

Abstract
Breast cancer is no longer considered a single disease, but instead is made up of multiple subtypes with genetically and most likely epigenetically heterogeneous tumors composed of numerous clones. Both the hierarchical cancer stem cell and clonal evolution models have been invoked to help explain this intratumoral heterogeneity. Several recent studies have helped define the functional interactions among the different cellular subpopulations necessary for the evolution of this complex ecosystem. These interactions involve paracrine interactions that include locally acting Wnt family members, reminiscent of the signaling pathways important for normal mammary gland development and stem cell self-renewal. In this review, we discuss the interactions among various cell populations in both normal and tumor tissues. A better understanding of these interactions, especially in the metastatic setting, will be important for the development of improved combinatorial therapies designed to prevent relapse and to ultimately decrease mortality.
Understanding tumorigenesis: lessons from the normal gland development
Over the past decade and a half, our concepts of tumor heterogeneity have evolved, driven in part by major conceptual advances that invoked a cancer stem cell (CSC) hierarchy, as well as technical advances in DNA sequencing. Rather than mutually exclusive CSC and clonal evolution models, it is generally accepted that branched Darwinian evolution integrated with a CSC hierarchy, which may include more than a single CSC population, may help explain intratumoral heterogeneity (ITH) [1]. With the advent of deep sequencing and recent single cell DNA and RNA sequencing applications, the enormous clonal heterogeneity and complexity of many solid cancers, including breast cancer, has become apparent [2, 3]. In fact, in a seminal paper by Nik-Sainal and colleagues on “The Life History of 21 Breast Cancers,” the authors concluded, “The cancer genome is like a palimpsest, an ancient parchment that was frequently reused, each time retaining traces of what had previously been written…Our model has an obvious similitude with the concept of CSCs-infrequent, self-renewing, metabolically quiescent cells capable of reconstituting a tumor” [4]. Cancers are now starting to be viewed as complex ecosystems that evolve due to both mutational and epigenetic mechanisms [5, 6]. With thousands of mutations and epigenetic changes, many of which occur at low frequencies, an understanding of their functional importance is a daunting task. Furthermore, recent studies suggest that selection of major and minor alleles is not a stochastic process, but may be dependent upon cell-cell and paracrine interactions [7]. An understanding of these interactions in normal development can provide the foundation for deciphering their enormous complexity in cancer. The following review will highlight some of the concepts that have emerged from studying these interactions in normal mammary gland development and how insights about signaling pathways have provided the groundwork for understanding the heterogeneity in breast cancer, one of the future challenges for cancer therapeutics.
Mammary gland architecture
The mammary gland is particularly amenable to studies defining paracrine and cell-cell interactions, because the majority of development occurs postnatally. Postnatal development is regulated by the systemic hormones estrogen, progesterone and prolactin, which relay local cues via growth factors. Like many epithelial tissues, the mammary gland is comprised of luminal and basal epithelial cell populations, with the basal compartment composed primarily of contractile myoepithelial cells. These epithelial cell populations are able to self-organize into duct-like structures when grown in matrigel or collagen. Furthermore, mammary gland reconstitution experiments can be performed in vivo by transplantation of isolated cells into the cleared mammary fad pad [8]. Transplanted cells respond appropriately to the hormonal environment and undergo ductal morphogenesis during puberty and estrous cycling, and alveologenesis during pregnancy.
Surprisingly, only a subset of the luminal cells express estrogen, progesterone and prolactin receptors, and a precise patterning of these receptors established after the onset of puberty is necessary for proper development [9, 10]. In fact, contrary to expectation, based upon studies in breast cancer cell lines, the vast majority of the estrogen receptor alpha (ERα)- and progesterone receptor (PR)-positive cells do not proliferate in mature ducts [11, 12]. Proliferative cells instead are in close proximity to steroid receptor-positive cells, suggestive of possible paracrine mediators, rather than the textbook model where steroid receptors translocate into the nucleus and directly regulate transcription of cyclin D1 and other genes involved in cell autonomous proliferation. Formal proof required the development of genetic knockout mice lacking ER and PR together with elegant chimeric transplantation experiments, where ERα– and PR–null cells were mixed together with wild-type cells. PR wild-type cells rescued PR-null cells, defective in alveologenesis [13], and ERα wild-type cells restored ductal morphogenesis of ERα-null epithelium [14]. These genetic chimera approaches are analogous to classical experiments in the Drosophila eye used to define non-cell autonomous effects [15].
Ovarian hormones also play a major role in guiding mammary stem cell (MaSC) homeostasis within the basal compartment and can result in major changes in the stem cell pool during the reproductive cycle [16]. Evidence has suggested that in the mammary gland, as in the skin [17], there are multiple populations of stem cells [18–21] (see a detailed description in Review [22]) located in different sites and with different features, possibly due to cell plasticity induced by developmental cues, hormones and the environment [23, 24]. Isolation and characterization of MaSCs using cell surface markers and fluorescence-activated cell sorting (FACS) revealed that MaSCs lack ERα and PR, suggesting that they must respond to systemic hormones through the action of paracrine mediators [16, 25]. Furthermore, this implied that MaSCs must divide asymmetrically to ultimately give rise to steroid-receptor positive cells.
