. (2015) show that PKC-mediated phosphorylation of Glut1 S266 increases glucose uptake and may regulate cell surface trafficking.
The mechanism by which S226 phosphorylation regulates Glut1 and glucose uptake remains largely uncertain. An increase in cell surface localized, endogenous Glut1 was detected after treatment with phorbol esters, which was blocked by PKC inhibitors and may suggest a key role for phosphorylation in Glut1 trafficking. Cell surface trafficking of Glut1 can be stimulated by activation of the phosphatidylinositol 3-kinase/Akt pathway, which leads to rapid transport of intracellular Glut1 to the plasma membrane as well as stabilization on the cell surface (Wieman et al., 2007). The relative roles of PKC and Akt-mediated regulation of Glut1 trafficking remain unclear and may indeed be complementary (Figure 1). In addition, the precise mechanism by which Akt may control Glut1 trafficking is uncertain, and it will be interesting in future studies to examine cooperativity between these two signaling pathways. In addition to trafficking, the transport activity of Glut1 is regulated in rapid fashion (Cura and Carruthers, 2010). While AMPK has been implicated in metabolic stress-induced rapid glucose uptake, it is feasible that PKC may also mediate a growth factor-regulated increase in Glut1 activity.
This new Glut1 S226 modification is particularly exciting, as this phosphorylation motif was found mutated in some cases of G1D. While no mutations of S226 itself were identified, several in surrounding amino acids were found that could impair Glut1 S226 phosphorylation. Additional patients must now be examined to determine if S226 itself can be mutated to directly confirm that this site itself will be G1D associated. G1D mutations are varied but often lead to truncations that disrupt Glut1 protein (Gras et al., 2014). Mutations of the S226 phosphorylation site or motif did not significantly alter total Glut1 protein levels, demonstrating that signaling mechanisms that modify this site impact Glut1 function independent of overall expression to play essential roles in growth factor-induced glucose uptake in vivo.
A key question that is presented in this study is how S226 phosphorylation modifies Glut1 to control glucose transport. The structure of Glut1 suggests that this intracellular loop will not directly modify the basic transport glucose mechanism (Deng et al., 2014). However, Glut1 can oligomerize and bind additional factors, such as ATP, and it may be that phosphorylation affects these Glut1 mechanisms and as such regulates Glut1 activity. In addition, association of Glut1 with trafficking proteins, such as GIPC, is critical for Glut1 cell surface localization, and this may also be S226 regulated (Reed et al., 2005).
The identification by Lee et al. (2015) of disease-associated Glut1 mutations linked to altered phosphorylation demonstrates for the first time that direct signaling mechanisms are essential for rapid regulation of Glut1 and glucose uptake in vivo. G1D is a severe disease with profound neurological consequences. It is striking, however, that despite the prevalence of Glut1 expression, most other tissues appear unaffected. Indeed, Glut1 is essential for rapid T cell activation and proliferation in vivo (Macintyre et al., 2014). The PKC regulation of Glut1 may thus be predominantly neuron or blood-brain barrier endothelial cell specific. More likely, however, this mode of Glut1 regulation is widespread and contributes to the rapid increase in glucose uptake observed in other acutely stimulated cell types and tissues. In these cases, however, it is also likely that other cell types are more capable of adaptation or have greater tolerance for low glucose uptake than neurons. The biological consequences and role for Glut1 phosphorylation by PKCs or other kinases, in neurons and other tissues, therefore remains uncertain and an important area for future inquiry.
Acknowledgments
This work was supported by grant R01DK105550 (J.C.R.).
References
- Cura AJ, Carruthers A. J Biol Chem. 2010;285:15430–15439. doi: 10.1074/jbc.M110.110593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- Gras D, Roze E, Caillet S, Méneret A, Doummar D, Billette de Villemeur T, Vidailhet M, Mochel F. Rev Neurol (Paris) 2014;170:91–99. doi: 10.1016/j.neurol.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, Jiang Y, Pascual JM, North PE, Shaul PW, et al. Mol Cell. 2015;58:845–853. doi: 10.1016/j.molcel.2015.04.015. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueckler M, Thorens B. Mol Aspects Med. 2013;34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BC, Cefalu C, Bellaire BH, Cardelli JA, Louis T, Salamon J, Bloecher MA, Bunn RC. Mol Biol Cell. 2005;16:4183–4201. doi: 10.1091/mbc.E04-11-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters LA, Vater CA, Lienhard GE. Nature. 1985;315:777–778. doi: 10.1038/315777a0. [DOI] [PubMed] [Google Scholar]