Independent of ovarian hormonal input and the expression of ER and PR, the embryonic mammary gland instead relies on local paracrine interactions between the mammary epithelial bud and underlying mesenchyme. Classic reconstitution experiments between epithelium and mesenchyme using specific mouse mutants have revealed the importance of key signaling nodes between these two compartments [26]. Several notable examples are the expression of fibroblast growth factor (FGF) 10 in the mesenchyme and its receptor FGFR2b in the epithelium [14] and reciprocally, parathyroid hormone related protein (PTHrP) in the epithelium and its receptor, PTHR1 in the mesenchyme. In the latter case, a mesenchymal, canonical Wnt pathway appears to mediate the PTHrP specification of the mesenchyme [27]. The signaling between the mesenchyme and epithelium in embryonic development is in many ways analogous to the stromal-epithelial crosstalk that occurs postnatally. Thus, every facet of mammary gland development requires an intricate spectrum of cellular interactions guided by both systemic and local cues.
Paracrine mediators of normal development
Gene expression studies performed first in PR-null and later in ERα-null mammary epithelial cells (MECs) in comparison to wild-type MECs identified several putative paracrine mediators involved in both MaSC self-renewal [16, 25] and hormone-induced proliferation [28]. These factors included amphiregulin (AREG) and epidermal growth factor ligand [29], the TNF-α family member RANKL, IGF-II and Wnt4 [30]. Several genetic studies have helped establish the functional importance of these mediators. For example, Wnt signaling plays a critical role in MaSC self-renewal [31]. Both Wnt4 and RANKL are induced by progesterone in luminal cells and regulate MaSC self-renewal [32]. R-spondin1 (Rspo1, see Glossary), a modifier of Wnt signaling, is also hormonally regulated in luminal epithelial cells by estrogen and progesterone, and acts in concert with Wnt4 to expand MaSCs [33, 34] (Figure 1a). Furthermore, RANK signaling amplifies Wnt-responsive mammary progenitors and stem cells through Rspo1 [32]. Thus, there appears to be a combined progesterone-regulated RANKL/Wnt signaling axis that is instrumental for the regulation of both proliferation and self-renewal within the mammary epithelium [35]. Interestingly, based upon elegant studies in embryonic stem cells, Wnt proteins appear to act locally to orient asymmetric stem cell division [36] (see Glossary). By inference these studies in normal development suggest that localized Wnt signaling might also play a critical role in the self-renewal of breast cancer stem cells (BCSCs).
Figure 1. Paracrine interactions in normal mammary gland development.
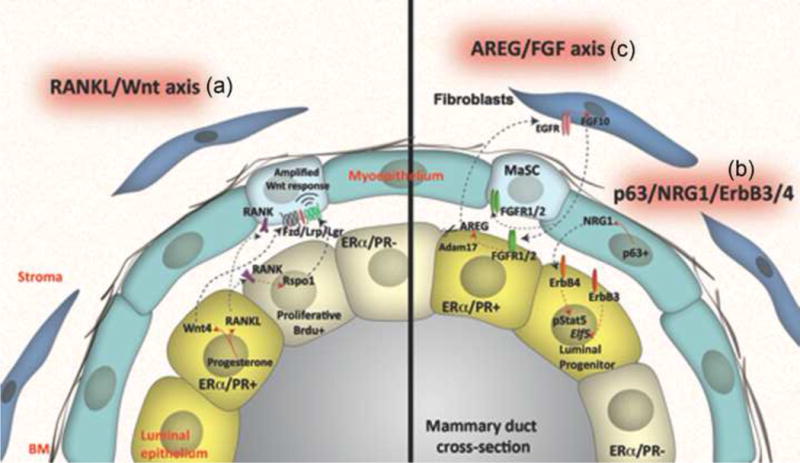
(a) R-spondin 1 (Rspo1) and Wingless-type MMTV integration site family, member 4 (Wnt4) secreted by the luminal epithelial cells upon estrogen and progesterone regulation are able to expand mammary stem cell (MaSC) and maintain their full developmental potential. Despite lacking of expression of estrogen receptor (ER) and progesterone receptor (PR), MaSCs respond to hormonal stimulations possibly through receptor activator of nuclear factor κB ligand (RANKL) mediated cell signaling. Similarly, the tumor necrosis factor (TNF)-α family member, RANKL, has also been shown to amplify Wnt-responsive mammary progenitors and stem cells through Rspo1. (b) Erb-B2 receptor tyrosine kinase 3 (ErbB3) expression is necessary for luminal epithelial differentiation and for maintaining the balance between luminal and basal epithelium. Neuregulin 1 (NRG1), one of the ErbB family ligands, has been reported to signal from basal p63 positive cells to luminal ErbB3 positive cells. The expression of basal p63 is critical for the development and lactation potential of the luminal epithelial cells. (c) Fibroblast growth factor receptor 1 (FGFR1) has been shown to induce the expression of epidermal growth factor receptor (EGFR) ligand amphiregulin (AREG) in luminal epithelial cells. In addition, FGFR1/R2-null cells were rescued in chimeric outgrowths containing wildtype mammary epithelial cells (MECs), indicating the maintaining of basal epithelial stem cell pool was partially depending on the paracrine feedback loop among factors of FGF, FGFR, EGFR and AREG. Modified with permission from [34].
Several ErbB family ligands also play a role in paracrine signaling. For example, neuregulin 1 (NRG1) has been shown to signal from basal p63 positive cells to luminal ErbB4 positive cells (Figure 1b). Genetic deletion of p63 leads to dramatic defects in luminal cell proliferation and differentiation, ultimately resulting in lactation failure [37]. Furthermore, an additional ErbB receptor, ErbB3, is necessary for luminal epithelial differentiation and to maintain the balance between luminal and basal epithelium [38]. AREG has been shown to be an essential mediator of ERα function in pubertal mice for ductal morphogenesis, including epithelial proliferation and terminal end bud formation [29]. Intriguingly, earlier studies suggested that the epidermal growth factor receptor (EGFR) in the stroma was required to mediate the effects of AREG on ductal morphogenesis, perhaps via induction of reciprocal FGF10 signaling from the stroma back to the epithelium [39, 40] (Figure 1c). These reconstitution experiments do not rule out, however, possible direct effects of EGFR on epithelial cells [29]. FGFR1 has also been shown to induce AREG expression in luminal epithelial cells, suggesting the possibility of a positive feedback loop [41]. This may help explain the rescue of FGFR1 and FGFR2 null cells by surrounding wild-type cells in chimeric mammary outgrowths, presumably due to AREG acting through EGFR [42]. These studies in normal mammary gland development have been informative for designing therapeutic interventions in breast cancer, as combinatorial targeting of FGF and ErbB receptors has been shown to inhibit both growth and metastasis in some breast cancer models [43]. In addition, they highlight the importance of critical paracrine signaling pathways that when disrupted in breast cancer progression due presumably to loss of critical feedback mechanisms may lead to uncontrolled growth, invasion and metastasis. A number of these pathways, e.g. those driven by Wnt, Il-6R, RANKL, FGFR etc. are now active therapeutic targets of ongoing and presumably future clinical trials [44].
Heterogeneity in breast cancer: inter- and intratumoral heterogeneity
More than a decade ago, Perou and colleagues performed groundbreaking studies of gene expression profiling using cDNA microarrays, representing 8,102 human genes on 65 human breast tumor samples [45]. An intrinsic gene subset of 496 genes demonstrating significantly greater variation between individual tumors than between paired samples from the same tumor was further used to cluster tumors into different subtypes with distinct molecular features. These subtypes include luminal-like, basal-like, ErbB2/HER2+ and normal breast. Similar hierarchical clustering studies with a more extended sample size confirmed the original studies and were able to further separate luminal tumors into luminal A and B based upon the expression of ER-regulated genes and prognostic factors [46–48]. More recent studies suggest the existence of even a greater diversity of subtypes with as many as 10 integrative clusters each associated with distinct clinical outcomes [49, 50].
Such intertumoral heterogeneity can be correlated with specific genetic alterations within individual tumor subtypes. For example, selected somatic mutations have been observed to drive the tumor heterogeneity of luminal tumors defined by prediction analysis of microarray 50 (PAM50), a 50 gene set used to classify breast cancer subtypes [51]. Similarly, TP53 mutations were associated with high-grade histology and high proliferation rates of the luminal B subtype, whereas loss-of-function mutations in MAP3K1 were associated with luminal A status of low proliferation rates and low-grade histology [52].
Although ITH has been observed by breast cancer pathologists for years, only recently has this been examined in primary breast cancers by next-generation sequencing (NGS)-analysis [53] (see Glossary). Cancer-derived DNAs extracted from many tumor cells of each of 21 individual breast cancers were assessed to identify copy number variations and gene mutations using NGS [4, 54]. Both clonal mutations (e.g. mutations in TP53 and PIK3CA), which occurred in all tumor cells, and subclonal mutations, which only occurred in a subset of tumor cells, were observed. The number of subclonal mutations was much greater than the number of clonal mutations, suggestive of an early complex subclone expansion process within this collection of breast cancers. A dominant clone was found in 50 to 95% of the tumor cells in all tumors studied, and expansion of this dominant clone presumably triggered the diagnosis. Shah et al [55] also investigated the clonal heterogeneity of basal-like triple negative breast cancer (TNBC) using similar NGS mutation profiling methods. They demonstrated that these tumors showed a wide variation in their clonal frequencies. Mutations in TP53 and PIK3CA identified in all samples from the study of 21 individual breast cancers [4, 54] were only present in most samples in the largest clone of TNBC, but not present in all tumor cells, indicating that a more complex early clonal expansion occurred in some of the TNBC.
Definition of the luminal ER subtype of breast tumors requires that only 1% of the tumor cells need to be classified as ERα-positive, and furthermore ER mutations need not be present in all cells of endocrine-resistant tumors. In addition, variations in DNA copy number from distinct cell populations in close proximity within a tumor may be more significant than copy numbers detected among different tumors [56]. Therefore, analyzing the genomic feature of single cells, if possible without amplification, may provide more detailed insights into ITH.
With the advancement of the isolation methods and analytical techniques to analyze both RNA and DNA at the single cell level, genomic, epigenetic, and transcriptomic heterogeneity within an organism can be investigated to decipher the full extent of ITH [57]. For example, three distinct clonal subpopulations were identified when 100 single cells from a polygenomic breast tumor were analyzed following FACS, whole genome amplification and NGS. Analysis of 100 single cells from another monogenomic primary tumor and its liver metastasis revealed that a single clonal expansion in the primary tumor appeared to be responsible for liver metastasis [58]. Furthermore, studies using a barcoding system to track individual clones showed that the majority of resistant clones after cancer therapies were pre-existing, indicating that a combinational therapeutic pre-treatment targeting non-overlapping resistance may be an effective approach [59]. Various barcoding systems to label and trace individual subpopulations of cells are also ideal tools for studying the earliest stages of tumorigenesis and the clonal contribution to tumor progression[60, 61].
Polyak and colleagues analyzed the copy number variations in both stem-like cells and more differentiated cells based on the expression of cell surface markers (CD24, CD44, HER2, etc.) and uncovered a high level of genetic heterogeneity and dynamic conversion between different cancer cell populations [62]. Additional studies also revealed that the extent of diversity was correlated with breast tumor subtypes, and the individual populations of cells with different invasive potential were observed [62]. These observations supported not only the existence of ITH, but also its importance as a factor to be considered in a tumor’s initial response to treatment and for selecting drug regimens for effective treatment.
The origins of ITH: clonal evolution vs. CSC models
Tumorigenesis is a multistep process involving acquired genetic and epigenetic alterations that generate individual cell populations with aberrant differentiation and proliferation potential, leading to ITH [6, 63, 64]. It is generally accepted that a branched Darwinian evolution integrated with a CSC hierarchy that may include more than a single CSC population may help explain ITH [1, 65, 66] (see Glossary).
The clonal evolution model suggests that a single clone with accumulated malignant genetic and epigenetic hereditary changes may outcompete other clones with different mutations and initiate carcinogenesis [5]. The co-existence of multiple clones of different genotypes, phenotypes, therapeutic sensitivities, as well as the potential to metastasize, all contribute to ITH. Readers are referred to several recent reviews by Swanton and colleagues [67, 68] for a more in depth discussion of tumor evolution and its impact on ITH.
In normal tissues, multipotent stem cells are capable of self-renewal and give rise to hierarchical cell populations through sequential cell divisions, consisting of daughter stem cells and other cells with varying differentiation potential. When a tissue stem cell or progenitor undergoes oncogenic mutations or loss of tumor suppressors, it can give rise to a tumor initiating CSC, which further produces the heterogeneity of cells that also bear these genetic alterations [69–72]. The CSC hierarchical model has been validated in a number of specific solid cancers in addition to acute myelogenous leukemia [73], and may account at least in part for the observed ITH that appears to be a property of many cancers, including breast cancer [74]. The clonal evolution and CSC models are not mutually exclusive. Malignant transformation may occur in both normal multipotent stem cells as well as more differentiated progenitors through clonal evolution, which then results in the existence of multiple cell lineages [5, 72, 75]. Accumulating evidence, both in experimental models and in the clinic, suggests that CSCs with intrinsic resistance to radiation and chemotherapy may be responsible for tumor recurrence and metastasis [76–80]. A better understanding of the functional interactions among such a diverse network of tumor cells, especially in the metastatic setting, will be important for the development of improved combinatorial therapies designed to prevent relapse and to ultimately decrease mortality.
Signaling and potential interactions among various subclones in tumors
The genetic and epigenetic mechanisms for subclonal diversity and functional interactions among various tumor cell clones remain unclear. There are a few examples of non-cell autonomous interactions in normal mammary gland development, many of which are involved in the breast cancer. Mice that expressed a constitutively activated Smoothened (SMO), the primary effector of Hedgehog (Hh) signaling, showed paracrine stimulation of mammary epithelial proliferation [81]. While the nature of the paracrine effectors has not been elucidated, SMO signaling appears to be mediated through G proteins, specifically the pertussis toxin sensitive Gαi class [82]. Furthermore, although some of these interactions may involve diffusible morphogens, others may be dependent on short-range local interactions.
The lessons learned from the studies in normal development have helped provide a foundation for deciphering the complex interactions that exist in the breast cancer ecosystem. Like those required for normal development, the effects of estrogen on BCSCs also appear to be regulated by paracrine mediators and local interactions. For example, the Notch ligands Jagged and Delta in the mouse mammary gland can signal from basal cells to the Notch receptors on luminal cells, and these local interactions play an important role in mammary gland development [83]. Accordingly, estrogen can increase the activity of ERα-negative BCSCs through both paracrine EGFR and Notch signaling [84] (Figure 2). Analogous to studies in embryonic mammary gland development, estrogen has also been shown to regulate BCSCs through paracrine FGF/TBX3 signaling [85]. The NF-κB signaling in the non-CSC population of cells was shown to regulate self-renewal of the CSC through jagged-1 mediated Notch signaling activation in CSCs in TNBC cell lines [86]. In addition, Hh signaling is required for stem cell maintenance and tissue homeostasis and aberrant expression has been observed in breast cancer. A model of progesterone and Wnt4, downstream of PR signaling, in promoting tumorigenesis by expanding both the mutated luminal progenitor and stem cell compartments has also been proposed (Figure 2).
Figure 2. Potential tumor cell-cell interactions in mammary tumors.
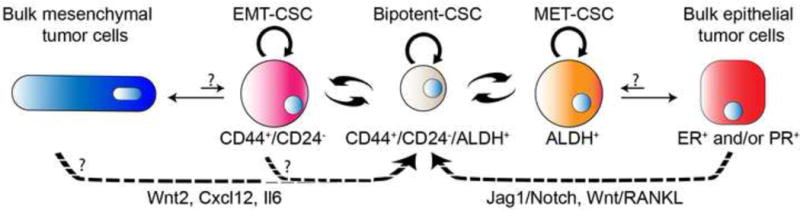
The tumorigenic CSCs are cells that can initiate a tumor through both self-renewal to generate more CSCs and differentiation to produce the bulk of tumor cells with less tumorigenic potential. More than one single population of CSC may exist within a tumor. Both CD44+/CD24− and aldehyde dehydrogenase positive (ALDH+) cells have been shown to be more tumorigenic than the non-CD44+CD24− and ALDH – cells, respectively. Conversion between the bipotent CD44+CD24−/ALDH+ cells and the CD44+/CD24− cells with epithelial-mesenchymal transition (EMT) features, and the ALDH+ cells with mesenchymal-epithelial transition (MET) features are illustrated, but at present are hypothetical and will need to be validated by lineage tracing. Similar to the signaling between the various cell types in the normal mammary gland development, WNT ligands, RANKL, Jagged1/Notch signaling and interleukin (IL) family members that are produced by the non-CSC tumor cells can act on the receptors of the CSCs, and regulate CSC activities through a positive feedback loop. Modified with permission from [34].
Only a few studies to date have been reported that demonstrate the importance of functional ITH. Gunther and colleagues [87] reported that both the basal and luminal cells were required for efficient tumor formation in their mouse mammary tumor virus (MMTV)-driven Wnt1 transgenic mouse model, which was dependent on Wnt1 expression in luminal cells. Polyak and colleagues [7] recently demonstrated that competition triggered clonal expansion within tumors derived from transplanting various artificially engineered clones of weakly tumorigenic MDA-MB-468 cells. These clonal populations were engineered to individually overexpress secreted factors that have been implicated in tumor progression, such as CCL5, IL11, and VEGFB. Interestingly, a minor cell population was capable of facilitating tumor growth by overpowering environmental constraints to allow the rapid proliferation of all tumor cells in a non-cell autonomous manner. During tumor development, heterogeneous clones of tumor cells remain dynamic and are highly competitive. Cross-talk between subpopulations of tumor cells has also been reported in metastasis [88]. Similarly, a minor subclone within glioblastomas was able to drive tumorigenesis and maintain tumor heterogeneity by accelerating tumor growth [89].
Using a p53-null mouse model, our group described a novel feedback loop in which the breast CSC functions through the action of paracrine mediators secreted by the CSC progeny [90], similar to what is observed in the normal mammary gland [16, 25–27]. We demonstrated the existence of cross-talk between tumor cells via Wnt2, Cxcl12 and IL6 signaling, indicating the non-cell-autonomous properties and importance of cooperativity between tumor subpopulations in tumor initiation and progression. The progeny of CSCs that display mesenchymal features produced through the asymmetric division of CSCs were both less proliferative and tumorigenic as compared to the CSCs. However, the functional interactions between CSCs and this subset of CSCs with mesenchymal features may provide an improved niche microenvironment, especially for those tumors initiated from extremely low numbers of the CSCs. These studies indicate that similar regulatory mechanisms may exist for the maintenance of both normal and tumor stem cells with respect to their symmetric and asymmetric division. However, unique mutations within the CSCs may cause them to increase the percentage of symmetric divisions to alter both their frequency and population dynamics. Other studies have shown that the CSCs may dynamically switch from differentiated tumor cells [44, 91].
Due to limitations on studies of tumor initiation and progression in humans, findings from mouse tumors and human patient derived xenograft (PDX) models have a high relevance for human breast cancer. PDX models developed by serial transplantation into the orthotopic site, the mammary fat pad, in immunodeficient mice have been shown to closely recapitulate the molecular features of human tumors. Using whole genome sequencing and reverse phase protein arrays, Li et al [92] thoroughly characterized 13 PDX lines along with their advanced primary breast tumors. These studies showed that while PDXs have relatively stable genomes without a significant accumulation of DNA structural rearrangements, minor mutant clones are retained in PDXs during multiple transplants, indicating the possibility of cooperation of clones during tumor evolution.
Using deep-genome and single-cell sequencing methods, Eirew and colleagues [93] analyzed DNAs from PDX lines as well as their matched patient samples to identify any variations in clonal dynamics between patients samples, and initial as well as subsequent engraftments at the single-cell level. Varying degrees of clonal selection on engraftment were observed in all 10 primary and 5 metastatic breast tumors, ranging from rare clone (<5% of starting population) to moderate, polyclonal engraftment. This clonal heterogeneity was conserved in PDX models with some drift. These investigators suggested that the reproducible clonal dynamics were due to both selective pre-existing clones and variations in clonal fitness.
Concluding Remarks
Tremendous technical advances have helped decipher the biological impact of ITH in cancer evolution, treatment response and resistance, and metastasis. These advances, especially the utilization of genomic analyses at the single cell level, should provide both biologically and clinically relevant information in the near future. Of particular importance will be the comparison of primary tumors and paired metastases, since more than 90% of patients succumb to metastatic disease. Future progress involving combinatorial therapies will require a better understanding of ITH. For example, whether the cells that display mesenchymal features and CSCs represent cells of different states as well as their relative spatial distribution remains to be determined. Similarly, the likely epigenetic determinants that regulate the equilibrium between multiple clones remain largely unknown. Monitoring multiple clone dynamics in PDX models would be a valuable tool in deciphering both their drug response and cancer biology [93]. However, whether functional studies can be performed in PDX models in both the primary and metastatic settings in the absence of the innate immune system needs to be carefully characterized (see Outstanding Questions Box).
Trends Box.
Mammary gland stem cell self-renewal is regulated by paracrine signaling involving Wnt4 and RSPO1 secreted by differentiated mammary luminal cells under the influence of systemic steroid hormones.
Members of the Wnt signaling pathway as well as a number of other secreted cytokines are involved in the communication between CSCs and their progeny and influence their tumorigenicity.
Intratumoral genetic and epigenetic heterogeneity and cooperativity between tumor subpopulations have important biological and clinical implications.
While PDXs have relatively stable genomes during tumor evolution, minor mutant clones are retained, indicating the possibility of cooperation of clones during serial transplantation.
Outstanding Questions Box.
What are the most likely epigenetic determinants that regulate the equilibrium between CSCs and their progeny?
What is the extent of plasticity among tumor subpopulations and how does this influence treatment response?
What is the spatial distribution of different clonal subpopulations in breast cancer?
What new combinatorial approaches and treatment paradigms will be required to inhibit cooperativity among tumor subpopulations?
Can functional studies be performed in PDX models in both the primary and metastatic settings in the absence of the innate immune system?
Glossary Box.
Asymmetric stem cell division, the ability to generate additional stem cells, i.e. self-renewal, as well as a more differentiated progeny.
Cancer stem cell (CSC) model of intratumoral heterogeneity, a tissue stem cell or progenitor undergoes oncogenic mutations or loss of tumor suppressors, thus giving rise to a tumor initiating CSC, which further produces the heterogeneity of cells that also bear these genetic alterations.
Clonal model of intratumoral heterogeneity, a single clone with accumulated malignant genetic and epigenetic hereditary changes may outcompete other clones with different mutations, leading to heterogeneity in tumors. The clonal and CSC models are not mutually exclusive in cancers that follow a stem cell model.
Intratumoral heterogeneity, the variability among tumor cells within a single tumor.
Next-generation sequencing, is a cheaper and faster sequencing method, as compared to first-generation Sanger sequencing, that performs massively parallel sequencing on millions of fragments of (c)DNA so that to determine the exact order of nucleotides in a given DNA or RNA molecule.
RSPO1, R-spondin 1, one of a family of small secreted proteins the enhance Wnt/ß-catenin signaling through interaction with the Lgr family of seven transmembrane receptors.
Wnt4, one of the families of Wnt ligands regulated by progesterone in luminal mammary epithelial cells.
Acknowledgments
We apologize to those authors whose work we were unable to cite due to space limitations. We would like to especially thank Dr. Kevin Roarty for his critical comments and help with the graphics. This work was supported by NIH/NCI Pathway to Independence (PI) Award CA142898 (to M. Zhang); and CA016303 (to J.M. Rosen).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easwaran H, et al. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marusyk A, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deome KB, et al. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer research. 1959;19:515–520. [PubMed] [Google Scholar]
- 9.Seagroves TN, et al. C/EBPbeta (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Molecular endocrinology. 2000;14:359–368. doi: 10.1210/mend.14.3.0434. [DOI] [PubMed] [Google Scholar]
- 10.Rosen JM. On hormone action in the mammary gland. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a013086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke RB, et al. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer research. 1997;57:4987–4991. [PubMed] [Google Scholar]
- 12.Russo J, et al. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast cancer research and treatment. 1999;53:217–227. doi: 10.1023/a:1006186719322. [DOI] [PubMed] [Google Scholar]
- 13.Brisken C, et al. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5076–5081. doi: 10.1073/pnas.95.9.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailleux AA, et al. Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development. 2002;129:53–60. doi: 10.1242/dev.129.1.53. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds-Kenneally J, Mlodzik M. Notch signaling controls proliferation through cell-autonomous and non-autonomous mechanisms in the Drosophila eye. Developmental biology. 2005;285:38–48. doi: 10.1016/j.ydbio.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 16.Joshi PA, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–807. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spike BT, et al. A mammary stem cell population identified and characterized in late embryogenesis reveals similarities to human breast cancer. Cell stem cell. 2012;10:183–197. doi: 10.1016/j.stem.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai L, Rohrschneider LR. s-SHIP promoter expression marks activated stem cells in developing mouse mammary tissue. Genes & development. 2010;24:1882–1892. doi: 10.1101/gad.1932810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, et al. Identification of multipotent mammary stem cells by protein C receptor expression. Nature. 2015;517:81–84. doi: 10.1038/nature13851. [DOI] [PubMed] [Google Scholar]
- 21.dos Santos CO, et al. Molecular hierarchy of mammary differentiation yields refined markers of mammary stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7123–7130. doi: 10.1073/pnas.1303919110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sreekumar A, et al. The mammary stem cell hierarchy: a looking glass into heterogeneous breast cancer landscapes. Endocrine-related cancer. 2015 doi: 10.1530/ERC-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prater MD, et al. Mammary stem cells have myoepithelial cell properties. Nature cell biology. 2014;16:942–950. 941–947. doi: 10.1038/ncb3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asselin-Labat ML, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 26.Robinson GW. Cooperation of signalling pathways in embryonic mammary gland development. Nature reviews. Genetics. 2007;8:963–972. doi: 10.1038/nrg2227. [DOI] [PubMed] [Google Scholar]
- 27.Hiremath M, et al. Parathyroid hormone-related protein activates Wnt signaling to specify the embryonic mammary mesenchyme. Development. 2012;139:4239–4249. doi: 10.1242/dev.080671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beleut M, et al. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2989–2994. doi: 10.1073/pnas.0915148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciarloni L, et al. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5455–5460. doi: 10.1073/pnas.0611647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brisken C, O’Malley B. Hormone action in the mammary gland. Cold Spring Harbor perspectives in biology. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell stem cell. 2010;6:568–577. doi: 10.1016/j.stem.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi PA, et al. RANK Signaling Amplifies WNT-Responsive Mammary Progenitors through R-SPONDIN1. Stem cell reports. 2015;5:31–44. doi: 10.1016/j.stemcr.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai C, et al. R-spondin1 is a novel hormone mediator for mammary stem cell self-renewal. Genes & development. 2014;28:2205–2218. doi: 10.1101/gad.245142.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosen JM, Roarty K. Paracrine signaling in mammary gland development: what can we learn about intratumoral heterogeneity? Breast cancer research : BCR. 2014;16:202. doi: 10.1186/bcr3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajaram RD, et al. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. The EMBO journal. 2015;34:641–652. doi: 10.15252/embj.201490434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Habib SJ, et al. A localized Wnt signal orients asymmetric stem cell division in vitro. Science. 2013;339:1445–1448. doi: 10.1126/science.1231077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forster N, et al. Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Developmental cell. 2014;28:147–160. doi: 10.1016/j.devcel.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balko JM, et al. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:221–226. doi: 10.1073/pnas.1115802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, et al. TGF-beta2 inhibits AKT activation and FGF-2-induced corneal endothelial cell proliferation. Experimental cell research. 2006;312:3631–3640. doi: 10.1016/j.yexcr.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Sternlicht MD, et al. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bade LK, et al. Mammary tumorigenesis induced by fibroblast growth factor receptor 1 requires activation of the epidermal growth factor receptor. Journal of cell science. 2011;124:3106–3117. doi: 10.1242/jcs.082651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pond AC, et al. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem cells. 2013;31:178–189. doi: 10.1002/stem.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Issa A, et al. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast cancer research : BCR. 2013;15:R8. doi: 10.1186/bcr3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brooks MD, Wicha MS. Tumor twitter: cellular communication in the breast cancer stem cell niche. Cancer discovery. 2015;5:469–471. doi: 10.1158/2159-8290.CD-15-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 46.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorlie T, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldhirsch A, et al. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2011;22:1736–1747. doi: 10.1093/annonc/mdr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dawson SJ, et al. A new genome-driven integrated classification of breast cancer and its implications. The EMBO journal. 2013;32:617–628. doi: 10.1038/emboj.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ali HR, et al. Genome-driven integrated classification of breast cancer validated in over 7,500 samples. Genome biology. 2014;15:431. doi: 10.1186/s13059-014-0431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellis MJ, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desmedt C, et al. Next-generation sequencing in breast cancer: first take home messages. Current opinion in oncology. 2012;24:597–604. doi: 10.1097/CCO.0b013e328359554e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nik-Zainal S, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah SP, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez P, et al. Parallel evolution of tumour subclones mimics diversity between tumours. The Journal of pathology. 2013;230:356–364. doi: 10.1002/path.4214. [DOI] [PubMed] [Google Scholar]
- 57.Macaulay IC, Voet T. Single cell genomics: advances and future perspectives. PLoS genetics. 2014;10:e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhang HE, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nature medicine. 2015;21:440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen LV, et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nature communications. 2014;5:5871. doi: 10.1038/ncomms6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nolan-Stevaux O, et al. Measurement of Cancer Cell Growth Heterogeneity through Lentiviral Barcoding Identifies Clonal Dominance as a Characteristic of Tumor Engraftment. PloS one. 2013;8:e67316. doi: 10.1371/journal.pone.0067316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park SY, et al. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. The Journal of clinical investigation. 2010;120:636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burrell RA, et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 64.Martelotto LG, et al. Breast cancer intra-tumor heterogeneity. Breast cancer research : BCR. 2014;16:210. doi: 10.1186/bcr3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merlo LM, et al. Cancer as an evolutionary and ecological process. Nature reviews. Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 66.Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell cycle. 2007;6:2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]
- 67.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 68.Yap TA, et al. Intratumor heterogeneity: seeing the wood for the trees. Science translational medicine. 2012;4:127ps110. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 69.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reya T, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 71.Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes & development. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. BioEssays : news and reviews in molecular, cellular and developmental biology. 2013;35:253–260. doi: 10.1002/bies.201200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 74.Al-Hajj M, et al. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature reviews. Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 76.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 77.Atkinson RL, et al. Thermal enhancement with optically activated gold nanoshells sensitizes breast cancer stem cells to radiation therapy. Science translational medicine. 2010;2:55ra79. doi: 10.1126/scitranslmed.3001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang M, et al. Selective targeting of radiation-resistant tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3522–3527. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rich JN. Cancer stem cells in radiation resistance. Cancer research. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 80.Rich JN, Bao S. Chemotherapy and cancer stem cells. Cell stem cell. 2007;1:353–355. doi: 10.1016/j.stem.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 81.Visbal AP, et al. Altered differentiation and paracrine stimulation of mammary epithelial cell proliferation by conditionally activated Smoothened. Developmental biology. 2011;352:116–127. doi: 10.1016/j.ydbio.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Villanueva H, et al. Science Signaling. in press. [Google Scholar]
- 83.Raafat A, et al. Expression of Notch receptors, ligands, and target genes during development of the mouse mammary gland. Journal of cellular physiology. 2011;226:1940–1952. doi: 10.1002/jcp.22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harrison H, et al. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast cancer research : BCR. 2013;15:R21. doi: 10.1186/bcr3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fillmore CM, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto M, et al. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nature communications. 2013;4:2299. doi: 10.1038/ncomms3299. [DOI] [PubMed] [Google Scholar]
- 87.Cleary AS, et al. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–117. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Calbo J, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer cell. 2011;19:244–256. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 89.Inda MM, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes & development. 2010;24:1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang M, et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer discovery. 2015;5:520–533. doi: 10.1158/2159-8290.CD-14-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Klevebring D, et al. Sequencing of breast cancer stem cell populations indicates a dynamic conversion between differentiation states in vivo. Breast cancer research : BCR. 2014;16:R72. doi: 10.1186/bcr3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li S, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eirew P, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]